Chemistry and biology of resorcylic acid lactones
Nicolas
Winssinger
* and
Sofia
Barluenga
Organic and Bioorganic Chemistry Laboratory, Institut de Science et Ingénierie Supramoléculaires, Université Louis Pasteur, 8 allée Gaspard Monge, 67000, Strasbourg, France. E-mail: winssinger@isis-u.strasbg.fr; Fax: +33 (0)3 89 24 51 12; Tel: +33 (0)3 89 24 51 13
First published on 25th September 2006
Abstract
While resorcylic acid lactones (RALs) have been known for a long time, the more recent discoveries that radicicol is a potent and selective HSP90 inhibitor while other members such as hypothemycin, LL-Z1640-2 and LL-783,277 are potent kinase inhibitorshave stimulated a renewed interest in this family of natural products. The recent developments regarding the chemistry and biology of RALs are reviewed.
 Nicolas Winssinger | Nicolas Winssinger was born in Belgium. He received his BSc from Tufts university and then joined the group of KC Nicolaou at the Scripps Research Institute for his PhD. He then remained at Scripps as an NIH postdoctoral fellow in the group of Peter G. Schultz. In 2002 he moved to the Institut de Science et Ingénierie Supramoléculaires, université Louis Pasteur in Strasbourg as an assistant professor and was promoted to full professor in 2005. His current interests lie in chemical biology, total and combinatorial synthesis, oligosaccharide chemistry and templated reactions. |
 Sofia Barluenga | Sofia Barluenga was born in Mulheim Ruhr, Germany. She received her BSc and PhD from the university of Oviedo where she worked under the guidance of F. Aznar and J. Barluenga. After a postdoctoral training in the group of K. C. Nicolaou, she joined Anadys pharmaceuticals in 2001. She then moved to the Institut de Science et Ingénierie Supramoléculaires at the Université Louis Pasteur in Strasbourg where she currently holds a position as a CNRS researcher (CR1). Her interests include organometallic chemistry, natural product synthesis as well as diversity-oriented synthesis and their application to chemical biology. |
Introduction
Resorcylic acid lactones (RALs) have been known for decades with the first isolation of radicicol (monorden) in 19531 followed by zearalenone in 1962,2LL-Z1640-2 in 19783 and hypothemycin in 1980.4 Aside from zearalenone which was shown to have estrogen agonistic properties, the original biological activity ascribed to radicicol and other RALs did not solicit much interest from the organic chemistry community. It should also be noted that the original structures proposed for radicicol and hypothemycin were erroneous. In the early 1990s, reports of inhibition by radicicol of a kinase revived interest in this molecule. A few years later, several publications appeared regarding antiproliferative activities of hypothemycin along with reports of several selective kinase inhibitorsamongst the RALs.The prevalence of polyketide synthases (PKS) responsible for RAL biosynthesis amongst fungi has led to the reisolation of the same RAL products from different fungal strains. Radicicol was first isolated from Monocillium nordinii and named initially monorden.1 The same molecule was independently isolated from Nectria radicicola and given the name of radicicol.5 As the original structure of monorden was incorrect, the name radicicol has prevailed. Likewise, hypothemycin (Fig. 1) was originally isolated4 from Hypomyces tricothecoides and reisolated from lignicolous mangrove fungus Aigialus parvus along with new RALs.6 More recently, radicicol and some of its derivatives were also found to be synthesized by fungi associated with a Sonoran desert plant.7 Improved analytical techniques in combination with modified fermentation conditions will undoubtedly lead to the isolation of new RALs further extending the diversity of this important class of natural products.
 | ||
| Fig. 1 Selected members of the resorcylic acid lactone family of natural products. | ||
Biosynthesis of RALs
Resorcylic acid lactones (RALs) are mycotoxins produced by a variety of different fungal strains viapolyketide biosynthesis (Fig. 2). The type I fungal polyketide synthases (PKSs) involved in RAL biosynthesis are large multidomain enzymes that iteratively catalyze the condensation of nine units of thioacetates or malonates. Different modules can further process the product of each condensation to reduce or dehydrate the β-ketone. The proposed biosynthesis of zearalenone involves two PKSs, the first one is responsible for the assembly of the first five acetate units with the further processing to arrive at the adequate oxidation state at each carbon, while the second one performs the remaining three rounds of condensation without carbonyl reduction. The unreduced ketones are highly reactive and engage in a cyclisation/aromatization. The lactone is then released via a cyclisation module on the second PKS.8 Different combinatorial arrangements of the modules involved in the processing of the β-ketones in the first five condensations can account for the diversity of functionality present around the RAL macrocycles. | ||
| Fig. 2 Biosynthesis of resorcylic acid lactones. The red carbons represent the two-carbon units that are added in each reiterative condensation. | ||
Biological activity
Zearalenone was isolated from moulded corn fed to swine which led to vaginal eversion in female animals and growth of mammary glands in both males and females.2 It was later shown that the estrogen agonistic properties of zearalenone were the product of a direct interaction on the estrogen receptor in competition with 17-estradiol9 and that the macrocycle of zearalenone was able to adopt a conformation which mimics the one of the steroid.10 The anabolic properties of zearalenone and its derivatives have been used as a bovine growth stimulant and have been extensively reviewed.Radicicol (monorden, Fig. 1) was originally reported to have a mild sedative activity along with moderate antibiotic activity.11 In 1992, Kwon et al. reported that radicicol reverse the Src-transformed morphology of fibroblast and attributed this effect to the inhibition of the oncogenic kinase Src.12,13 It was later revealed that radicicol is in fact a potent and selective inhibitor of HSP90,14,15 a molecular chaperone responsible for the maturation and stability of a number of oncogenic proteins including Src. In the absence of HSP90 chaperoning activity, clients of HSP90 are unfunctional and targeted for degradation by the proteosome. Despite the fairly ubiquitous role of chaperones, clients of HSP90 include proteins that are involved in the tumourgenesis (growth factors, angiogenesis, metastasis and apoptosis evasion) rather than tumour suppression and, HSP90 is believed to be in an activated form in cancer cells.16 These two features have brought HSP90 inhibition on the front stage of new chemotherapeutic development.17,18 Pearl and co-workers solved the cocrystal structure of radicicol bound to HSP90 and showed that despite the lack of structural similarity between radicicol and ATP, radicicol was a competitive ligand for the ATP binding site of HSP90.19 It is important to note that although radicicol has two different reactive sites (Michael acceptor and epoxide), it does not react covalently with HSP90.
While radicicol does not have any notable kinase inhibitory activity, several RALs containing a cis-enone (hypothemycin, LL-Z1640-2 and LL-783,277, Fig. 1) have been reported to inhibit irreversibly mitogen activated protein kinases (MAP kinases) and be competitive with ATP (i.e. as radicicol, these compounds target the ATP-binding pocket of kinases). Inhibitors of MAP kinases are particularly interesting as the MAP kinases relay, amplify and integrate signals from a variety of extracellular stimuli thereby regulating a cell's response to its environment. The fidelity and amplitude of the signal is controlled by a phosphorelay system composed of three sequentially activated kinases. In a generic fashion, a stimulus turns on the activator which phosphorylates the first kinase (MAPKKK) which then phosphorylates the second kinase (MAPKK), which in turn phosphorylates the third kinase (MAPK) that finally phosphorylates a cytosolic protein or transcription factor.20 In mammalian organisms, at least three subfamillies of MAP kinases have been identified which include the extracellular signal-regulated kinases(ERK); the c-JUN NH2-terminal kinases(JNK); and the p38 enzymes. There are at least seventeen MAPKKKs, seven MAPKKs and twelve MAPKs. The specificity of these cascades is also regulated by scaffolding proteins which specifically organize and localize these kinase cascades to provide a unique combinatorial arrangement and down stream signal to a given stimuli.21 The importance of MAP kinases in regulating cellular response to stimuli and translating such environmental cues into gene expression, cell growth and apoptosis has made the MAP kinases primary targets in drug discovery. Aside from their potential therapeutic value, MAP kinase inhibitors are also important to identify and dissect the function of individual MAP kinases in these complex networks.
The first RAL reported to inhibit a kinase was radicicol A (F87-25909.04, Fig. 1) which was identified from a screen for inhibition of IL1β activity.22 Investigation in the mode of action of radicicol A revealed that it accelerated the degradation of specific mRNA sequences containing AU-rich elements (AREs).22,23 This effect did not come from a disruption of direct protein/RNA interaction or on the state of phosphorylation of protein/RNA complex at the ARE. It was found that radicicol A did inhibit tyrosine phosphorylation of several proteins and it was proposed that the phosphorylation level of the substrates of radicicol A's target affect their interaction with proteins that bind mRNA. The authors noted that although a number of radicicol analogues were found to inhibit Il1β secretion most of them had no effect on mRNA stability suggesting that structural differences in the radicicol family are able to change the mode of action and presumably the target of inhibition. In light of the activity of closely related cis-enone RAL (vide infra), it can be speculated that radicicol A is an inhibitor of mitogen-activated protein kinases (MAPKs).
LL-Z1640-2, another cis-enone RAL (Fig. 1), was first reported in 1978 however the authors noted that this compound was devoid of anabolic and estrogen-like activity and presented no particularly interesting activities.3 In 2003, it was rediscovered in a screen for TAK1 inhibition.24 Importantly, it was shown that radicicol and zearalenone had no appreciable activity in this assay while LL-Z1640-2 had an IC50 of 8.1 nM. The authors further showed this compound to be competitive with ATP and to irreversibly inhibit TAK1. TAK1 is a MAPKKK involved in the p38 signalling cascade for proinflammation signals such as cytokines. In evaluating the selectivity of this compound for TAK1, the authors showed this compound to be 50-fold less active against MEK1 (411 nM), another MAP kinase and having no inhibitory effect on other MAP kinase such as MEKK1, ASKN and MKK4. The authors also demonstrated LL-Z1640-2 to effectively prevent inflammation in an animal model (topical application).
LL-783,277, a third cis-enone RAL (Fig. 1), was found by researchers at Merck to be a potent and irreversible inhibitor of MEK1 (4 nM).25 The authors reported that hypothemcin (another cis-enone RAL, Fig. 1) was also an inhibitor of MEK (15 nM) and showed that the cis-enone was essential for their activities. It was later shown that this irreversible inhibition can be attributed to a Michael addition onto the cis-enone of a cysteine residue present in the ATP-binding pocket of a subset of kinases.26 A structure-bioinformatics analysis of the kinome revealed that 46 out of the 510 identified kinasescontain this cysteine residue. Amongst 16 tested kinasescontaining this conserved cysteine, hypothemycin had a Ki in the low nanomolar range for five kinases (MEK1, MEK2, FLT1, FLT3 and KDR), a high nanomolar Ki for one kinase (TRKB), low micromolar Ki for seven of them (ERK1, ERK2, PDGFRα and β, PKD1, MAPKAP5, TRKA and SRC) while it inhibited the remaining two kinases only weakly (GSK2α and β). The difference in a Ki clearly reflects the fact that while the ATP binding pockets are highly conserved and contain the required cysteine, hypothemycin is able to discriminate with some efficiency amongst the 16 tested kinases. These results provide a mechanism for earlier observations that hypothemycin is an inhibitor of the ras-signalling pathway27 and inhibits the production of several cytokines (IL2, Il6, Il10, IFNγ and TNFα).28Hypothemycin was found to inhibit several oncogenic cell lines dependent on kinase activation26 as well as inhibit tumour growth in animal models.27 As both MEK and TAK kinasesare part of the 46 kinasescontaining the suitably positioned cysteine, the irreversible inhibition previously observed can be rationalized by the same mechanism. While the target of radicicol A has not been defined, based on the structural similarity with hypothemycin, LL-783,277 and LL-Z1640-2, it can be speculated that it is too a MAP kinase inhibitor. Despite the fact that these four compounds are irreversible inhibitors, the detailed profile of kinase inhibition for hypothemycin clearly showed selectivity for certain cysteine-containing kinasesand it would not be surprising that the subtle difference in structure amongst these four compounds could lead to differences in selectivity. Clearly, the nature of the benzylic position (saturated, unsaturated or epoxidised) does affect the conformation of these different compounds and may affect their respective selectivities.
The pochonins were identified in a high throughput screen for inhibition of the herpes simplex virus (HSV) replication.29 While radicicol was a potent inhibitor in this assay, it was found to be cytotoxic at the concentration necessary for HSV inhibition. Although it can be speculated that the HSV inhibitory activity may originate from HSP90 inhibition, pochonin D (Fig. 1) which is an HSP90 inhibitor30 had no activity in the assay. On the other hand, pochonin C (Fig. 1) which is a poor HSP90 inhibitor retained activity in the HSV replication assay while being the least cytotoxic compound (90 µM). As many of the more substituted RALs, the pochonins were also found to be devoid of estrogenic activity. A library based on the pochonin D scaffold31 led to the identification of moderate kinase inhibitors(low µM) for Src, VEGFR, Aurora A and B. Although pochonin D is an HSP90 inhibitor, the analogues that showed kinase activitywere poor HSP90 inhibitors and vice versa.
Aigialomycins were recently discovered from a screen for anti-malarial activity.6 Both aigialomycin D (Fig. 1) and hypothemycin were found to have moderate antimalarial activity (low µM) and be cytotoxic at similar concentration, whereas other closely related aigialomycins were inactive in these assays. Synthetic aigialomycin D was reported to be devoid of HSP90 inhibition32,33 but to be a moderate kinase inhibitor (low µM) of CDK1 and CDK5 as well as GSK3 which could account for its cytotoxicity.33 Interestingly, it was not an inhibitor of the plasmodium analogue of GSK3.
The diverse biological activity of RALs is impressive considering the rather small changes of functional groups and stereochemistries amongst them. While these changes can account for the gain or loss of interactions, they can also have dramatic impacts on the conformation of these macrocycles. Although zearalenone and its derivatives are potent estrogen agonist, none of the more substituted RALs have been shown to have estrogenic activity. On the other hand, the fact that several RALs inhibit kinasesand ATPase raises the possibility that the RAL may be a good scaffold to discover new inhibitors in these important classes of enzymes.
Chemical synthesis
The widespread interest in zearalenone's anabolic properties stimulated a number of syntheses of this molecule with the first synthesis reported in 1968.34 It has also served as a testing ground for new cyclisation methodologies as exemplified by the development of the Corey–Nicolaou macrolactonization,35 Masemune's thioester-lactonization,36 and more recently the ring-closing metathesis.37 However, this review focuses on the synthesis of the more substituted resorcylides.Radicicol
The first synthesis of radicicol was reported by Lampilas and Lett in 199238 (Scheme 1) which aside from providing access to modified analogues served to confirm the absolute stereochemical assignment. Two challenging aspects in the synthesis of radicicol are the presence of a strained and thus sensitive allylic epoxide as well as the ketone functionality which is readily enolised at the benzylic position to yield an isocoumarin. Lampilas and Lett addressed both of these challenges by exploiting the isocoumarin to achieve a palladium-catalyzed (Stille) coupling (2 + 3 → 7) only to reveal the ketone and the conjugated diene functionality at a late stage in the synthesis. A Mitsunobu reaction was used very efficiently for the macrocyclisation (71% yield) which led to the important observation that such reactions proceed more efficiently if the ortho-phenol is unprotected. While the final MOM elimination used to form the conjugated diene proved very efficient in a model system, it afforded disappointing results in the actual synthesis of radicicol (25–30%). Tinchkowski and Lett39 reported a second-generation synthesis wherein the Stille coupling was replaced by a Suzuki coupling in order to avoid tin contamination and more importantly, the MOM group was replaced by a PMB which could be selectively remove and exchange for a mesylate. The elimination of the mesylate now proceeded in excellent yield (83–91%). | ||
| Scheme 1 First- and second-generation syntheses of radicicol by Lett and co-workers.38,39 | ||
Several derivatives of radicicol have also been prepared from the natural product itself in efforts to improve its activity in vivo. While radicicol is very active in cellular assays, it lacks activity in animal models presumably due to metabolic instability. In fact it has been shown that thiols such as DTT can participate in a 1,6-Michael addition to the conjugated diene yielding an inactive product.13,40 Nevertheless, it has been found that the electrophilicity of the Michael acceptor can be reduced by forming oximes (Scheme 2). These radicicol analogues have prolonged half life in sera and have been shown to be active in mouse xenographs.40–43 The oxime formation is usually in competition with conjugate addition and affords the desired product in moderate yield as an E/Z mixture. It has been shown that at least in the case of one oxime (KF58333), the E isomer was more active in vivo.43
 | ||
| Scheme 2 Conversion of the carbonyl group of radicicol to an oxime.41–43 | ||
Inspired by the potential of HSP90 inhibition, the Danishefsky group developed a highly convergent synthesis of radicicol from three key intermediates (Scheme 3).44 The choice of dithiane methodology was motivated by the fact that it could serve as an acyl anion equivalent as well as prevent isocoumarin formation. While the esterification of benzoic acid 12 bearing methyl groups on the phenols proceeded very smoothly via the acid chloride,45 the same transformation proved to be challenging with other protecting groups. As previously observed by Lett, the esterification using a Mitsunobu reaction worked best with the ortho-phenol unprotected. However, standard Mitsunobu conditions (DEAD, Ph3P) afforded poor results due to the formation of undesired phthalide. In this case, the used of a trifuryl phosphine was essential to suppress the competing phthalide formation. Subsequent alkylation with lithiated dienyl dithiane 13 afforded the metathesis precursor 18. It is important to note that the nature of the protecting group on the ortho-phenol had a dramatic impact on the α : γ selectivity in this alkylation. Once again, the unprotected ortho-phenol gave the best results. Ring-closing metathesis using the second-generation Grubbs' catalyst afforded the product in good yield and excellent selectivity. It was noted that this metathesis proceeded more efficiently if the free ortho-phenol was protected as a silyl ether. Conversion of the dithiane to the ketone via an oxidation/Pummerer rearrangement with desilylation followed by chlorination afforded radicicol. The expedient nature of the chemistry allowed Danishefsky and co-workers to explore the biological activity of the different stereoisomers of radicicol thus showing that the correct stereochemistry is necessary at each centre for HSP90 inhibition. Based on their findings in their epothilone program,46,47 it was hypothesized that the epoxide moiety of radicicol might contribute to non-specific toxicity narrowing the therapeutic window and compromising its stability in vivo. Using the same chemistry the cyclopropane analogue of radicicol was prepared and shown to be nearly as potent as radicicol.48 This was a significant observation since it was predicted based on the cocrystal structure of radicicol bound to HSP90 that the epoxide was implicated in an interaction with a lysine residue from HSP90.19 However, the synthesis of cycloproparadicicol suffered from several low-yielding steps compromising its availability for further studies. Interestingly, the same ring-closing metathesis conditions afforded more dimeric product in the case of cycloproparadicicol than in radicicol itself. Only under high-temperature conditions the yields became acceptable.49 In order to gain access to cycloproparadicicol in larger quantities, a second-generation synthesis was developed.32 The key feature of this approach is the generation of the aromatic moiety through a Diels–Alder reaction involving the macrolide bearing an alkyne as the dieneophile and a suitably protected diene (20 + 21, Scheme 4). It is noteworthy that nonactivated alkynes are considered poor dienophiles. Nevertheless, this so-called ynolide strategy proved to be productive and provided a viable solution to acess cycloproparadicicol in gram quantities. Innovative features of this second-generation synthesis include the protection of the alkyne function for the metathesis reaction using a cobalt complexation (25 → 26) and the use of dimedone derived diene (20) to assemble the aryl ring. Several analogues of cycloproparadicicol were also evaluated for their inhibition of HSP90. While cycloproparadicicol was the most potent inhibitor in the series, it was found that the carbonyl could be converted to an oxime while keeping its activity, corroborating earlier findings with radicicol and that the carbonyl could be reduced (only the α epimer was active).32
 | ||
| Scheme 3 Synthesis of radicicol by Danishefsky and co-workers.44 | ||
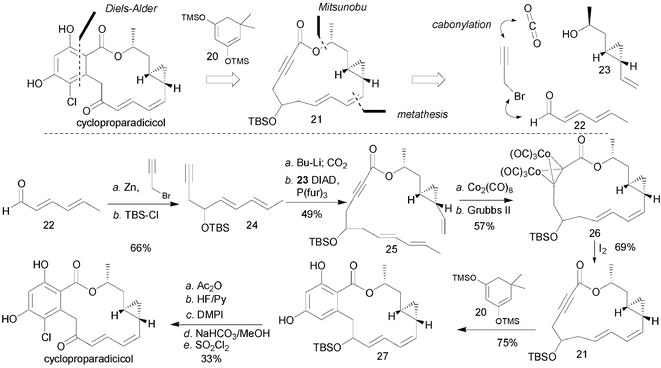 | ||
| Scheme 4 Synthesis of cycloproparadicicol by Danishefsky and co-workers.32 | ||
Hypothemycin and LL-Z1640-2
The first synthesis of LL-Z1640-2 was reported by Tatsuta and et al.50 The authors envisioned closing the macrocycle via a Mukaiyama lactonisation (Scheme 5). The required stereochemistry of the two alcohols present in the final product were envisioned to come from D-ribose. Thus, compound 30 derived from D-ribose in six steps was engaged in a Sonagashira coupling to iodobenzoate 31. While a Lindlar reduction of this alkyne afforded the Z-olefin, reduction of the allylic carbonate under Tsuji's conditions led to its isomerisation thus affording the desired E-olefin. The primary hydroxyl group was then deprotected and elaborated into a lithiated alkyne via the Corey–Fuchs protocol which was used to open S-propylene oxide. It is noteworthy that the transmetallation and alkylation reactions were done in the presence of the methyl benzoate. A second Lindlar reduction was used to control the geometry of the olefin followed by a saponification to afford the cyclisation precursor. Treatment of this compound under the Mukaiyama conditions provided the desired macrocyle in 47% yield (reduction, saponification and macrocyclisation). The success of this macrolactonisation is impressive considering that the secondary alcohol has to react with a 2,6-disubstituted benzoate. Final deprotection and selective oxidation of the allylic alcohol afforded LL-Z1640-2. It is noteworthy that only the Dess–Martin periodinane reagent proved effective for this transformation. Revealing the cis-enone at a late stage is important as this olefin readily epimerizes to the more stable trans isomer.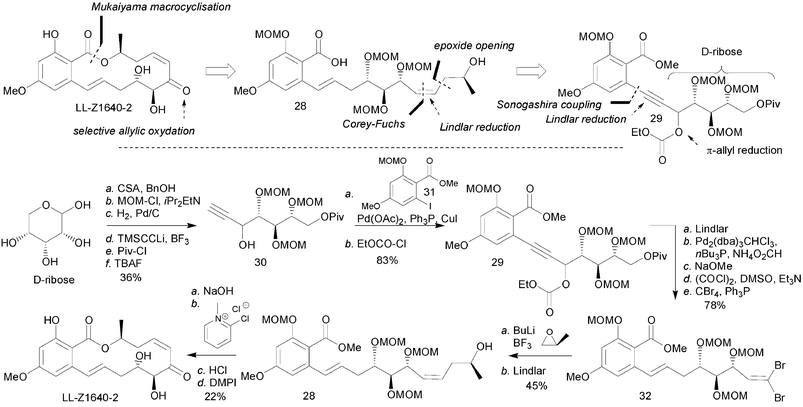 | ||
| Scheme 5 Synthesis of LL-Z1640-2 by Tatsuta et al.50 | ||
The second synthesis of LL-Z1640-2 was reported by Selles and Lett51,52 recognizing the potential of these compounds as kinase inhibitors. While preliminary work to convert readily available zearalenone into LL-Z1640-2 was unfruitful, a stereospecific synthesis of this molecule and its conversion into hypothemycin was achieved. The key features of the synthesis are selective epoxidation of the benzylic olefin to convert LL-Z1640-2 into hypothemycin, the use of a Mitsunobu reaction for the macrolactonisation and a Suzuki coupling to form the benzylic olefin. Intermediate 37 was obtained in eight steps from 1,4-butynediol and converted to the key aldehyde 38 as shown in Scheme 6. Transmetallation of the vinyl iodide 39 (obtained in five steps from propylene oxide) and reaction with aldehyde 38 afforded the alcohol product as a diastereomeric mixture which is inconsequential as this allylic alcohol will ultimately be oxidised to the α,β-conjugated ketone. Following protecting group manipulation, the alkyne was hydroborated and the product was engaged in a Suzuki coupling with aryl bromide 33 to obtain the protected macrolactonisation precursor 40. Deprotection of the silyl ether and hydrolysis of the methyl benzoate followed by Mitsunobu macrolactonisation afforded the desired macrocycle. As in their synthesis of radicicol, the Mitsunobu proved to be efficient (67% yield) for macrolactonisation. It is interesting to note that inversion of the reaction sequence, namely Mitsunobu esterification followed by a palladium-mediated Suzuki macrocyclisation was not effective. Selective removal of the allylic PMB and oxidation to the α,β-conjugated ketone with PCC in the presence of 2,5-dimethylpyrazole (Parish conditions) afforded the desired ketone in excellent yield however, only one of the diastereoisomers was found to react under these conditions. Nevertheless, the other diastereoisomer could be oxidised using a fast Jones oxidation. Careful deprotection of the acetonide (to avoid isomerisation of the cis-enone) afforded LL-Z1640-2. Meticulous analysis of the crystal structure of hypothemycin53 had led the authors to speculate that epoxidation of LL-Z1640-2 should be diastereoselective leading to the desired hypothemycin. This proved to be the case and a diastereoselective epoxidation was achieved, albeit in poor yield (17%), due to the extraordinary unreactivity of this benzylic olefin and lability of the final product.
 | ||
| Scheme 6 Synthesis of LL-Z1640-2 and hypothemycin by Selles and Lett.51,52 | ||
Pochonins
While pochonin C is structurally related to radicicol, it was shown to be much less cytotoxic while still being an inhibitor of HSV (vide supra). In fact, amongst all the pochonins, pochonin C has the highest selectivity index for HSV inhibition. The subtle changes in biological activity stemming from such similar compounds prompted us to develop synthetic methodologies towards the pochonins that would be amenable to combinatorial chemistry allowing us to expand the diversity of this class of compounds beyond the natural analogues in the hope not only to hone in on the activity of pochonins and radicicol but also to expand on the rich biological activity of RALs. It was clear from the onset that the halohydrin of pochonin C could be derived from an epoxide such as radicicol and vice versa (Scheme 7). However, the stereochemistry at the carbon bearing the chlorine atom in pochonin C had not been defined. We reasoned that pochonin C could be disconnected into three fragments as shown in Scheme 7.54 A key feature of this approach is the use of a thioether which masks the sensitive conjugated olefin and could serve as an attachment point to a resin for solid phase synthesis. As we needed access to both potential stereochemistries of the carbon center which ultimately bears the chlorine atom, a divergent synthesis of both cis and trans epoxides 41 was necessary. A Brown–Oehlschlager allylation afforded the halohydrin 43 which could be converted to the required cis and trans epoxide 41 in good yields. As it had been previously noted,38 Mitsunobu esterification works best with the ortho-hydroxyl group unprotected. However, if both phenols are unprotected, the esterification reaction is in competition with alkylation of the para-phenol. We found that if the reaction was carried out with tris(3-chlorophenyl)phosphine, the alkylation of the para-phenol could be suppressed which minimized protecting group manipulation. Protection of both phenols and deprotonation of the benzylic position with LDA followed by reaction with Weinreb amide 42 gave the cyclisation precursors 45. The metathesis was then investigated on the compound bearing the α,β-conjugated olefin masked as a thioether as well as the α,β-conjugated diene. Interestingly, under the same reaction conditions, an E/Z mixture was obtained in the first case whereas only the desired Z olefin was formed in the second case. This result clearly testifies to the conformation restrictions of the radicicol macrocycle. A further testimony of the conformational rigidity of these highly substituted macrocycles is the fact that the metathesis with the cis-epoxide under the same conditions gave much poorer results arguably due to a higher strain in forming the product. Conversion of the epoxide 46 to the halohydrin was then achieved using SO2Cl2 with concomitant chlorination of the aryl ring. Deprotection of the MOM group afforded a compound which was identical to pochonin C thereby assigning the stereochemistry at the carbon bearing the chlorine atom as S. Pochonin C could be converted smoothly into radicicol using mild bases such as K2CO3. Importantly, it was shown that the phenylthio ether could be substituted for a polymer-bound version and that the chemistry could be carried out on solid phase.55 | ||
| Scheme 7 Synthesis of pochonin C and radicicol by Winssinger and co-workers.54,55 | ||
The comparison of NMR data between the closely related pochonin C and radicicol clearly showed that despite their similar structures, their conformations are very different.55 In a collaboration with the Karplus group, the conformation profile of radicicol and several other analogues and related natural products were analyzed computationally.30 This analysis led to the identification of three main conformations for radicicol: an L-shape conformation (Fig. 3), which is the bioactive one, a planar conformation or P-shape (with an energy of 2.4 kcal mol–1 relative to the L-shape), and a L′-shape, in which the macrocycle is positioned in the opposite side of the aromatic ring than the one present in the L-shape (with an energy of 3.3 kcal mol–1 relative to the L-shape). Interestingly, the same analysis for inactive radicicol analogues such as the one having the wrong stereochemistry of the epoxide or the epoxide opened showed that these compounds would suffer a high energetic penalty while adopting the bioactive L-shape conformation. These results suggested that the wrong conformational bias could be involved in the lack of activity towards HSP90 of analogues in which the stereochemistry of specific centers was altered. Based on the presumed importance of maintaining the bioactive (L-shape) conformation, the conformation space of a series of compounds with modifications of the epoxide and olefin region was profiled leading to the identification of pochonin D (Fig. 1) as a potential HSP90 inhibitor. While pochonin D would incur a small energetic penalty in adopting the bioactive conformation (1.2 kcal mol–1), the fact that it did not contain the labile epoxide and was synthetically much more accessible were encouraging.
 | ||
| Fig. 3 Representation of the three main conformers of radicicol and their relative energies (kcal mol–1). | ||
The synthesis of pochonin D followed the same logic as the synthesis of pochonin C, namely a disconnection into three fragments (Scheme 8). The Weinreb amide fragment 48 was prepared in four steps using a polymer bound version of the phenylthio ether. Selective esterification of 2,4-dihydroxy-6-methybenzoic acid or its chlorinated analogue using a polymer-bound version of DEAD followed by protection of both phenols with EOM-Cl afforded compound 49. The use of polymer-bound reagents obviated the need for regular chromatography and the product could be treated directly with LDA and engaged in a reaction with the aforementioned Weinreb amide 48 obtained directly from solid phase. This last reaction was quenched with a polymer bound acid which also sequestered all the diisopropyl amine. Cyclisation with the second-generation Grubbs' catalyst followed by deprotection of the EOMs with sulfonic acid resin in MeOH afforded pochonin D in 31% overall yield for six steps. Importantly, despite its simpler structure, pochonin D was indeed a good ligand for HSP90 with a 80 nM affinity (compared to 20 nM for radicicol). The difference in affinity is consistent with the calculated 1.2 kcal change in internal energy of pochonin D upon binding. The C14–15 olefin of pochonin D could be regioselectively epoxidised using dimethyldioxirane to obtain pochonin A which was also found to be an HSP90 ligand however it was not superior to pochonin D and presents a labile epoxide.56
 | ||
| Scheme 8 Synthesis of pochonin D by Winssinger and co-workers.30 | ||
The fact that the synthesis of pochonin D required only six steps and one conventional purification rendered it amenable to combinatorial synthesis. As shown in Scheme 9, a library represented by the general structure 50 which bears five points of diversity was prepared31 from intermediates 51 obtained through the aforementioned chemistry. The diversity of the library included the following modifications: the group on C17 (R1, both stereochemistry are present in natural resorcylides, however only with a methyl substituent); the meta position on the aryl ring (R2, a number of natural resorcylides bear a chlorine at that position); the substitution of the para phenol (R3, a number of natural resorcylides bear a methyl group at that position); the C14–15 olefin (R4) which was converted to a diol or an epoxide; the C9 carbonyl (R5) which was reduced or converted to oximes and the olefin C10–C11 which was reduced. While not all permutations of the library were pursued, a total of 113 compounds were prepared using combinations of the chemistry shown in Scheme 9. A representative subset of the library (84 compounds) was tested for its inhibition in a panel of 24 kinaseswhich led to the identification of twelve compounds which had greater than 50% inhibition for one kinase at 10 µM. The high hit rate in this resorcylide library (14%) testifies to the potential of the RAL scaffold for kinase inhibition. Importantly, the promising kinase leads that were identified were not HSP90 inhibitors and showed diverse selectivity profiles of kinase inhibition amongst the 24 tested kinases.
 | ||
| Scheme 9 Synthesis of a library of pochonins by Winssinger and co-workers.31 | ||
Radicicol A
Based on the premise that natural products have been an excellent source of novel structures covering a different chemical space than most synthetic libraries, researchers at Novartis prepared a library using trans-radicicol A as a starting material (Scheme 10).57 This product was obtained in multigram quantities by fermentation and was converted to intermediate 60 with a free phenol and a suitably protected amine to generate a library with two points of diversity. Loading of compound 60 on a resin via ketal formation and alkylation of the phenol using a phosphazene base afforded polymer bound intermediate 63 with the first point of diversity. Deprotection of the Teoc group afforded the amine which was used as the second point of diversity and reacted with different acid chlorides and isocyanates followed by a release from the resin under acidic conditions. While a matrix of only five products was reported, this chemistry is clearly amenable to larger libraries. Unfortunately, evaluation of the library for its biological activity was not reported. | ||
| Scheme 10 Synthesis of radicicol A library by Marzinzik and co-workers.57 | ||
Aigialomycin
The first synthesis of aigialomycin D was reported by Danishefsky and co-workers.32,58 Inspired by their previous success with the ynolide protocol in the context of cycloproparadicicol whereby the aromatic ring is formed at a late stage via a Diels–Alder reaction, the authors wished to expend the scope of this methodology to other resorcylic macrolides (Scheme 11). This approach proved to be highly effective once again. Recognizing that the required stereochemistry of the two alcohols could be derived from D-deoxyribose, the key alcohol 67 was obtained in four steps. Oxidation of the alcohol and coupling to propargyl bromide activated with zinc afforded a mixture of diastereoisomers which was unimportant since this alcohol had to be eliminated. Protecting group manipulation afforded compound 68 which was oxidised and engaged in an olefination reaction. Carboxylation of the alkyne followed by a Mitsunobu esterification afforded the macrocyclisation precursor 69. Protection of the alkyne (dicobalt hexacarbonyl complex) followed by ring-closing metathesis and deprotection of the alkyne (CAN) afforded the ynolide which was reacted with the diene 20 to afford compound 70 in good yield. Protection of the phenols followed by selective deprotection of the homobenzylic alcohol which was dehydrate using Martin's sulfurane conditions and global deprotection yielded aigialomycin D. The authors noted that although aigialomycin D did bind to HSP90, it's affinity was >10 µM. | ||
| Scheme 11 Synthesis of aigialomycin D by Danishefsky and co-workers.32,58 | ||
As part of our program to further the diversity of RAL beyond the naturally available RAL analogues in the hope of finding new ATPase and kinase inhibitors, we became interested in the aigialomycins. As for the pochonins, our goal was to develop synthetic protocols which were sufficiently flexible as to be carried out in a combinatorial fashion. As shown in Scheme 12, we reasoned that the molecule could be disconnected in two fragments using a metathesis to close the macrocycle.33 Importantly, the use of the seleno or thioether at the benzylic position should facilitate alkylation chemistry at that position in addition to providing an attachment point to a resin for solid phase synthesis. Furthermore, by virtue of the different modes of cleavage of seleno or thio-ethers, either the benzylic olefin product or the corresponding reduced product should be accessible. The key allylic epoxide 72 or acetonide protected diol 73 were prepared in four and six steps respectively. The aromatic fragment 74 was obtained in three steps from commercially available products. Deprotonation of the benzylic position with LDA followed by addition of bromide73 afforded the alkylated product which was cyclised using Grubbs' second-generation catalyst to obtain macrocycles 75 in excellent yield. Oxidation/elimination of the selenide followed by deprotection afforded aigialomycin D in ten steps from bromobutene. The same chemistry was found equally productive with alkyl bromide 72 containing the allylic epoxide, however opening of the epoxide in macrocycle 76 under a variety of conditions failed to give the 1,2-cis diol and led in all cases to SN2′ addition. While this route can not be used to access aigialomycin D, this diversion proved quite general for different nucleophiles such as alcohols, cyanide or azide thus obtaining new RAL analogues 77. Importantly, as in the case of the pochonins, it was shown that the chemistry could be carried out on solid phase by replacing the phenyl selenide with a polymer-bound thioether. Thus benzyl chloride78 was loaded on a thiol resin and both phenols were protected to obtain polymer-bound intermediate 79. Alkylation of the benzylic position with five different alkyl bromides (80) followed by RCM afforded macrocycles which could be released from the resin either via oxidation/elimination or under reductive conditions to obtain aigialomycin analogues 82 and 83, respectively, after deprotection with sulfonic acid resin. It is interesting to note that the metathesis conditions that were successfully used with the selenoether in solution (toluene, 80 °C) proved to be ineffective on solid phase with the thioether. Excellent yields were nevertheless obtain by carrying out the RCM in CH2Cl2 at 120 °C using microwave irradiation. This pilot library was screened against a panel of kinases revealing that aigialomycin D is a moderate inhibitor of CDK1/cyclin B and CDK5/p25 at 5.7 and 5.8 µm, respectively, as well as GSK-3 at 14 µm but much less of PfGSK-3, the Plasmodium homologue of GSK-3. Closely related analogues were not inhibitors of these kinasessuggesting that the particular functionalities present on aigialomycin D are important for this activity. It was further confirmed that aigialomycin D is not an inhibitor of HSP90.
 | ||
| Scheme 12 Diverted synthesis of aigialomycin D and analogues by Winssinger and co-workers.33 | ||
A third synthesis of aigialomycin was reported by Pan and co-workers (Scheme 13).59 The key features of the synthesis are the use of two Julia-Kocienski couplings to establish the E geometry of both olefins and a Yamagushi macrocyclisation. Thus, the key fragment 87 was obtained in ten steps from propargylic alcohol and coupled to the functionalized benzaldehyde 85via a first Julia–Kocienski coupling. Deprotection of the Piv and oxidation of the resulting alcohol provided the aldehyde which was engaged in a second Julia–Kocienski coupling with 86 (prepared in five steps). Removal of the silyl protecting group and conversion of the aryl bromide into the carboxylate via transmetalation (nBuLi) and reaction with CO2 afforded the macrolactonisation precursor. Treatment of this compound to the Yamagushi conditions afforded the macrocycle in 51% yield after three days at reflux. The result of this reaction is notable as there is not a considerable steric difference between the carbonyl groups of the anhydride formed with the Yamagushi reagent (both aryl rings are 2,6 substituted). Final deprotection under the same conditions as were previously used by Danishefsky afforded aigialomycin D.
 | ||
| Scheme 13 Synthesis of aigialomycin D by Pan and co-workers.59 | ||
Conclusions
The more recent discoveries that resorcylic acid lactones can mimic ATP and be potent inhibitors of ATPases such as HSP90 or kinasessuch as MEK, TAK and VGFR have raised interest in this family of natural products. From a chemical biology perspective, kinasesare involved in numerous signalling cascades and selective inhibitors are particularly useful to dissect the implication of individual kinasesin complex networks. The fact that some RALs have been shown to be irreversible inhibitors may prove to be an asset as they can be used to selectively label given kinasesor as probes for activity based profiling. To which extend the selectivity of given inhibitors can be modulated with changes in the functionalities around the macrocycle remains to be defined, but it is clear that small changes of the functional groups around the macrocycle can have a dramatic impact on the conformation of these compounds. From a chemical synthesis perspective, while several elegant approaches to important RALs have already been reported, synthesizing such compounds through concise and modular routes which can be used to extend the diversity of this family remains challenging. From a therapeutic perspective, the targets of many RALs (HSP90 and MAP kinases) are considered amongst the most promising targets for chemotherapy as well as inflammation treatment and several RALs have already been shown to be effective in animal models (hypothemycin and radicicol derivatives).Abbreviations
| AIBN | Azobis(isobutytonitrile) |
| BBN | Borabicyclononane |
| CAN | Ceric ammonium nitrate |
| CSA | Camphorsulfonic acid |
| DDQ | 2,3-Dichloro-5,6-dicyanobenzoquinone |
| DEAD | Diethylazodicarboxylate |
| DIAD | Diisopropylazodicarboxylate |
| Dibal-H | Diisobutylaluminium hydride |
| DMDO | Dimethyldioxirane |
| DMPI | Dess–Martin periodinane |
| EOM | Ethoxymethyl |
| HFIP | Hexafluoroisopropanol |
| Grubbs II | Second-generation Grubbs' catalyst (ruthenium[1,3-bis(2,4,6-trimethylphenyl)-2-imidazolidinylidene]dichloro(phenylmethylene)(tricyclohexylphosphane) |
| KHMDS | Potassium hexamethyldisilylamide |
| LDA | Lithium diisopropylamide |
| mCPBA | meta-Chloroperbenzoic acid |
| MOM | Methoxymethyl |
| NMO | N-Methylmorpholine-N-oxide |
| PCC | Pyridinium chlorochromate |
| Piv | Pivaloyl |
| PMB | para-Methoxybenzyl |
| Py | Pyridine |
| RCM | Ring-closing metathesis |
| RedAl | Sodium bis(methoxyethoxy) aluminium hydride |
| pTSA | para-Toluenesulfonic acid |
| SAE | Sharpless asymmetric epoxidation |
| TBS | tert-Butyldimethylsilyl |
| TBDPS | tert-Butyldiphenylsilyl |
| Teoc | 2-(Trimethylsilyl)ethoxycarbonyl |
| TBAF | Tetrabutylammonium fluoride |
| TFA | Trifluoroacetic acid |
| TMS | Trimethylsilyl |
Acknowledgements
We thank our collaborators who have contributed to our work in the area of RAL synthesis (P. Lopez, E. Moulin, P.-Y Dakas, L. Brethous, N. Reichardt and M. Boulifa) and the funding agencies (Human Frontier Science Program – HFSP, Association pour la Recherche sur le Cancer – ARC, Agence Nationale de la Recherche – ANR and Marie Curie). This review was not intended to be comprehensive but rather a highlight of some recent work and we apologize for arbitrary omissions.References
- P. Delmotte and J. Delmotte-Plaquee, Nature, 1953, 171, 344 CrossRef CAS.
- M. Stob, R. S. Baldwin, J. Tuite, F. N. Andrews and K. G. Gillette, Nature, 1962, 196, 1318 CAS.
- G. A. Ellestad, F. M. Lovell, N. A. Perkinson, R. T. Hargreaves and W. J. McGahren, J. Org. Chem., 1978, 43, 2339–2343 CrossRef CAS.
- M. S. R. Nair and S. T. Carey, Tetrahedron Lett., 1980, 21, 2011–2012 CrossRef CAS.
- R. N. Mirrington, E. Ritchie, C. W. Shoppee, W. C. Taylor and S. Sternhell, Tetrahedron Lett., 1964, 365–370 CrossRef CAS.
- M. Isaka, C. Suyarnsestakorn, M. Tanticharoen, P. Kongsaeree and Y. Thebtaranonth, J. Org. Chem., 2002, 67, 1561–1566 CrossRef CAS.
- T. J. Turbyville, E. M. Wijeratne, M. X. Liu, A. M. Burns, C. J. Seliga, L. A. Luevano, C. L. David, S. H. Faeth, L. Whitesell and A. A. Gunatilaka, J. Nat. Prod., 2006, 69, 178–184 CrossRef CAS.
- I. Gaffoor and F. Trail, Appl. Environ. Microbiol., 2006, 72, 1793–1799 CrossRef CAS.
- R. J. Miksicek, J. Steroid Biochem. Mol. Biol., 1994, 49, 153–160 CrossRef CAS.
- W. L. W. Duax and M. Charles, Dev. Toxicol. Environ. Sci., 1980, 5, 11–31 Search PubMed.
- F. McCapra, A. I. Scott, P. Delmotte, J. Delmotte-Plaquee and N. S. Bhacca, Tetrahedron Lett., 1964, 869–875 CrossRef.
- H. J. Kwon, M. Yoshida, K. Abe, S. Horinouchi and T. Beppu, Biosci. Biotechnol. Biochem., 1992, 56, 538–539 CrossRef CAS.
- H. J. Kwon, M. Yoshida, Y. Fukui, S. Horinouchi and T. Beppu, Cancer Res., 1992, 52, 6926–6930 CAS.
- T. W. Schulte, S. Akinaga, S. Soga, W. Sullivan, B. Stensgard, D. Toft and L. M. Neckers, Cell Stress Chaperones, 1998, 3, 100–108 CrossRef CAS.
- S. V. Sharma, T. Agatsuma and H. Nakano, Oncogene, 1998, 16, 2639–2645 CrossRef CAS.
- A. Kamal, L. Thao, J. Sensintaffar, L. Zhang, M. F. Boehm, L. C. Fritz and F. J. Burrows, Nature, 2003, 425, 407–410 CrossRef CAS.
- Y. L. Janin, J. Med. Chem., 2005, 48, 7503–7512 CrossRef CAS.
- G. Chiosis, A. Rodina and K. Moulick, Anticancer Agents Med. Chem., 2006, 6, 1–8 Search PubMed.
- S. M. Roe, C. Prodromou, R. O'Brien, J. E. Ladbury, P. W. Piper and L. H. Pearl, J. Med. Chem., 1999, 42, 260–266 CrossRef CAS.
- L. Chang and M. Karin, Nature, 2001, 410, 37–40 CrossRef CAS.
- G. L. Johnson and R. Lapadat, Science, 2002, 298, 1911–1912 CrossRef CAS.
- T. Kastelic, J. Schnyder, A. Leutwiler, R. Traber, B. Streit, H. Niggli, A. Mackenzie and D. Cheneval, Cytokine, 1996, 8, 751–761 CrossRef CAS.
- D. Cheneval, P. Ramage, T. Kastelic, T. Szelestenyi, H. Niggli, R. Hemmig, M. Bachmann and A. Mackenzie, J. Biol. Chem., 1998, 273, 17846–17851 CrossRef CAS.
- J. Ninomiya-Tsuji, T. Kajino, K. Ono, T. Ohtomo, M. Matsumoto, M. Shiina, M. Mihara, M. Tsuchiya and K. Matsumoto, J. Biol. Chem., 2003, 278, 18485–18490 CrossRef CAS.
- A. Zhao, S. H. Lee, M. Mojena, R. G. Jenkins, D. R. Patrick, H. E. Huber, M. A. Goetz, O. D. Hensens, D. L. Zink, D. Vilella, A. W. Dombrowski, R. B. Lingham and L. Huang, J. Antibiot., 1999, 52, 1086–1094 CAS.
- A. Schirmer, J. Kennedy, S. Murli, R. Reid and D. V. Santi, Proc. Natl. Acad. Sci. USA, 2006, 103, 4234–4239 CrossRef CAS.
- H. Tanaka, K. Nishida, K. Sugita and T. Yoshioka, Jpn. J. Cancer Res., 1999, 90, 1139–1145 CAS.
- R. Camacho, M. J. Staruch, C. DaSilva, S. Koprak, T. Sewell, G. Salituro and F. J. Dumont, Immunopharmacology, 1999, 44, 255–265 CrossRef CAS.
- V. Hellwig, A. Mayer-Bartschmid, H. Mueller, G. Greif, G. Kleymann, W. Zitzmann, H.-V. Tichy and M. Stadler, J. Nat. Prod., 2003, 66, 829–837 CrossRef CAS.
- E. Moulin, V. Zoete, S. Barluenga, M. Karplus and N. Winssinger, J. Am. Chem. Soc., 2005, 127, 6999–7004 CrossRef CAS.
- E. Moulin, S. Barluenga, F. Totzke and N. Winssinger, Chem. Eur. J., 2006, 12 DOI:10.1002/chem.200600553.
- Z.-Q. Yang, X. Geng, D. Solit, C. A. Pratilas, N. Rosen and S. J. Danishefsky, J. Am. Chem. Soc., 2004, 126, 7881–7889 CrossRef CAS.
- S. Barluenga, P. Y. Dakas, Y. Ferandin, L. Meijer and N. Winssinger, Angew. Chem., Int. Ed., 2006, 45, 3951–3954 CrossRef CAS.
- D. Taub, N. N. Girotra, R. D. Hoffsommer, Jr., C.-H. Kuo, H. L. Slates, S. Weber and N. L. Wendler, Tetrahedron, 1968, 24, 2443–2461 CrossRef CAS.
- E. J. Corey and K. C. Nicolaou, J. Am. Chem. Soc., 1974, 96, 5614–5616 CrossRef CAS.
- S. Masamune, S. Kamata and W. Schilling, J. Am. Chem. Soc., 1975, 97, 3515–3516 CrossRef CAS.
- A. Furstner, O. R. Thiel, N. Kindler and B. Bartkowska, J. Org. Chem., 2000, 65, 7990–7995 CrossRef CAS.
- M. Lampilas and R. Lett, Tetrahedron Lett., 1992, 33, 773–776 CrossRef CAS; M. Lampilas and R. Lett, Tetrahedron Lett., 1992, 33, 777–780 CrossRef CAS.
- I. Tinchkowski and R. Lett, Tetrahedron Lett., 2002, 43, 3997–4001 CrossRef; I. Tinchkowski and R. Lett, Tetrahedron Lett., 2002, 43, 4003–4007 CrossRef CAS.
- T. Agatsuma, H. Ogawa, K. Akasaka, A. Asai, Y. Yamashita, T. Mizukami, S. Akinaga and Y. Saitoh, Bioorg. Med. Chem., 2002, 10, 3445–3454 CrossRef CAS.
- S. Soga, L. M. Neckers, T. W. Schulte, Y. Shiotsu, K. Akasaka, H. Narumi, T. Agatsuma, Y. Ikuina, C. Murakata, T. Tamaoki and S. Akinaga, Cancer Res., 1999, 59, 2931–2938 CAS.
- J. Kurebayashi, T. Otsuki, M. Kurosumi, S. Soga, S. Akinaga and H. Sonoo, Jpn. J. Cancer Res., 2001, 92, 1342–1351 CAS.
- S. Soga, S. V. Sharma, Y. Shiotsu, M. Shimizu, H. Tahara, K. Yamaguchi, Y. Ikuina, C. Murakata, T. Tamaoki, J. Kurebayashi, T. W. Schulte, L. M. Neckers and S. Akinaga, Cancer Chemother. Pharmacol., 2001, 48, 435–445 CrossRef CAS.
- R. M. Garbaccio, S. J. Stachel, D. K. Baeschlin and S. J. Danishefsky, J. Am. Chem. Soc., 2001, 123, 10903–10908 CrossRef CAS.
- R. M. Garbaccio and S. J. Danishefsky, Org. Lett., 2000, 2, 3127–3129 CrossRef CAS.
- A. Rivkin, T. C. Chou and S. J. Danishefsky, Angew. Chem., Int. Ed., 2005, 44, 2838–2850 CrossRef CAS.
- D. T. Bergstralh, D. J. Taxman, T. C. Chou, S. J. Danishefsky and J. P. Ting, J. Chemother., 2004, 16, 563–576 Search PubMed.
- K. Yamamoto, R. M. Garbaccio, S. J. Stachel, D. B. Solit, G. Chiosis, N. Rosen and S. J. Danishefsky, Angew. Chem., Int. Ed., 2003, 42, 1280–1284 CrossRef CAS.
- X. Geng, Z.-Q. Yang and S. J. Danishefsky, Synlett, 2004, 1325–1333 CAS.
- K. Tatsuta, S. Takano, T. Sato and S. Nakano, Chem. Lett., 2001, 172–173 CrossRef CAS.
- P. Selles and R. Lett, Tetrahedron Lett., 2002, 43, 4621–4625 CrossRef CAS.
- P. Selles and R. Lett, Tetrahedron Lett., 2002, 43, 4627–4631 CrossRef CAS.
- T. Agatsuma, A. Takahashi, C. Kabuto and S. Nozoe, Chem. Pharm. Bull., 1993, 41, 373–375 CAS.
- S. Barluenga, P. Lopez, E. Moulin and N. Winssinger, Angew. Chem., Int. Ed., 2004, 43, 3467–3470 CrossRef CAS.
- S. Barluenga, E. Moulin, P. Lopez and N. Winssinger, Chem. Eur. J., 2005, 11, 4935–4952 CrossRef CAS.
- E. Moulin, S. Barluenga and N. Winssinger, Org. Lett., 2005, 7, 5637–5639 CrossRef CAS.
- P. Grosche, K. Akyel and A. Marzinzik, Synthesis, 2005, 2015–2021 CAS.
- Z. Q. Yang, X. Geng, D. Solit, C. A. Pratilas, N. Rosen and S. J. Danishefsky, J. Am. Chem. Soc., 2004, 126, 7881–7889 CrossRef CAS.
- J. Lu, J. Ma, X. Xie, B. Chen, X. She and X. Pan, Tetrahedron: Asymmetry, 2006, 17, 1066–1073 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2007 |