Highly stable electrochemical oxidation of phenolic compounds at carbon ionic liquid electrode
Afsaneh
Safavi
*,
Norouz
Maleki
* and
Fariba
Tajabadi
Department of Chemistry, College of Sciences, Shiraz University, Shiraz, 71454 Iran. E-mail: safavi@ chem.susc.ac.ir; Fax: +98-711-2286008; Tel: +98-711-6305881
First published on 14th November 2006
Abstract
A carbon ionic liquid electrode (CILE) was used for the investigation of the electrochemical oxidation of phenolic compounds in acidic media using cyclic voltammetry, chronoamperometry and square wave voltammetry techniques. The results indicate that, contrary to many other electrodes, the oxidation of phenolic compounds on CILE is highly stable and does not result in electrode fouling. Cyclic voltammetry showed that phenolic compounds such as phenol, 2,4-dichlorophenol and catechol were oxidized at CILE and remained electroactive after multiple cycles and at high concentrations of phenol. The cyclic voltammetric response of the CILE is very stable with more than 99% of the initial activity remaining after 20 s of stirring of a 0.5 mM solution of phenol.
Introduction
Phenols and related compounds are one of the major and persistent pollutants of the environment.1–3 In terms of phenols and phenolic compounds, waste treatment and detection are two major research directions. In both fields electrochemical methods are of prime importance.4–10Phenolic species can readily be oxidized at solid electrodes. It is well known that due to production of the phenoxy radical and its subsequent reaction with phenol a polymeric adherent film deposits on the electrode surface. This film prevents further electron transfer and causes electrode fouling. Another pathway is the reaction of phenoxy radical with hydroxyl radicals to produce ortho- and para-benzoquinone, and in some cases further oxidation is also observed.11–16
Electrochemical passivating film is strongly adherent and cannot be removed by washing with organic solvents. This film must be removed mechanically by polishing the electrode surface. Wang and Lin used in situ repetitive electrochemical treatment to prevent solid electrode fouling in the presence of various deactivating compounds. The optimum parameters for the electrochemical treatments of glassy carbon, however, are dependent on the electroactive reactants.17 Activation of an electrode surface by using lasers is another method that has been presented for solving fouling problems. Laser ablation was used to remove physically a fouling layer from platinum and gold electrodes. McCreery and colleagues used high-intensity infrared laser pulses to activate the surface and to increase the rate of the heterogeneous electron-transfer kinetics at glassy carbon and platinum electrodes.18–21 Oltra et al. additionally used laser pulses to de-passivate an iron electrode under hydrodynamic conditions in a flow cell.22 Fulian and Compton applied laser-activated voltammetry to the measurement of the diffusion coefficient of phenol.23
Passivation can be prevented by using an enzyme-based sensor. In this case the signal is not due to the oxidation at the electrode and thus electrode fouling does not occur at all. These methods are not in widespread use because of associated problems with using biological compounds.24–27
Carbon nanotube electrodes can be used to enhance the sensitivity and stability of the electrodes towards phenol oxidation. Chronoamperometric and cyclic voltammetric experiments show that surface-confined layers promote the phenol oxidation.28
Dimensionally stable anodes (DSAs) prepared by the deposition of a thin layer of metal oxides on a base metal, usually titanium29,30 and boron doped diamond (BDD) electrodes, are found to have resistance to phenol fouling,31–34 especially when treated at high anodic potentials.
Based on the above background information, it is still of prime importance to search for a new electrode material that can overcome the above problems and does not need treatment or pretreatment for prevention of electrode fouling.
In the present work, a carbon ionic liquid electrode (CILE) based on the use of pyridinium–n-octylpyridinum hexafluorophosphate (OPFP) as a binder is employed for phenol oxidation. In our previous works,35 CILE was introduced as a high performance electrode with many good features such as resistivity towards biomolecules (NADH, dopamine, catechol, ascorbic acid) fouling and provision of high rates of electron transfer. Cyclic voltammetry, chronoamperometry and square wave voltammetry were used to elucidate the phenol oxidation process on CILE. Similar experiments were conducted on a glassy carbon electrode (GCE), for comparison. The results illustrate that CILE exhibits excellent resistance to fouling, even in the presence of high concentrations of phenol.
Experimental
Reagents
1-Iodooctane, pyridine, catechol, hydroquinone, phenol, 2,4-dichlorophenol (2,4-DCP) and paraffin oil were obtained from Merck. Ammonium hexafluorophosphate and graphite powder (mesh size <100 µm) were supplied by Fluka. Phenolic compounds solutions were prepared in Britton–Robinson buffer (pH = 2). The ionic liquid, octylpyridinum iodide was synthesized as described elsewhere.36 The OPFP was obtained by anion exchange of octylpyridinum iodide with ammonium hexafluorophosphate.Electrode preparation
The carbon ionic liquid electrode (CILE), 1.8 mm diameter, was prepared using graphite powder and OPFP with a ratio of 50/50 (w/w), as described previously.35 A new surface was obtained by smoothing the electrode onto a weighing paper. Carbon paste electrode (CPE) was prepared by hand-mixing paraffin oil and graphite powder with 70:30 graphite–paraffin oil (w/w). The paste was packed into the cavity of a Teflon tube (1.8 mm diameter). An electrical contact was established via a stainless steel handle.Apparatus
Voltammetric measurements were performed using an Autolab electrochemical system (Eco-Chemie, Utrecht, The Netherlands) equipped with PGSTAT-12 and GPES software (Eco-Chemie, Utrecht, The Netherlands). The electrochemical cell was assembled with a conventional three-electrode system: an Ag/AgCl/KCl (3 M) reference electrode (Metrohm) and a platinum disk as a counter electrode. Different working electrodes used in this study were a carbon ionic liquid electrode (CILE), a carbon paste electrode (CPE) and a glassy carbon electrode (GCE) (2 mm diameter) (Metrohm). The cell was a one-compartment cell with an internal volume of 10 mL. All experiments were typically conducted at 25 ± 2 °C. Pure nitrogen was used for deaeration of solutions.Results and discussion
The comparative electrochemical behaviour of phenol (in Britton–Robinson buffer of pH 2) at GCE and CILE has been studied by cyclic voltammetry. Fig. 1 shows repetitive cyclic voltammograms recorded at GCE, traditional carbon paste electrode (CPE) and CILE. As expected in the case of GCE and CPE, the shape and height of the oxidation peak dramatically change after the first scan and the signal is not detectable after several scans. Reduction signals in the reverse scan are also hardly detectable. Thus, as suggested previously,37,38 the GCE and CPE display a rapid loss of activity due to the formation of a polymeric passivating layer. Such fouling is not observed at CILE. Fig. 1C shows cyclic voltammetric scans for phenol at a CILE surface. It can be seen that the CILE showed different behaviour towards the oxidation of phenol compared with GCE and CPE. It displays a sharper single anodic peak at about +0.98 V in the first scan, corresponding to phenol oxidation. The peak current for 0.5 mM phenol was linearly proportional to ν1/2 within the range of 25–600 mV s−1, indicating that the oxidation peak current is diffusion controlled. This peak was stabilized after the second scan, along with the appearance of other oxidation and reduction peaks (due to the products of phenol oxidation) which had a gradual growth with the increase in the number of cyclic scans. Fig. 1C shows the assignment of the products of phenol oxidation at CILE. It has been reported previously that the main products in phenol oxidation are hydroquinone (p-benzoquinone) and catechol (o-benzoquinone).14 The evolution of these products was confirmed by comparing the voltammograms obtained for authentic compounds in the presence and the absence of phenol. | ||
| Fig. 1 Successive scans of 0.5 mM phenol in Britton–Robinson buffer (pH 2). (A): GCE, (B): CPE, (C): CILE. a: first scan, b: fifth scan, c: baseline in buffer solution. | ||
It is also obvious from Fig. 1C that more repeatable responses are, in fact, observed in the case of CILE and the electrode surface is very resistant to fouling. The excellent behaviour after several voltammetric scans implies negligible polymerization of phenoxy radicals. Such a good state of the surface is attributed to the use of a different substrate material (ionic liquid) at which the phenoxy products do not polymerize and thus do not block the surface.
Fig. 2 shows the first scan and the fifth scan of 0.5 mM solution of phenol at the CILE. Now, if after the fifth scan the solution is stirred for about 20 s (at a rate of 1500 min−1) and then another scan is taken, the oxidation peak current is recovered. At other electrodes, such as GCE or BDD, a passivating and strongly adherent polymer layer is formed on the surface which cannot be washed away with organic solvents. The GCE electrode is recovered by scrubbing33 and the BDD electrode can be regenerated by high anodic potential treatment.33,39,40 This point again confirms the absence of any polymerized phenol on CILE.
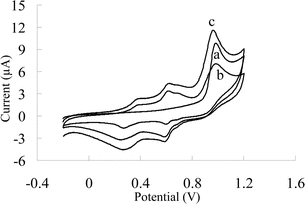 | ||
| Fig. 2 Cyclic voltammograms of 0.5 mM phenol in Britton–Robinson buffer (pH 2) at CILE. (a) First scan, (b) fifth scan, (c) first scan after fifth scan with 20 s stirring the solution (stirring rate 1500 min−1). | ||
2,4-Dichlorophenol (2,4-DCP) was studied as a representative of chlorophenol compounds. The cyclic voltammetric oxidation peak potential was found to be 0.895 V versus Ag/AgCl reference electrode (Fig. 3), which is less positive than that of phenol (Fig. 1C). This trend is in agreement with that obtained on GCE41 and BDD electrodes.33 After the first cycle, new peaks appeared at CILE similar to the case of phenol. This behaviour is similar to that of the BDD electrode towards 2,4-DCP. These peaks have been attributed to chlorinated benzoquinone.33,42 Here again, the CILE shows good resistance towards fouling.
 | ||
| Fig. 3 Successive scans of 0.5 mM 2,4-dichlorophenol in Britton–Robinson buffer (pH 2) on CILE. | ||
Another phenolic compound which was studied was catechol. Catechol showed one quasi-reversible redox couple with a ΔEp of 0.165 V in Britton–Robinson buffer (pH 2). Repetitive scans showed no fouling of the electrode.
Chronoamperometry was used further to show the stability of the CILE. Fig. 4 displays potentiostatic I-t curves in a stirred solution of 0.1 mM of phenol and 2,4-DCP at an applied potential of 1.2 V on GCE and CILE. It can be seen that the CILE is active after 15 min, while GCE shows a chronoamperometric curve which decays with time, indicating the fact that GCE has been fouled in the presence of these phenolic compounds.
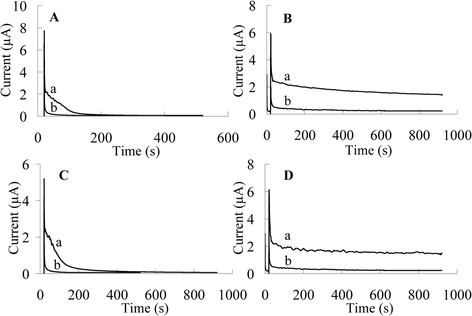 | ||
| Fig. 4 Chronoamperometric curves of 0.1 mM: (A) and (B) phenol, (C) and (D) 2,4-dichlorophenol in Britton–Robinson buffer (pH 2) on (A) and (C) GCE, (B) and (D) CILE at an applied potential of 1.2 V (stirring rate: 500 min−1). | ||
CILE stability was also examined by repetitive measurements at high phenol concentrations. This was examined because it has been observed previously that fouling of the electrode is pronounced at higher phenol concentrations.37 For 5 mM phenol at GCE, the current falls to baseline after the second scan (not shown). Fig. 5 displays the cyclic voltammogram of 5 mM phenol at CILE. After five scans the signal deteriorates. However, if the solution is stirred for 60 s, the signal recovers by 85%.
 | ||
| Fig. 5 Cyclic voltammograms of 5 mM phenol in Britton–Robinson buffer (pH 2) at CILE. (a) First scan, (b) fifth scan, (c) first scan after fifth scan (60 s stirring solution between (b) and (c)). | ||
The stability of the electrode can be attributed to the ionic liquid content of the electrode surface and its polarity. The ionic liquid content of the electrode seems to dissolve the reaction products and holds them at the electrode for reduction and oxidation, as is shown in Fig. 1C. In addition, as was described previously, the electrode kinetics are faster on the CILE than other electrodes.
The voltammogram of phenol at carbon electrodes is known to exhibit a linear dependence of the anodic peak potential on pH, with a slope of 58 mV per pH under pH 10.43 At CILE, the Epversus pH plot is linear below pH 10 with a slope of 59.8 mV per pH, indicating the participation of equal numbers of protons and electrons in the oxidation reaction. From this plot, pKa was found to be 10.2, which is in good agreement with the pKa of phenol reported previously (10.3).44
Square wave voltammetric determination of phenol
In view of their high toxicity, reliable analytical procedures are required for the sensitive determination of phenolic compounds at low levels in various matrices. The electroanalytical techniques applied so far, for the direct determination of these compounds, suffer a number of drawbacks due to electrode fouling.14,37 In this work, we examined the direct electroanalysis of phenol at CILE surface. Fig. 6 shows square wave voltammamograms of different concentrations of phenol and the calibration curves obtained. | ||
| Fig. 6 Calibration curves for different concentrations of phenol. | ||
As is obvious, two calibration ranges for low levels, 25–80 (ng ml−1), Fig. 6 (inset), and higher levels, 80–200 (ng ml−1), were obtained. The equations for the calibration lines were Y = 0.0106X − 0.275, R2 = 0.998, and Y = 0.0596X − 4.143, R2 = 0.992, for low and high levels, respectively (where X in the above equations is the phenol concentration in ng ml−1).
Conclusions
The voltammetric investigation described in this study demonstrates that electrooxidation of phenolic compounds at the surface of CILE showed very distinct characteristics.Results demonstrated that by using a CILE as the working electrode, the problem of electrode fouling reported for most electrodes in phenolics oxidation processes can be circumvented without the need for any electrode pretreatment. The outstanding stability of CILE compared with other electrodes such as GCE is mainly due to the presence of ionic liquid used as the binder in the electrode. Based on the data reported here, a possible reaction mechanism was proposed for the oxidation of phenol. The proposed mechanism indicates that the oxidation product of phenol is not polymerized and not deposited at the electrode surface; instead hydroquinone and catechol are formed as the main oxidation products of phenol.
In order to extend the electrochemical investigation to chlorophenol derivatives, 2,4-dichlorophenol (2,4-DCP) was studied as a representative of chlorophenol compounds. The results showed similar mechanism as phenol for 2,4-DCP.
Highly stable responses are achieved even at high concentrations of phenol. This property of the CILE makes it an ideal sensor for quantitative determination of traces of phenol and chlorophenol derivatives.
Acknowledgements
The authors wish to acknowledge the support of this work by the Iranian Ministry of Sciences, Research and Technology.References
- European Community directive (80/778/EEC).
- US Environmental Protection Agency EPA/600/x-87/12.
- Mutagenicity Testing in Environmental Pollution Control, eds. F. Zimmermann and R. Tayler-Mayer, Wiley, New York, 1985 Search PubMed.
- J. Grimm, D. Bessarabov and R. Sanderson, Desalination, 1998, 115, 285 CrossRef CAS.
- G. Chen, Sep. Purif. Technol., 2004, 38, 11 CrossRef CAS.
- K. B. Korbahti and A. Tanyolac, Water Res., 2003, 37, 1505 CrossRef.
- K. Yamada, Y. Akiba, T. Shibuya, A. Kashiwada, K. Matsuda and M. Hirata, Biotechnol. Prog., 2005, 21, 823 CrossRef CAS.
- Z. Wu and M. Zhou, Environ. Sci. Technol., 2001, 35, 2698 CrossRef CAS.
- C. Nistor, J. Emne'us, L. Gorton and A. Ciucu, Anal. Chim. Acta, 1999, 387, 309 CrossRef CAS.
- G. Marko-Varga, J. Emne'us, L. Gorton and T. Ruzgas, Trends Anal. Chem., 1995, 14, 319 CrossRef CAS.
- M. Pham, P. Lacaze and J. Dubois, J. Electroanal. Chem., 1978, 86, 147 CrossRef.
- J. Wang and T. Martinez, J. Electroanal. Chem., 1991, 313, 129 CrossRef CAS.
- J. Wang, M. Jiang and L. Lu, J. Electroanal. Chem., 1998, 444, 127 CrossRef CAS.
- M. Gattrell and D. W. Kirk, J. Electrochem. Soc., 1993, 140, 903 CrossRef.
- P. I. Iotov and S. V. Kalcheva, J. Electroanal. Chem., 1998, 442, 19 CrossRef CAS.
- S. Andreescu, D. Andreescu and O. A. Sadik, Electrochem. Commun., 2003, 5, 681 CrossRef CAS.
- J. Wang and M. S. Lin, Anal. Chem., 1988, 60, 499 CrossRef CAS.
- M. Poon and R. L. McCreery, Anal. Chem., 1986, 58, 2256 CrossRef.
- M. Poon and R. L. McCreery, Anal. Chem., 1987, 59, 1615 CrossRef CAS.
- M. Poon, R. L. McCreery and R. Engstrom, Anal. Chem., 1988, 60, 1725 CrossRef CAS.
- R. K. Jaworski and R. L. McCreery, J. Electrochem. Soc., 1993, 140, 1360 CAS.
- R. Oltra, G. M. Indrianjafy, M. Keddam and H. Takenouti, Corros. Sci., 1993, 35, 827 CrossRef CAS.
- Q. Fulian and R. G. Compton, Anal. Chem., 2000, 72, 1830 CrossRef.
- C. Saby, K. B. Male and J. H. T. Luong, Anal. Chem., 1997, 69, 4324 CrossRef CAS.
- F. D. Munteanu, A. Lindgren, J. Emneus, L. Gorton, T. Ruzgas, E. Csoregi, A. Ciucu, R. B. van Huystee, I. G. Gazaryan and L. M. Lagrimini, Anal. Chem., 1998, 70, 2596 CrossRef CAS.
- Z. Liu, B. Liu and J. Deng, Anal. Chem., 2000, 72, 4707 CrossRef CAS.
- S. S. Rosatto, L. T. Kubota and G. O. Neto, Anal. Chim. Acta, 1999, 390, 65 CrossRef CAS.
- J. Wang, R. P. Deo and M. Musameh, Electroanalysis, 2003, 15, 1830 CrossRef CAS.
- Y. J. Feng and X. Y. Li, Water Res., 2003, 37, 2399 CrossRef CAS.
- X. Y. Li, Y. H. Cui, Y. J. Feng, Z. M. Xie and J. Y. Gu, Water Res., 2005, 39, 1972 CrossRef CAS.
- J. Iniesta, P. A. Michaud, M. Panizza, G. Cerisola, A. Aldaz and A. C. Comninellis, Electrochim. Acta, 2001, 46, 3573 CrossRef CAS.
- P. L. Hagans, P. M. Natishan, B. R. Stoner and W. E. O'Grady, J. Electrochem. Soc., 2001, 148, E298 CrossRef CAS.
- C. Terashima, T. N. Rao, B. V. Sarada, D. A. Tryk and A. Fujishima, Anal. Chem., 2002, 74, 895 CrossRef CAS.
- A. Morao, A. Lopes, M. T. Pessoa de Amorim and I. C. Gonçalves, Electrochim. Acta, 2004, 49, 1587 CAS.
- N. Maleki, A. Safavi and F. Tajabadi, Anal. Chem., 2006, 78, 3820 CrossRef CAS; A. Safavi, N. Maleki, O. Moradlou and F. Tajabadi, Anal. Biochem., 2006 DOI:10.1016/j.ab.2006.09.008.
- E. Fisicaro, A. Ghiozzo, E. Pelizzetti, G. Viscaradi and P. L. Quagliotto, J. Colloid Interface Sci., 1996, 182, 549 CrossRef CAS.
- J. Wang and R. Li, Anal. Chem., 1989, 61, 2809 CrossRef CAS.
- I. Rodrıguez, J. A. Munoz Leyva and J. L. Hidalgo Hidalgo de Cisneros, Analyst, 1997, 122, 601 RSC.
- M. Panizza, I. Duo, P. A. Michaud, G. Cerisola and C. Comninellis, Electrochem. Solid-State Lett., 2000, 3, 429 CAS.
- G. Foti, D. Gandini, C. Comninellis, A. Perret and W. Haenni, Electrochem. Solid-State Lett., 1999, 2, 228 CrossRef CAS.
- J. D. Rodgers, W. Jedral and N. J. Bunce, J. Environ. Sci. Technol., 1999, 33, 1453 Search PubMed.
- A. M. Polcaro and S. Palmas, Ind. Eng. Chem. Res., 1997, 36, 1791 CrossRef CAS.
- V. F. P. Bub, K. Wisser, W. J. Lorenz and W. Heimann, Ber. Bunsen-Ges. Phys. Chem., 1973, 77, 823.
- J. R. A. Pollock and R. Stevens, Dictionary of Organic Compounds, Eyre and Spottiswoode, London, 4th edn, 1965 Search PubMed.
| This journal is © The Royal Society of Chemistry 2007 |