Dihydroxylation of 2-vinylaziridine: efficient synthesis of D-ribo-phytosphingosine†
Hyo Jae
Yoon
a,
Yong-Woo
Kim
a,
Baeck Kyoung
Lee
a,
Won Koo
Lee
*a,
Yongeun
Kim
b and
Hyun-Joon
Ha
*b
aDepartment of Chemistry and Program of Integrated Biotechnology, Sogang University, Seoul, 121-742, Korea. E-mail: wonkoo@sogang.ac.kr; Fax: +82 2-701-0967; Tel: +82 2-705-8449
bDepartment of Chemistry, Hankuk University of Foreign Studies, Yongin, 449-791, Korea. E-mail: hjha@hufs.ac.kr; Fax: +82 31-3304566; Tel: +82 31-3304369
First published on 17th November 2006
Abstract
An efficient and highly stereoselective synthesis of D-ribo-(2S,3S,4R)-phytosphingosine was accomplished in 62% overall yield starting from commercially available (2S)-hydroxymethylaziridine via osmium-catalyzed asymmetric dihydroxylation as a key step.
Sphingolipids along with glycerides are the main constituents of cell membranes and their metabolism is closely related to many inherited human diseases and to cell regulation. Sphingosine and phytosphingosine are two most important long-chain backbones of sphingolipids in the realm of nature (Fig. 1).1 Recently, it has been revealed that phytosphingosine shows a great variety of biological activities such as cell differentiation, signal transduction, regulation, proliferation and induction of apoptosis.2 Furthermore, phytosphingosine was shown to be widely distributed as one of the molecular species of sphingolipids in microorganisms, plants, and many mammalian tissues such as brain, hair, kidney, skin, liver, uterus, intestine and in blood plasma.3
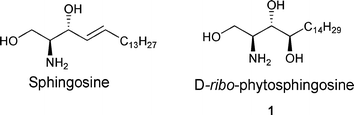 | ||
| Fig. 1 Structure of sphingosine and D-ribo-phytosphingosine (1). | ||
Among all eight possible stereoisomers of phytosphingosine, (2S,3S,4R)-2-aminooctadecane-1,3,4-triol with D-ribo-stereochemistry, is the most common. Owing to interests in these vital biological activities many synthetic approaches to enantiomerically pure phytosphingosines have been reported in the literature.4 However, most of them suffer from long synthetic steps and low overall yield. Recent success in the synthesis of sphingosine5 and ceramide analogues6 prompted us to aim for the efficient synthesis of phytosphingosine. In this communication we would like to report a remarkably efficient synthetic route to yield D-ribo-phytosphingosine from commercially available (2S)-hydroxymethylaziridine through a simple five-step transformations including stereoselective dihydroxylation.
Aziridine-2(S)-carbaldehyde was readily prepared via Swern oxidation of the enantiomerically pure aziridine-2(S)-methanol which is commercially available.7 The bromopentadecane was converted to the corresponding phosphonium salt in 80% yield by stirring with triphenylphosphine in acetonitrile at reflux for 30 h.8 Treatment of the phosphonium salt with LiHMDS at −78 °C generated the ylide which was then reacted with aziridine-2(S)-carbaldehyde to provide 2(S)-[(Z)-hexadec-1-enyl]aziridine 2a in 93% yield with the ratio of 99 : 1 (Z : E).
We studied the stereoselective dihydroxylations of the (Z)-2-alkenylaziridine (2a) under a variety of conditions (Scheme 1 and Table 1). The Sharpless asymmetric dihydroxylation with AD-mix α and AD-mix β was not completed after two days. Furthermore, the ratio of the vicinal diols 3Aa and 3Ba was poor in terms of stereoselectivity. We concluded that the reluctance of the dihydroxylation of the 2-vinylaziridine using AD-mix reagents is due to the steric congestion around the double bond of the aziridine which hinders the approach of the bulky oxidation catalyst to the double bond. This interaction between the catalyst and bulkiness of the substrate in stereoselective dihydroxylation has been reported in the literature.9 When the 2-vinylaziridine was oxidized with 10 mol% of osmium tetraoxide in aqueous acetone with N-methylmorpholine oxide (NMO) as re-oxidant, we obtained the two vicinal diols 3Aa and 3Ba with high stereoselectivity (93 : 7) and high yield. A better ratio (99 : 1) was obtained by lowering the reaction temperature to −10 °C. The diastereomeric mixture of the diols was easily separated by flash column chromatography to remove trace amount of the other isomer. The identity of each diastereoisomeric diol was confirmed by 1H NMR spectra in the literature after regioselective aziridine ring opening with AcOH and removing the benzyl group from the nitrogen. Once we obtained 2-(1,2-dihydroxyhexadecanyl)aziridine (3Aa) in high yield, we looked into the origin of the stereoselectivity and the scope of dihydroxylation reactions of the 2-vinylaziridine in general.
 | ||
| Scheme 1 Dihydroxylation reactions of 2a. | ||
| Entry | Reagents | Solvent | Temp | Time | 3Aa : 3Bac | Yieldd (%) |
|---|---|---|---|---|---|---|
| a 5 wt% in H2O. b N,N,N′,N′-Tetramethylethylenediamine. c Based on the integration of the crude 1H NMR (300 MHz) spectra. d Total isolated yields. | ||||||
| 1 | AD-mix α (1.4 g/1 mmol) | t-BuOH–H2O (1 : 1) | RT | 2 days | 2 : 3 | <40 |
| 2 | AD-mix β (1.4 g/1 mmol) | t-BuOH–H2O (1 : 1) | RT | 2 days | 3 : 2 | <40 |
| 3 | OsO4a (1.1 equiv.)–TMEDAb (1.1 equiv.) | Acetone–H2O (1 : 1) | RT | 2 days | ND | Trace |
| 4 | OsO4a (1.1 equiv.) | Acetone | RT | 2 days | ND | Trace |
| 5 | OsO4a (0.1 equiv.)–NMO (3 equiv.) | Acetone–H2O (9 : 1) | RT | 2 h | 5 : 1 | 80 |
| 6 | OsO4a (0.1 equiv.)–NMO (3 equiv.) | Acetone/H2O (9 : 1) | 0 °C | 4 h | 93 : 7 | 88 |
| 7 | OsO4a (0.1 equiv.)–NMO (3 equiv.) | Acetone/H2O (9 : 1) | −10 °C | 10 h | 99 : 1 | 87 |
The origin of this high stereoselectivity was investigated by changing the substrate structures to address the following questions. Is there any steric influence of the vinyl substituents R in 2a–h? Will stereoselectivity be changed by double bond configuration? Is there any influence by the group X attached on the aziridine ring nitrogen? Thereby various 2-vinylaziridines (2b–h) were used to investigate the stereoselectivities of dihydroxylation reactions (Scheme 2) and the results were shown in Table 2.
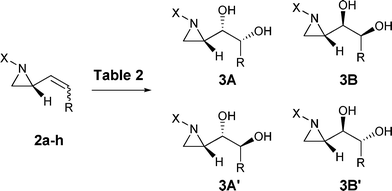 | ||
| Scheme 2 Dihydroxylation reactions and their stereoselectivities of 2. | ||
| Subst. | X | R | Isomer | 3A : 3Bb | 3A′ : 3B′b | Yield (%) |
|---|---|---|---|---|---|---|
| a Under the reaction conditions of entry 6 in Table 1. b Based on the integration of the crude 1H NMR (300 MHz) spectra. | ||||||
| 2a | (R)-Phenylethyl | C14H29 | Z | 93 : 7 | 88 | |
| 2b | (R)-Phenylethyl | C14H29 | E | 72 : 28 | 90 | |
| 2c | (R)-Phenylethyl | C5H11 | Z | 89 : 11 | 92 | |
| 2d | (R)-Phenylethyl | Phenyl | Z | 70 : 30 | 84 | |
| 2e | (R)-Phenylethyl | Phenyl | E | 58 : 42 | 79 | |
| 2f | (R)-Phenylethyl | Benzyl | Z | 84 : 16 | 88 | |
| 2g | (R)-Phenylethyl | Benzyl | E | 71 : 29 | 82 | |
| 2h | Benzyl | C14H29 | Z | 83 : 17 | 85 | |
The Z isomer of the olefins (2a, 2d and 2f) always showed better selectivity than E (2b, 2e and 2g). When we compare the reactions of Z isomeric substrates (2a, 2c, 2d and 2f) we find that a longer alkyl chain gives better selectivity. Decrease of selectivity, from 93 : 7 to 83 : 17, was also observed by changing the alkyl group X attached on the nitrogen of the aziridine from phenylethyl (2a) to benzyl (2h). These observations can be explained by the conformational preference during the hydroxylation. In the literature, it has been reported that some ligands, especially tertiary amine group for asymmetric dihydroxylation, play an important role in controlling enantioselectivity by coordinating with OsO4.10 We assume that the two substituents on the nitrogen and the vinyl group at C-2 have an anti relationship11 and OsO4 may coordinate to the aziridine ring nitrogen. Two conformations (A and B, Fig. 2) from the Z-stereoisomer (2a, 2d and 2f) are possible by rotating the σ-bond for the double bond to get close to the oxygen atoms of OsO4 for the dihydroxylation reactions. One pathway “b” leading to the conformer B is unfavorable due to nonbonding interaction between X and the bulky R groups while the other pathway “a” toward the conformer A is easily accessible. Therefore, the nitrogen bound OsO4 can readily deliver oxygen atoms to the double bond with A conformer to result in the major dihydroxylation product 3Aa. In the case of E-stereoisomers (2b, 2e and 2g) the rotation barrier difference to go to the possible conformers A′ and B′ are not as large as for A and B of the Z-isomers since the steric interaction between the bulky substituent of the E-double bond and X group is less than that of the Z-stereoisomer. Therefore, the E-stereoisomer shows decreased diastereoselectivity in the asymmetric dihydroxylation reaction. The decrease of the selectivity by changing the alkyl group X attached on the nitrogen of the aziridine from phenylethyl (2a) to the smaller benzyl (2h) also can be explained by the decrease of the nonbonding interaction of the reactive conformation A or B.
 | ||
| Fig. 2 Dihydroxylations of (Z)- and (E)-2-alkenylaziridine (2). | ||
Regioselective ring opening reaction of the aziridine diol 3Aa by AcOH and hydrolysis of the acetate by KOH in ethanol provided the aminotriol 4 (Scheme 3). AcOH was used for the activation of the basic ring nitrogen of the aziridine and was also used as the source of the acetate nucleophile to attack the less sterically hindered C-3 position of the aziridine.5 Finally, enantiomerically pure D-ribo-phytosphingosine 1 was obtained by palladium catalyzed debenzylation reaction. We further synthesized and characterized N-Boc derivative 5 and tetraacetate 6 to confirm the absolute configuration of the C-3 and C-4 hydroxy group in D-ribo-phytosphingosine.
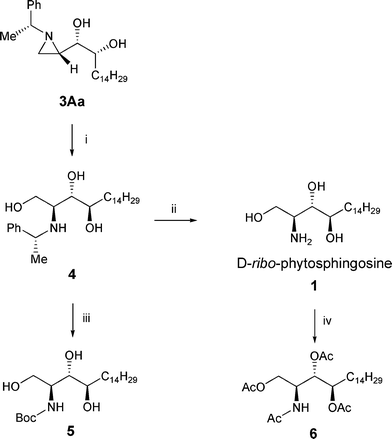 | ||
| Scheme 3 Conversion of dihydroxyaziridine (3Aa) to D-ribo-phytosphingosine (1). Reagents and conditions: (i) (1) AcOH, CH2Cl2; (2) KOH, EtOH, rt, 93%; (ii) Pd(OH)2, H2(g) 100 psi, EtOH, rt, 92%; (iii) Pd(OH)2, H2(g) 100 psi, EtOH, rt, (Boc)2O, 95%; (iv) acetic anhydride, pyridine, rt, 98%. | ||
In summary, we efficiently synthesized enantiomerically pure D-ribo-phytosphingosine through a five-step sequence in 62% overall yield from commercially available (2S)-hydroxymethylaziridine using asymmetric dihydroxylation of the Z-vinylaziridine as the key step. This methodology also provides an efficient way for the preparation of structurally modified phytosphingosine analogs. Additional study regarding the relation between the stereoselectivity in the dihydroxylation and the chain length of the substituents will be reported in due course.
This work was supported by the following institutions: The Korea Science and Engineering Foundation (R01-2005-000-10032-0 and the Center for Bioactive Molecular Hybrids to HJH), Korea Research Foundation (KRF-2002-070-C00060 to WKL) and Imagene for providing enantiomerically pure chiral aziridines.
Notes and references
- (a) A. H. Merrill, Jr. and C. C. Sweeley, Biochemistry of Lipids, Lipoproteins and Membranes, ed. D. E. Vance and J. E. Vance, Elsevier Science, Amsterdam, 1996, ch 12, pp. 309–339 Search PubMed; (b) J. Liao, J. Tao, G. Lin and D. Liu, Tetrahedron, 2005, 61, 4715 CrossRef CAS; Y. A. Hannun, J. Biol. Chem., 1994, 269, 3125 CAS 1; (c) T. Kolter and K. Sandhoff, Angew. Chem., Int. Ed., 1999, 38, 1532 CrossRef CAS; (d) R. C. Dickson, Annu. Rev. Biochem., 1998, 67, 27 CrossRef CAS; (e) T. Kolter and K. Sandhoff, Chem. Soc. Rev., 1996, 371 RSC; (f) Y. A. Hannun and R. M. Bell, Science, 1989, 243, 500 CAS.
- (a) C. A. Welsch, L. W. A. Roth, J. F. Goetschy and N. R. Movva, J. Biol. Chem., 2004, 279, 36720 CrossRef CAS; (b) M. T. Park, J. A. Kang, J. A. Choi, C. M. Kang, T. H. Kim, S. Bae, S. Kang, S. Kim, C. I. Choi, C. K. Cho, H. Y. Chung, Y. S. Lee and S. J. Lee, Clin. Cancer Res., 2003, 9, 878 CAS; (c) T. Natori, M. Morita, K. Akimoto and Y. Koezuka, Tetrahedron, 1994, 50, 2771 CrossRef CAS; (d) J. Turinsky and G. W. Nagel, Biochem. Biophys. Res. Commun., 1992, 188, 358 CrossRef CAS; (e) M. Honda, Y. Ueda, S. Sugiyama and T. Komori, Chem. Pharm. Bull., 1991, 39, 1385 CAS; (f) S. Dharmawardhane, B. Rubinstein and A. I. Stern, Plant Physiol., 1989, 89, 1345 CrossRef CAS; (g) A. H. Merrill, Jr., S. Nimkar, D. Menaldino, Y. A. Hannun, C. Loomis, R. M. Bell, S. R. Tyagi, J. D. Lambeth, V. L. Stevens, R. Hunter and D. C. Liotta, Biochemistry, 1989, 28, 3138 CrossRef.
- (a) K. Takamatsu, M. Mikami, K. Kikuchi, S. Nozawa and M. Iwamori, Biochim. Biophys. Acta, 1992, 1165, 177 CAS; (b) P. W. Wertz, M. C. Miethke, S. A. Long, J. S. Strauss and D. T. Downing, J. Invest. Dermatol., 1985, 84, 410 CrossRef CAS; (c) K. A. Karlsson, Acta Chem., Scand., 1964, 18, 2395 CrossRef CAS; (d) K. A. Karlsson, Acta Chem., Scand., 1964, 18, 2397 CrossRef CAS; (e) K. A. Karlsson, B. E. Samuelsson and G. O. Steen, Acta Chem., Scand., 1968, 22, 1361 CrossRef CAS; (f) K. Okabe, R. W. Keenan and G. Schmidt, Biochem. Biophys. Res. Commun., 1968, 31, 137 CrossRef CAS; (g) Y. Barenholz and S. Gatt, Biochem. Biophys. Res. Commun., 1967, 27, 319 CrossRef CAS; (h) D. E. Vance and C. C. Sweeley, J. Lipid Res., 1967, 8, 621 CAS.
- (a) M. Lombardo, M. G. Capdevila, F. Pasi and C. Trombini, Org. Lett., 2006, 8, 3303 CrossRef CAS; (b) D. Enders, J. Paleček and C. Grondal, Chem. Commun., 2006, 655 RSC; (c) D. Y. Jung, S. Kang, S. B. Chang and Y. H. Kim, Synlett, 2005, 14, 2183; (d) X. Lu, H. S. Byun and R. Bittman, J. Org. Chem., 2004, 69, 5433 CrossRef CAS; (e) S. Raghavan and A. Rajender, J. Org. Chem., 2003, 68, 7094 CrossRef CAS; (f) H. Y. Chiu, D. L. M. Tzou, J. N. Patkar and C. C. Lin, J. Org. Chem., 2003, 68, 5788 CrossRef CAS; (g) S. Raghavan, A. Rajender and J. S. Yadav, Tetrahedron: Asymmetry, 2003, 14, 2093 CrossRef CAS; (h) A. J. Ndakala, M. Hashemzadeh, R. C. So and A. R. Howell, Org. Lett., 2002, 4, 1719 CrossRef CAS; (i) T. Nakamura and M. Shiozaki, Tetrahedron, 2001, 57, 9087 CrossRef CAS; (j) L. He, H. S. Byun and R. Bittman, J. Org. Chem., 2000, 65, 7618 CrossRef CAS; (k) C. Martin, W. Prünck, M. Bortolussi and R. Bloch, Tetrahedron: Asymmetry, 2000, 11, 1585 CrossRef CAS; (l) O. Shirota, K. Nakanishi and N. Berova, Tetrahedron, 1999, 55, 13643 CrossRef CAS; (m) R. Imashiro, O. Sakurai, T. Yamashita and H. Horikawa, Tetrahedron, 1998, 54, 10657 CrossRef CAS.
- Y. M. Yun, T. B. Sim, H. S. Hahm, W. K. W. K. Lee and H.-J. Ha, J. Org. Chem., 2003, 68, 7675 CrossRef CAS.
- (a) H.-J. Ha, M. C. Hong, S. W. Ko, Y. W. Kim, W. K. Lee and J. Park, Bioorg. Med. Chem. Lett., 2006, 16, 1880 CrossRef CAS; (b) J. W. Kim, Y. W. Kim, Y. Inagaki, Y. A. Hwang, S. Mitsutake, Y. W. Ryu, W. K. Lee, H.-J. Ha, C. S. Park and Y. Igarashi, Bioorg. Med. Chem., 2005, 13, 3475 CrossRef CAS.
- W. K. Lee and H.-J. Ha, Aldrichimica Acta, 2003, 36, 57 CAS.
- V. Kumar and S. Dev, Tetrahedron, 1987, 43, 5933 CrossRef CAS.
- R. Imashiro, O. Sakurai, T. Yamashita and H. Horikawa, Tetrahedron, 1998, 54, 10657–10670 CrossRef CAS.
- (a) H. C. Kolb, M. S. VanNieuwenhze and K. B. Sharpless, Chem. Rev., 1994, 94, 2483 CrossRef CAS; (b) M. Tokles and J. K. Snyder, Tetrahedron Lett., 1986, 27, 3951 CrossRef CAS.
- (a) K. D. Lee, J. M. Suh, J. H. Park, H.-J. Ha, H. G. Choi, C. S. Park, J. W. Chang, W. K. Lee, Y. Dong and H. Yun, Tetrahedron, 2001, 57, 8267 CrossRef CAS; (b) Y. Dong, Y. H. Yun, C. S. Park, W. K. Lee and H.-J. Ha, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2003, 59, 659; (c) H.-Y. Noh, S. W. Kim, S. I. Paek, H.-J. Ha, H. Yun and W. K. Lee, Tetrahedron, 2005, 61, 9281 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details of all reactions and the calculated rotational energy barriers of 2a and 2b. See DOI: 10.1039/b612740a |
| This journal is © The Royal Society of Chemistry 2007 |