Freezing transitions in mixed surfactant/alkane monolayers at the air–solution interface
Katherine M.
Wilkinson
,
Lei
Qunfang†
and
Colin D.
Bain‡
*
Department of Chemistry, University of Oxford, Chemistry Research Laboratory, Mansfield Road, Oxford, UK OX1 3TA. E-mail: c.d.bain@durham.ac.uk
First published on 14th November 2005
Abstract
Mixed monolayers at the air–water interface of the cationic surfactant, hexadecyltrimethylammonium bromide (CTAB), with alkanes show a first-order freezing transition as a function of temperature. Sum-frequency spectra and ellipsometric measurements are consistent with a structure in which the high-temperature phase is liquid-like and the low-temperature phase has all-trans, upright chains. There are strong structural similarities between the low-temperature phase in the mixed monolayers and frozen monolayers at the alkane–air and alkane–CTAB solution interfaces. The difference between the surface freezing point Ts and the freezing point of the bulk alkane Tb ranges from 1 °C for alkane chain length m = 17, to 28 °C for m = 11. Surface freezing is more favourable in mixed monolayers at the air–water interface than at the bulk alkane–water interface for the same surfactant concentration. Long-chain alkanes do not wet water, but it is postulated that if they did, they would also show surface freezing analogously to alkanes on silica. The surfactant plays the dual role of enhancing wetting and surface freezing of the alkane on water.
Introduction
Surface phase transitions in insoluble monolayers of organic molecules on the surface of water have been studied intensively in the century since the pioneering work of Pockels, Langmuir and Harkins.1 The development of grazing incidence X-ray diffraction in the 1980's permitted the structural determination of a wide range of mesophases differing in the lattice distortion, direction of tilt of the hydrocarbon chains, and long-range correlations in the twist of the chains.2 Many of these phases are metastable with respect to bulk 3-dimensional crystallites, but can be formed by careful spreading and compression of the amphiphile on the surface of water. In contrast to the rich phase behaviour of insoluble (Langmuir) monolayers, the adsorbed films of most soluble surfactants (Gibbs monolayers) have uninteresting phase diagrams, showing only a single fluid phase over the whole range of concentrations. There is no hard and fast distinction between soluble and insoluble monolayers: the classification depends as much on the timescale of the experiment as on the absolute solubility of the surfactant. It is therefore almost inevitable that surfactants exist that are sufficiently soluble to form monolayers at equilibrium with a bulk solution, yet which have strong enough intermolecular interactions to show a liquid–solid monolayer phase transition at the air–water interface. The simplest of these molecules are the normal alcohols, which for chain lengths m > 9 show a liquid–solid phase transition, analogous to the L1–LS phase transition in Langmuir monolayers, at a temperature about 15 K above the bulk melting point of the alcohols.3 (Since the 1990s, phase transitions have been studied in a number of other sparingly soluble surfactants, such as monoethyleneglycol tetradecyl ether (C14E1),4,5 monoethyleneglycol dodecyl ether (C12E1),5n-hexadecyl phosphate,6–8n-dodecyl-γ-hydroxybutyric acid amide (DHBAA),9,10 2-hydroxyethyl laurate (2-HEL),11 and the catanionic surfactants dodecyltrimethylammonium dodecylsulfate (DTA.DS) and decyltrimethylammonium decylsulfate (DeTA.DeS).12Common, short-chain, micelle-forming surfactants such as alkyltrimethylammonium bromides (CnTABs) and sodium dodecylsulfate (SDS) do not, on their own, form monolayers in condensed phases. These ionic surfactants do, however, form 2D solid phases in the presence of n-alcohols. The best known example is SDS, which when mixed with dodecanol at very low levels (≪1%) forms a condensed monolayer. Since commercial samples of SDS are always contaminated with dodecanol, SDS solutions show anomalous properties, such as increased foam stability, due to the formation of mixed monolayers of SDS and dodecanol in a solid phase. We have previously studied mixtures of CnTAB (n = 12, 14, 16) and SDS with dodecanol and characterised the 2D phase transition in these mixed monolayers by sum-frequency spectroscopy (SFS) and ellipsometry.13,14
The focus of this paper is on freezing transitions in mixtures of CnTABs with linear alkanes, neither of which will form a condensed monolayer on water on their own. CnTABs form highly disordered, liquid-like monolayers at all coverages up to the saturation value, which is reached at the critical micelle concentration (cmc). Pure alkanes with chain lengths m > 7 do not spread on water, but instead form a lens in equilibrium with a dilute 2D gas of alkane molecules on the water surface.15 If surfactants are progressively added to the aqueous sub-phase a concentration is reached where the alkane undergoes a first-order wetting transition to form a mixed monolayer of surfactant and alkane, coexisting with a lens of alkane:16,17 a situation known as ‘pseudo-partial wetting’18 or ‘frustrated complete wetting’.15
We have shown in a preliminary communication that upon cooling a mixed monolayer of C16TAB and tetradecane in the pseudo-partial wetting regime, a further first-order phase transition is observed from a liquid-like, conformationally disordered monolayer to a highly conformationally ordered solid phase.19 We will refer to this change as a freezing transition. The discontinuous nature of the freezing transition is shown clearly by ellipsometric measurements of the coefficient of ellipticity, ![[small rho, Greek, macron]](https://www.rsc.org/images/entities/i_char_e0d1.gif) (see below for a definition), as a function of temperature (Fig. 1). The transition (indicated by a dashed line) occurs 12 °C above the bulk freezing point of tetradecane (Tb = 6 °C).
(see below for a definition), as a function of temperature (Fig. 1). The transition (indicated by a dashed line) occurs 12 °C above the bulk freezing point of tetradecane (Tb = 6 °C).
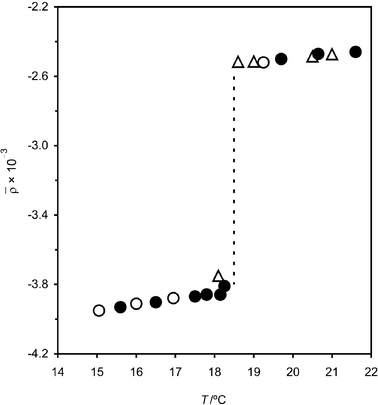 | ||
| Fig. 1 Variation in the coefficient of ellipticity, , with temperature, T, for the surface of a 0.60 mM solution of C16TAB in H2O in the presence of lenses of tetradecane: (solid symbols) heating cycles; (open symbols) cooling cycles. The triangles and circles refer to different samples. | ||
In this paper, we use sum-frequency spectroscopy and ellipsometry to elucidate the structure of the phases on either side of the freezing transition and explore the chain length dependence in mixed monolayers of C16TAB with alkanes CH3(CH2)m−2CH3 with m = 11–17 (denoted Cm). Qualitatively different behaviour is observed with longer alkanes, which will be the subject of a separate paper.
Although long-chain alkanes do not form frozen monolayers on water—for the simple reason that they do not spread—alkanes do have an unusual property that is of relevance to the phenomena we report here: they exhibit surface freezing.20–22 For chain lengths m = 16–50, a monolayer of alkane molecules at the vapour–liquid interface freezes at a temperature, Ts, up to 3 °C above the bulk freezing point, Tb. For lower values of m, this frozen monolayer is in an upright rotator phase with hexagonally packed chains: the same structure as the LS phase in insoluble monolayers on water. A single monolayer of solid alkane persists down to the bulk freezing point. The connection to the work in this paper is made clearer by the fact that surface freezing is not observed at the pure water–alkane interface23 but can be induced at the solution–alkane interface by the presence of the cationic surfactant, C16TAB.24 The entropy changes of the freezing transitions at the solution–alkane and alkane–air interfaces are in excellent agreement and ellipsometric measurements on the solution–alkane interface are well-explained by a frozen mixed monolayer in an upright, rotator phase, analogous to that observed at the alkane–air interface. A comparison of the freezing transitions in mixed monolayers of surfactant and alkane at the air–water and alkane–water interface concludes this paper.
The two experimental techniques used in this paper are sum-frequency spectroscopy and ellipsometry; a brief introduction to these two techniques is provided here.
Sum-frequency spectroscopy (SFS) is a form of surface-sensitive non-linear vibrational spectroscopy in which pulsed visible (frequency ωvis) and infrared laser beams (ωIR) are overlapped at a surface and light is emitted coherently at the sum of the two incident frequencies: ωSF = ωvis + ωIR.25–31
Within the electric dipole approximation, the intensity of the sum-frequency (SF) signal, I(ωSF), depends on the intensities of the visible and IR beams and the square of the second-order susceptibility tensor, χ(2):
| I(ωSF) ∝ |χ(2)|2I(ωvis)I(ωIR) | (1) |
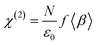 | (2) |
Three key features of SFS are highlighted here. First, χ(2) is a third-rank tensor, which vanishes by symmetry in centrosymmetric or isotropic environments. Therefore, there will be no SF signal from the bulk fluids on either side of a gas–liquid interface but only from molecules at the interface that have net orientation and hence break the symmetry of the bulk phases. Second, β is negligible except in the vicinity of a vibrational resonance that is both IR- and Raman-active. Varying the frequency of the infrared laser and recording the SF signal as a function of ωIR yields a vibrational spectrum. Third, the hyperpolarisability, β, is also a third-rank tensor which is zero by symmetry for a centrosymmetric molecule. An all-trans hydrocarbon chain is locally centrosymmetric (the middle of each C–C bond is a centre of inversion) and therefore the methylene vibrations in an all-trans chain are SF-inactive. Gauche defects break the symmetry and give rise to SF-allowed CH2 vibrations, provided that the overall symmetry of the interface is not isotropic (in which case β is non-zero but <β> vanishes). As a consequence of this orientational averaging, chemical groups that have a strong preferred orientation generally give stronger SF signals than those that have a broad distribution of orientations.
In ellipsometry, a polarised laser beam is reflected from an interface and the change in the state of polarisation is measured. In the form of ellipsometry used in this paper, the measured quantity is the coefficient of ellipticity, , which is defined as the imaginary part of rp/rs at the Brewster angle, where rp and rs are the complex amplitude reflection coefficients for p- and s-polarised light, respectively. The Brewster angle is defined as the angle where the real part of rp/rs is zero. The coefficient of ellipticity is a sensitive function of the profile of the optical dielectric constant (ε = n2, where n is the refractive index) through the interface. For a thin isotropic film between two semi-infinite media with dielectric constants ε1 and ε2, is given by the Drude equation:32
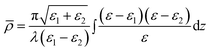 | (3) |
from each layer.
Monolayers on water are more accurately described as being optically uniaxial, with the extraordinary axis along the surface normal. In this case, the Drude equation has to be modified to read
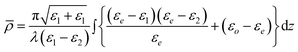 | (4) |
Experimental
Materials
Hexadecyltrimethylammonium bromide (C16TAB: Fluka, >99%) was recrystallised three times from an acetone–methanol mixture. Recrystallised, fully-deuterated hexadecyltrimethylammonium bromide (d-C16TAB) was a gift from Dr R. K. Thomas, University of Oxford. Tetradecyltrimethylammonium bromide (C14TAB), recrystallised three times from acetone with a small amount of ethanol, was kindly provided by Prof. P. D. I. Fletcher, University of Hull. Alkanes, Cm, with m = 11–16 (Aldrich, 99+%) or 17 (Sigma, 99%) were percolated 2–3 times through neutral alumina before use. Deuterated tetradecane (d-C14: Cambridge Isotope Laboratories, 98% D) was percolated once through neutral alumina. Solutions for ellipsometry were prepared in H2O (Elga, UHQ) and for sum-frequency spectroscopy in D2O (Aldrich, 99.9%).Ellipsometry
Ellipsometric measurements were performed on a Picometer Ellipsometer (Beaglehole Instruments, Wellington, NZ) equipped with a HeNe laser at 632.8 nm. Surfactant solutions were held in a 5 cm diameter glass dish in a sealed thermostatted cell with fused silica windows for the laser beams to pass through. Around 1 µL of alkane at room temperature was added to the surfaces of samples using a glass syringe with a stainless steel needle. The temperature was varied in discrete intervals with an equilibration time of ca. 45 min between each measurement. Increments of 1–2 °C were used away from the transition and <1 °C close to the transition. The temperature of the sample was measured directly to a precision of 0.1 °C with a thermocouple that was fixed in position throughout the experiment. Measurements were recorded every 1 s and averaged over 50 s. Readings were considered to be stable if varied by less than 2 × 10−5 over a period of 10 min. The precision of the averaged values of was typically better than ±1 × 10−5.
The precise value of the phase transition temperature is sensitive to impurities: freshly percolated alkanes and freshly recrystallised surfactants tended to give higher values of Ts. To provide an indication of reproducibility, multiple repeat measurements on the C16TAB + C14 system gave a range of values of Ts = 16.7–18.8 °C. Experiments in which one of the components was deuterated gave transition temperatures 2–3 °C lower. This difference may be ascribed partially to the lower purity of the deuterated compounds, but also reflects the thermodynamics of the phase transition: deuterated alkanes have lower melting points than normal alkanes.36
Sum-frequency spectroscopy
Sum-frequency spectroscopy (SFS) was carried out as described previously.29,37 A pulsed fixed-frequency visible beam (532 nm, s-polarised, 4 ns pulse length, 20 Hz repetition rate, 1 mm diameter, 10 mJ pulse−1) was overlapped with a tunable infrared beam (3000–2800 cm−1, p-polarised, ∼1 ns pulse length, 0.4 mm diameter, 0.4–0.7 mJ pulse−1) at the air–water interface. The beams were directed onto the surface at θvis = 55° and θIR = −50° (the negative sign indicating that two laser beams were counter propagating). The beams were incident at a point away from the centre of the sample and the sample was rotated at 2 rpm to reduce the effect of local heating by the IR laser beam. The use of D2O rather than H2O as a subphase further reduces local heating and decreases the non-resonant background signal from the subphase. The s-polarised sum-frequency (SF) light was emitted in a narrow beam at a well defined angle θSF = 37–38° depending on the IR wavelength and was detected with a liquid nitrogen-cooled CCD camera (Princeton Instruments). The SF signal from the sample was referenced to a signal obtained simultaneously from a GaAs crystal. After background subtraction, sample spectra were normalised to the reference spectra and fitted as described elsewhere.37,38Samples were held in a 6 cm glass dish in a thermostatted cell covered by a lid with slits for entry and exit of the laser beams. The temperature of samples was measured with a thermocouple before each spectrum to a precision of ±0.1 °C. Over the course of a spectrum the temperature of the sample varied by <0.5 °C.
Results
Sum-frequency spectroscopy (SFS)
In our preliminary communication, we presented SF spectra of the surface of a solution of C16TAB in the presence of tetradecane, above and below the freezing transition. Since both the surfactant and alkane contained hydrocarbon chains, these spectra did not distinguish between the two species. Here we use selective deuteration to look separately at the conformational order in the two components in the monolayer.Fig. 2 shows SF spectra in the C–H stretching region (2800–3000 cm−1) of monolayers of (A) h-C16TAB + h-C14 (denoted h + h), (B) h-C16TAB + d-C14 (denoted h + d), (C) d-C16TAB + h-C14 (denoted d + h) and (D) h-C16TAB alone, where h indicates a normal, protonated molecule and d a fully deuterated molecule. Fig. 2(A) thus contains contributions from both the surfactant and the alkane, while Fig. 2(B) shows just the surfactant and Fig. 2(C) just the alkane. The assignment of the peaks in Fig. 2 is well-established.39,40 The strongest peak at 2878 cm−1 is assigned to the symmetric methyl stretch (r+) and the peak at 2937 cm−1 is a Fermi resonance of the r+ mode with overtones of the methyl modes (r+FR). The behaviour of the r+FR peak parallels that of the r+ peak and will not be discussed further. The peak at 2850 cm−1, which is observed principally above Ts, is the symmetric methylene stretch (d+). The antisymmetric methylene (d−) and methyl (r−) stretches are weak in SSP-polarised spectra: the latter appears as a shoulder on the high frequency side of the r+FR mode. Previous SF studies on monolayers of partially deuterated C14TAB have shown that the vibrations of the trimethylammonium headgroup are not detected.41
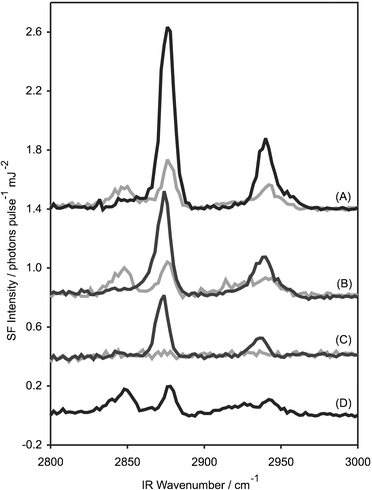 | ||
| Fig. 2 SF spectra at the air–solution interface of a 0.60 mM solution of C16TAB in D2O in the presence of lenses of tetradecane. T > Ts (grey lines) and T < Ts (black lines). (A) h-C16TAB and h-tetradecane, (B) h-C16TAB and d-tetradecane, (C) d-C16TAB and h-tetradecane, and (D) h-C16TAB without tetradecane at 15 °C. The visible and sum-frequency fields were s-polarised and the IR field was p-polarised (denoted SSP). | ||
For quantitative analysis of SF spectra, the spectra need to be deconvoluted into their constituent peaks. Each peak is assigned a line strength, Sν, resonant frequency, ων, and two linewidths corresponding to homogeneous and inhomogeneous broadening; these parameters are then minimised subject to some constraints. The detailed fitting procedure is described elsewhere,38 here we will merely discuss the line strengths, S, that are deduced from the fits (See Table 1). For an isolated peak, the SF intensity, I ∝ S2; however, interference between neighbouring peaks and with the weak non-resonant background from the subphase can significantly alter the shape and intensity of peaks in an SF spectrum.
| Below Ts | Above Ts | Pure | |||||||
|---|---|---|---|---|---|---|---|---|---|
| h + d | d + h | Sum | h + h | h + d | d + h | Sum | h + h | C16TAB | |
| S(r+) | 3.2 | 1.8 | 5.0 | 4.4 | 1.8 | 0.0 | 1.8 | 2.2 | 1.2 |
| S(d+) | 0.1 | 0.3 | 0.4 | 0.25 | 1.4 | 0.0 | 1.4 | 1.4 | 1.1 |
Below Ts, the d + h spectrum shows a sharp r+ mode and only a very weak d+ mode. The value of S(r+) is ∼60% of that of the same mode in the h + d spectrum. At first sight this result is very surprising: the low value of the ratio S(d+)/S(r+) would indicate a chain that was predominantly all-trans; but tetradecane in an all-trans configuration is centrosymmetric and hence sum-frequency inactive. The electric fields at ωSF from the two methyl groups are π out-of-phase and should cancel out in the far field. It is conceivable that <cos θ> might be different for the two ends of the alkane molecule; for example, if there were a greater population of chain defects at one end of the chain. Comparison of the Figs. 2(A) and (B) show that the CH3 groups of C16TAB and C14 contribute with the same phase and consequently the methyl group of the tetradecane at the outer side of the monolayer makes a larger contribution than the opposite terminus. Chain disorder tends to increase towards the air side of a monolayer, which would result in a larger contribution to the SF field from the inner methyl group, in contradiction to experiment. We believe that the net signal from the tetradecane molecules arises from a break-down in the electric dipole approximation, which assumes that the electric fields do not vary over the dimension of the molecule. Electric quadrupole SF transitions, which depend on gradients in the fields, are permitted for centrosymmetric molecules. As a result of the rapid changes in the dielectric constant across the interface, the local fields experienced at the air–monolayer and monolayer–water interfaces are significantly different. This effect has been observed previously by Sefler et al. for liquid eicosane (C20) below Ts, where a single frozen layer of eicosane molecules exists on the surface.45 The experimental data of Sefler et al. can be rationalised if the contribution of the lower methyl group to the sum-frequency field is 50–60% of that from the upper methyl group. If a similar degree of cancellation occurs in the mixed C16TAB + alkane monolayers, the composition of the mixed monolayer in the solid phase may be estimated from the line strengths of the methyl groups, with the tetradecane line strength corrected to allow for the (negative) contribution of the lower methyl group. We then find that the mixed monolayer contains 40–50% surfactant, which is in the same range as in mixed monolayers of CnTAB + dodecanol and SDS + dodecanol.13,14
Ellipsometry
The coefficient of ellipticity is plotted as a function of T for the C16TAB + C14 system in Fig. 1. A sharp phase transition is observed at a temperature of 18.5 °C. Careful heating and cooling cycles show that there is negligible hysteresis in the transition. Fig. 3 gathers together data for C16TAB + Cm monolayers with m = 11–17. In Fig. 3, the difference between heating and cooling cycles has been suppressed for clarity. The concentration of C16TAB was c = 0.60 mM except for m = 11, where we used 0.30 mM. The surfactant is below its Krafft temperature in all these experiments (TK = 25 °C) and crystals of C16TAB appeared at the lowest temperatures when c = 0.60 mM was used.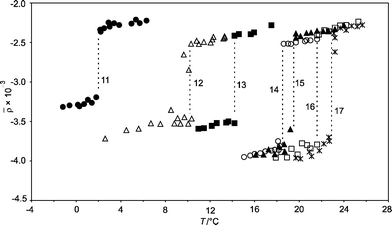 | ||
| Fig. 3 Coefficient of ellipticity, , against temperature, T, for the air–solution interfaces of C16TAB solutions in H2O in the presence of lenses of alkane with chain lengths, m (labelled on figure). c = 0.30 mM (m = 11) and 0.60 mM (m = 12–17). | ||
For every system there was a discontinuous change in (T) corresponding to a first-order transition between a liquid monolayer and a solid monolayer. Within error, the heating and cooling curves for each alkane were identical and the discontinuities occurred at the same temperatures, Ts(m) (marked by dashed vertical lines). The values of Ts(m) increased with increasing m and were always above the bulk melting point, Tb(m), of the alkane: just below Ts the solid monolayer was in equilibrium with a liquid lens. The transition temperatures are reported in Table 2.
| n | m | c/mM | T s/°C | Γ/µmol m−2 | σ/mN m−1 | d c(l)/Å | d c(s)/Å |
|---|---|---|---|---|---|---|---|
| a Approximated as Γ at 25 °C in the absence of oil.50 b From neutron reflection results at 25 °C.43 c Approximated as σaw at 25 °C for C16TAB + C12.49 d At 25 °C.43 | |||||||
| 16 | 11 | 0.30 | 2 | 2.8a | 45c | 12 | 18 |
| 16 | 12 | 0.60 | 10 | 3.4a | 40c | 12 | 18 |
| 16 | 13 | 0.60 | 14 | 3.4a | 40c | 11 | 19 |
| 16 | 14 | 0.60 | 18 | 3.4a | 40c | 12 | 21 |
| 16 | 15 | 0.60 | 19 | 3.4a | 40c | 11 | 20 |
| 16 | 16 | 0.60 | 22 | 3.4a | 40c | 11 | 21 |
| 16 | 17 | 0.60 | 22 | 3.4a | 40c | 11 | 20 |
| 14 | 12 | 1.20 | 0 | 2.7b | 43d | 9 | 16 |
We also measured the phase transition in a shorter homologue of C16TAB for one alkane. For dodecane lenses on a C14TAB solution (c = 1.2 mM), (T) decreased discontinuously at Ts = 0.2 °C from −1.9 × 10−3 to −2.8 × 10−3. Comparing C16TAB + C14 with C16TAB + C12, or C16TAB + C12 with C14TAB + C12 shows that reducing the chain length by two carbons lowered Ts by around 10 °C regardless of whether the length of the alkane or the surfactant chain were altered.
To interpret the values of quantitatively, we need to construct a model for the dielectric profile through the interface, which we describe in the following section. Readers who are not interested in the details of the model may wish to skip to Fig. 4 below and the Discussion.
Modelling ellipsometry of mixed surfactant/alkane monolayers
We divide the interface up into five parallel homogeneous layers: air; hydrocarbon chains of the surfactant + alkane (denoted by the subscript c); hydrated headgroups (subscript h); electrical double layer containing counterions (subscript i); and the bulk aqueous solution (approximated as pure water). The Drude equation (eqn. (3) or (4) as appropriate) is then used to calculate the contribution of each of the three internal layers to the coefficient of ellipticity. Roughness is incorporated by including an additional contribution to the coefficient of ellipticity from capillary waves (subscript r). We therefore write| = r + i + h + c | (5) |
Our approach is to estimate all the physical quantities involved in the calculation of the coefficient of ellipticity, other than the thickness of the chain region, which can then be determined by comparison of the model with the experimental values of . While the number of assumptions and approximations may appear to be large, the calculated coefficients of ellipticity are rather insensitive to the precise values of most of the parameters in the model. Two parameters to which displays particular sensitivity are the density of the hydrocarbon chain region in the liquid phase and the anisotropy of the hydrocarbon chains in the solid phase. We have shown previously that ellipsometric data of C16TAB monolayers can only be understood if the chain region has a density very close to that of a liquid hydrocarbon.46 The anisotropy in the hydrocarbon chains was determined ellipsometrically with similar assumptions to those in this paper; consequently errors in the assumptions are likely to cancel out.47 The model we use to calculate each of the terms in eqn. (5) is described below.
r, is proportional to √T and inversely proportional to √σ, where σ is the surface tension of the solution.48 The value of of pure water at 25 °C, 0.4 × 10−3, is ascribed entirely to capillary wave roughness. For pure water at 298 K, σ = 72 mN m−1. We estimated r of a solution from 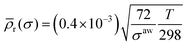 | (6) |
r on σ, we approximated the surface tensions of C16TAB solutions in the presence of alkanes by the literature values for C16TAB in the presence of dodecane at the same concentrations at 25 °C.49 For C14TAB solutions in the presence of dodecane σ was taken from ref. 43 (see Table 2).
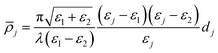 | (7) |
The optical dielectric constant, ε = n2, where n is the refractive index at the wavelength of the laser. For water, εw is a function of temperature. A third-order polynomial fit to literature values51 of εw in the range 0–40 °C was used to calculate εw at each transition temperature:
| εw(T) = 3.715 × 10−8T3 − 7.803 × 10−6T2 + 4.996 × 10−5T + 1.7756 | (8) |
For layers containing more than one component (e.g., water + ions, or water + headgroups), εj can be estimated by means of the Lorentz–Lorenz effective medium approximation (EMA) in terms of the dielectric constants and volume fractions, ϕ, of the individual components {α, β, …}.
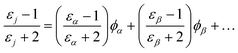 | (9) |
The values of the dielectric constants and molar volumes of different components are listed in Table 3.
| V m/cm3 mol−1 | ε | |
|---|---|---|
| a Calculated from the ionic radius.53 b Calculated from values of refractive index (ε = n2), 51 or density (Vm = 1/ρ).51 c V m(TMA) = Vm(N+(CH3)3Br−) − Vm(Br−) where Vm(N+(CH3)3Br−) = 81.3 cm3 mol−1.57 d Calculated with eqn. (11) with values of Rm from ref. 53. e Calculated with eqn. (11) with Rm equal to a sum of molar atomic refractivities.51 | ||
| Air | — | 1.00b |
| H2O | — | 1.77b |
| Br− | 19.0a | 6.29d |
| TMA | 62.3c | 2.38e |
| Undecane | 211b | 2.07b |
| Dodecane | 228b | 2.02b |
| Tridecane | 244b | 2.03b |
| Tetradecane/tetradecyl | 260b | 2.04b |
| Pentadecane | 276b | 2.05b |
| Hexadecane/hexadecyl | 293b | 2.06b |
| Heptadecane | 310b | 2.06b |
To calculate i (and h) we need to know the surface excess of surfactant, Γ (≅Nsurf/NA where NA is Avogadro's number). For C16TAB, we approximated Γ in the presence of an alkane at Ts by its value at 298 K in the absence of the alkane.50 The similarity between the SF spectra for pure C16TAB and for C16TAB + d-C14 (Fig. 2) together with previous neutron reflection43 and surface tension17 data for mixed CnTAB + alkane monolayers suggests that the presence of oil does not greatly affect the surface excess of C16TAB. For C14TAB, Γ has been measured directly by neutron reflection in the presence of dodecane at 25 °C;43 these values of Γ were employed as estimates for the surface excess of C14TAB at Ts. The values of Γ used in the modelling are listed in Table 2.
Charge neutrality requires that the surface excess of bromide counterions is equal to that of the surfactant. The bromide ions are treated as a homogeneous layer in water with a thickness equal to the Debye length, rD. The counterion term, i is, in fact, quite insensitive to the choice of thickness of the layer or the concentration profile within the layer. A solution of a 1 ∶ 1 ionic surfactant with ions of charge +1 and −1 at concentration c (moles per unit volume) has a Debye length
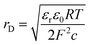 | (10) |
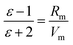 | (11) |
The dielectric constant of the counterion layer, εi, was then found from eqn. (9), with ϕw = 1 −
ϕBr−. Values of εi and di = rD were substituted into eqn. (7) to obtain i.
To estimate εTMA, values of the molar volume, Vm, and molar refractivity, Rm, are required. The molar volume of N(CH3)3Br is quoted as 81.3 cm3 mol−1 (135 Å3 per molecule) by Thomas and co-workers;57Vm(Br−) from the ionic radius is 19.0 cm3 mol−1,53 giving Vm(TMA) = 62.3 cm3 mol−1. To a good approximation molar refractivities are additive. The molar refractivity of the trimethylammonium headgroup, Rm(TMA), was estimated as a sum of molar atomic refractivities of C, H and N for a secondary amine (in the absence of reference values for quaternary amines),51εTMA was calculated from Rm(TMA) and Vm(TMA) using eqn. (11), εh from the Lorentz–Lorenz EMA (eqn. (9)), and finally h from eqn. (7), with dh = 8 Å.
The calculated values of r, i, and h are shown in Table 4.
r, i, and h for mixed monolayers of CnTAB + alkane for the conditions in Table 2. The experimental values of the coefficient of ellipticity exp used to calculate c are given
| n | m |
r
|
h
|
i
|
exp
|
c
|
|
|---|---|---|---|---|---|---|---|
| High T | 16 | 11 | 0.49 | −0.48 | −0.52 | −2.34 | −1.82 |
| 16 | 12 | 0.52 | −0.58 | −0.62 | −2.52 | −1.84 | |
| 16 | 13 | 0.53 | −0.58 | −0.62 | −2.42 | −1.74 | |
| 16 | 14 | 0.53 | −0.58 | −0.62 | −2.57 | −1.90 | |
| 16 | 15 | 0.53 | −0.58 | −0.62 | −2.39 | −1.72 | |
| 16 | 16 | 0.53 | −0.58 | −0.62 | −2.41 | −1.74 | |
| 16 | 17 | 0.53 | −0.58 | −0.62 | −2.39 | −1.72 | |
| 14 | 12 | 0.50 | −0.46 | −0.50 | −1.91 | −1.45 | |
| Low T | 16 | 11 | 0.49 | −0.48 | −0.52 | −3.19 | −2.68 |
| 16 | 12 | 0.52 | −0.58 | −0.62 | −3.46 | −2.78 | |
| 16 | 13 | 0.53 | −0.58 | −0.62 | −3.52 | −2.84 | |
| 16 | 14 | 0.53 | −0.58 | −0.62 | −3.80 | −3.13 | |
| 16 | 15 | 0.53 | −0.58 | −0.62 | −3.78 | −3.10 | |
| 16 | 16 | 0.53 | −0.58 | −0.62 | −3.79 | −3.12 | |
| 16 | 17 | 0.53 | −0.58 | −0.62 | −3.70 | −3.03 | |
| 14 | 12 | 0.50 | −0.46 | −0.50 | −2.79 | −2.32 |
From eqn. (7), the thickness of the hydrocarbon layer in the liquid phase, dc(l), is given by
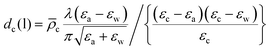 | (12) |
c = exp
−
h
−
i
−
r, (Table 4). The calculated values of dc(l) are tabulated in Table 2. The parameter in the model that has the greatest influence on dc(l) is the dielectric constant of the chain region. We have assumed the ‘oil-film’ model, which gave good agreement for monolayers of both cationic (CnTAB) and non-ionic (CnEm) surfactants.46,58 If the density of the chain region is less than that of a liquid alkane, the thicknesses will be larger than the calculated values. It is, however, unclear why oil molecules would spread from an oil droplet into an interphase of significantly lower density. The headgroup and counterion contributions are roughly linear functions of surface excess: a 10% error in Γ would change the calculated thickness by less than 1 Å.
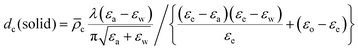 | (13) |
c is estimated as for the liquid phase. Values of dc(s) are shown in Table 2.
For the CnTAB + Cm monolayers, the isotropic contribution to c is negative and the anisotropic contribution is approximately half as large as the isotropic contribution but of opposite sign. For a given value of c, inclusion of the anisotropic contribution leads to an increase in the modelled thickness; neglecting anisotropy would lead to errors in the thickness of up to 40%.
The anisotropy of the hydrocarbon chain region is dependent on the tilt of the hydrocarbon chains. If the axis of the hydrocarbon chain were tilted from vertical by an angle ϕ = 15°, the anisotropy in the dielectric constants would decrease by 0.01 and the modelled thicknesses would be ∼1 Å smaller. A tilt of 56° from vertical would give εe = εo = 2.16 (the same as a completely disordered layer) and modelled thicknesses of 12–14 Å (30% shorter than with vertical chains). A large tilt is not consistent, however, with results from SFS. The intensity of the SF signal, ISF ∝ N2〈β〉2, from eqns. (1) and (2). For close-packed chains, N ∝ cosϕ, while in SSP-polarised spectra, 〈β(r+)〉 ∝ 〈cosϕ〉, Consequently, ISF(ϕ) ≈ ISF(ϕ = 0)(cosϕ)4 and is hence very sensitive to tilt. Comparing C16TAB + C14 and pure dodecanol monolayers in the low-T phase, we find that ISF(r+) for the surfactant–alkane mixture is ∼60% that of dodecanol. After correcting for the effect of the lower methyl group of the alkane, as discussed above, one infers that the tilts are very similar in the two monolayers.
The uniaxial dielectric constants are also very sensitive to the density of chains in the monolayer. If the area per molecule were chosen to be that of a crystalline phase (18.7 Å2) rather than a rotator phase (20.9 Å2), the Clausius–Mossotti equation would give values of εe = 2.43 and εo = 2.29, and the modelled thicknesses would be ∼40% smaller (11–12 Å). These thicknesses are significantly less than the extended chain lengths of alkane and surfactant and therefore not consistent with a crystalline layer of vertical or near vertical chains.
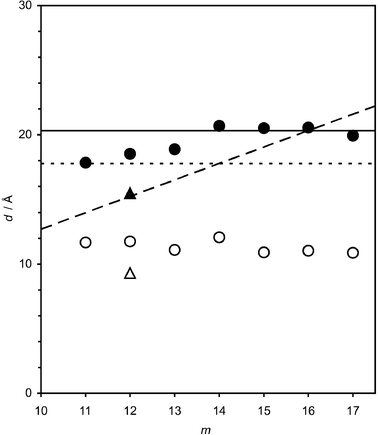 | ||
| Fig. 4 Modelled thicknesses, d, of C16TAB + Cm monolayers at the air-solution interface in the liquid phase (open circles) and solid phase (filled circles) at the transition temperature; and also of liquid and solid phase C14TAB + C12 monolayers (open and filled triangles). Values of d are plotted against the alkane chain length, m. The extended lengths of Cm, C16 and C14 are marked by dashed, solid and dotted lines respectively. | ||
Fig. 4 compares the experimentally determined thicknesses with the extended chain lengths of C16 and C14 alkyl chains and Cm alkanes. For C16TAB + Cm monolayers with m = 14–17, the calculated thicknesses are very close to the extended chain length of C16 (solid line in Fig. 4). For m = 11–13, dc(s) lies between the extended chain lengths of the alkane (dashed line) and the surfactant. For C14TAB + C12, the thickness also lies between the extended length of C14 (dotted line in Fig. 4) and C12. The ellipsometric data are thus consistent with a structure of the low-T phase comprising a monolayer of vertically oriented chains with the packing density of a rotator phase.
Discussion
Sum-frequency spectroscopy is a powerful technique for studying chain conformation in surfactant systems and provides clear evidence of the microscopic structural changes occurring at the phase transition. The conformation of the hydrocarbon chains changes from that characteristic of a liquid to that of a solid at the phase transition. The nature of the long-range correlations within the ‘solid’ monolayer is not addressed by spectroscopic techniques: to demonstrate that these monolayers show (quasi-) long-range translational order will require X-ray diffraction experiments, which are planned for the future. Isotopic substitution allows us to distinguish between the two components of the monolayer, to establish the conformational order in each and, within the solid phase, to estimate the composition of the monolayer. SFS is limited in the amount of quantitative information that it can provide about the composition or thickness of the surface film, partly due to uncertainty in the local electric fields experienced by the interfacial molecules (which we addressed earlier in the context of the non-vanishing signal from the alkane molecules) and partly because SFS is insensitive to molecules in an isotropic environment. Consequently, it provides no information on the composition of the high T, liquid phase.Conversely, ellipsometry is sensitive to both species at the interface regardless of the symmetry of the phase. It is also extremely sensitive (capable of detecting changes in coverage of <1% of a monolayer) and has good spatial resolution (<10 µm with imaging optics). However, the information content of ellipsometry is poor and it is only in conjunction with other measurements, such as SFS, neutron reflectometry or surface tensiometry, that one can build models to predict the coefficient of ellipticity of the air–water interface that do not have so many variable parameters as to be practically useless. While it is rare for ellipsometry alone to yield a unique structure, it can provide support for one structure and disprove another. Here we have used ellipsometry to determine the thickness of the high-T phase, knowing from SFS that it is liquid-like. We find that the thickness of the chain region of the monolayers is independent of chain length and about half the length of the fully extended surfactant chain (dc = 11–12 Å). In the solid phase, we showed that ellipsometry is consistent with an ordered mixed monolayer of densely packed upright chains, as inferred from SFS. Alternative models of tilted chains or multilayers would not be consistent with the combined SFS and ellipsometry data.3
We commented in the introduction on the connection between solid–liquid phase transitions in the mixed monolayers studied here and surface freezing at the pure alkane–air interface. While most solids exhibit surface melting, where the surface melts at a lower temperature than the bulk, alkanes and some related chain molecules (such as alcohols) are unusual in exhibiting surface freezing. For shorter chain lengths, 16 ≤ m < 44, the monolayers are rotator phases (lacking long-range correlations between the chain twists), while for longer chains, 44 ≤ m ≤ 50, they are crystalline.22 Rotator phases form even when a rotator structure is not a stable phase of the bulk alkane (e.g., even chain length alkanes with m < 20):62 the presence of the interface stabilises rotator phases compared to crystalline phases.
An X-ray reflectivity study by Tikhonov et al. found no interfacial freezing at the pure water–docosane (C22) interface.23 Lei and Bain showed that interfacial freezing at the aqueous solution–alkane interface can be induced by the presence of C16TAB.24 The transitions were identified from jumps in the coefficient of ellipticity, (T), of the solution–alkane interface and from discontinuous changes in the gradient of σ(T). The coefficients of ellipticity of the solid monolayers were successfully modelled with mixed monolayers of vertically oriented alkane and surfactant chains with an area per chain equal to that of a solid monolayer of dodecanol at the air–water interface, 21 Å2. The entropy change for the monolayer phase transition (determined from ∂σ/∂T) was in good agreement with the value expected from frozen monolayers at the alkane–air interface and with the melting of bulk rotator phases. The corresponding surface freezing range, ΔT = Ts
−
Tb, decreased with increasing alkane chain length, from 14 °C for C13 to 6 °C for C14 and 3 °C for C15. With hexadecane, the interface did not freeze.
Fig. 5 compares Ts(m) for the mixed C16TAB + Cm films at the air–solution interface with the bulk melting points, Tb, of the alkanes. Although Ts decreases with decreasing m, Tb decreases faster with the result that the ΔT = Ts − Tb increases from 1 °C for m = 17, to 28 °C for m = 11. The presence of a long-chain surfactant at the interface thus greatly increases the surface freezing range of an alkane. This may be a general phenomenon for alkanes at a variety of interfaces.
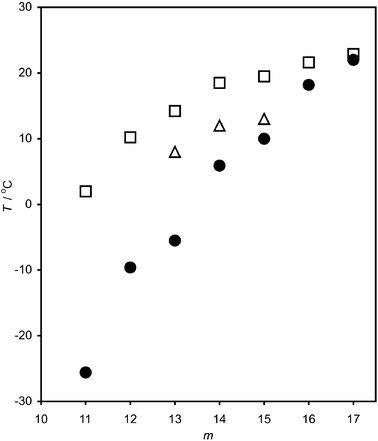 | ||
| Fig. 5 Comparison of the surface phase transition temperatures of mixed C16TAB + Cm monolayers at the air–solution interface, Ts (open squares) with the melting points of the bulk alkanes, Tb (filled circles) and the surface freezing temperatures at the interface between C16TAB solutions and bulk alkanes (open triangles). | ||
It is instructive to compare the low-T phase of mixed C16TAB + Cm monolayers at the air–water and alkane–water interfaces. In both cases, the same structure comprising upright, all-trans chains, can explain the available structural evidence. For C16TAB + C14, the compositions of the solid phase inferred from SFS (air–water) and tensiometry (alkane–water) are in agreement. The equivalence of the entropy change at the alkane–water interface with that at the alkane–air interface (where the structure has been determined unequivocally by X-ray diffraction)22 provides indirect support for the postulated structure. The fact that C16TAB + C16 monolayers undergo liquid–solid phase transitions at the air–water interface but not at the alkane–water interface suggests that the solid monolayers are more stable at the air–water interface. This increased stability is seen more generally in the values of ΔT, which are 6 °C higher for the monolayers at the air–water interface (see Fig. 5).
Several explanations for surface freezing in alkanes have been proposed, based upon capillarity,22 fluctuations of the molecules normal to the plane,63,64 and conformational disorder of molecules within the surface-frozen monolayer,65 but no consensus has emerged. The reason for increased stability of frozen C16TAB + Cm monolayers at the air–water interface compared to the alkane–water interface is also unclear. One possible explanation lies in the change in surface entropy on freezing.24 At the alkane–solution interface, the alkane molecules lose three degrees of translational freedom upon freezing but the surfactant molecules only lose two, as they are already constrained to the interface. On freezing at the air–solution interface, both surfactant and alkane molecules are already constrained to the interface, so the change in surface entropy on monolayer freezing is less negative than at the alkane–solution interface. The monolayer transition temperature, Ts = ΔHs/ΔSs, where ΔHs is the change in surface enthalpy on freezing and ΔSs is the change in surface entropy on freezing (both negative). If the enthalpic interactions at the two interfaces are comparable, then Ts will be higher at the air–water interface.
Alkanes (16 ≤ m ≤ 50) also exhibit surface-freezing behaviour at the air–silica interface.66–68 At temperatures above Ts the alkanes wet the solid surface. Surface-freezing coincides with a dewetting transition, since liquid alkanes do not wet low-energy (methyl-terminated) faces of their own solids. Below Ts (but above Tb) the SiO2 is covered by a frozen monolayer of vertically oriented alkane molecules in equilibrium with liquid alkane lenses.67 The variation of Ts with m closely matches that seen at the air–alkane interface.
No effect similar to alkanes on SiO2 can be observed for alkanes on water for the simple reason that long-chain alkanes do not wet water. One can pose the hypothetical question of whether a metastable monolayer of an alkane on water would show surface freezing. The probable answer is yes. For the bulk tetradecane–C16TAB solution interface, one can extrapolate the plot of Ts against surface excess, Γ, back to Γ = 0 and find that Ts for the pure tetradecane–water interface is only 1–2 °C below Tb. For C16TAB–tetradecane, the surface freezing temperature at the air–solution interface is 6 °C higher than at the alkane–solution interface, from which we infer that Ts would be above Tb for an alkane monolayer on pure water. Thus surfactant is needed to allow the alkane to spread on water (pseudo-partial wetting), but is not essential for the alkane to show surface freezing.
Conclusions
Monolayers of pure C16TAB at the air–solution interface do not show any phase transitions between condensed phases. In the presence of an alkane, with a chain length 11 ≤ m ≤ 17, a first-order phase transition is observed. This phase transition can be described in two different ways. Either, one can view the alkane as permitting the surfactant to crystallise, by filling in the free volume between the surfactant chains, or one can view the surfactant as inducing a surface freezing transition in a monolayer of the alkane. The latter perspective permits a comparison with surface freezing in alkanes at other interfaces. The same structural model for the low-T phase, consisting of a (mixed) monolayer of all-trans, upright chains in a rotator phase, explains the existing structural and spectroscopic information at the air–water, alkane–air and alkane–water interfaces. The frozen phase is significantly more stable at the air–water interface than at the alkane–water interface. Although a pure alkane monolayer cannot be studied on the surface of water, since long-chain alkanes do not wet water, we predict that if such a monolayer existed it too would show surface freezing, analogously to alkane monolayers on silica. A feature of the mixed monolayers of C16TAB + alkanes on water is that the alkane can freeze in the monolayer at temperatures well above (ΔT < 30 °C) the bulk melting point; a characteristic that might prove useful for controlling the surface properties of oil–water–surfactant systems.Acknowledgements
KMW would like to thank the EPSRC for a studentship. LQ would like to thank the Pao Yu-Kong & Pao Zhao-Long Foundation for support.References
- G. L. J. Gaines, Insoluble Monolayers at Liquid–Gas Interfaces, John Wiley & Sons Ltd., New York, 1966 Search PubMed.
- I. R. Petersen and R. M. Kenn, Langmuir, 1994, 10, 4645 CrossRef CAS.
- B. D. Casson, R. Braun and C. D. Bain, Faraday Discuss., 1996, 104, 209 RSC.
- M. L. Pollard, R. Pan, C. Steiner and C. Maldarelli, Langmuir, 1998, 14, 7222 CrossRef CAS.
- M. N. Islam and T. Kato, J. Phys. Chem. B, 2003, 107, 965 CrossRef CAS.
- M. M. Hossain, T. Suzuki and T. Kato, Colloids Surf., A, 1998, 198, 53.
- M. M. Hossain and T. Kato, Langmuir, 2000, 16, 10175 CrossRef CAS.
- M. M. Hossain, T. Suzuki and T. Kato, Langmuir, 2000, 16, 9109 CrossRef CAS.
- V. Melzer and D. Vollhardt, Phys. Rev. Lett., 1996, 76, 3770 CrossRef CAS.
- D. Vollhardt, Adv. Colloid Interface Sci., 1999, 79, 19 CrossRef CAS.
- M. M. Hossain, M. Yoshida, K.-i. Iimura, N. Suzuki and T. Kato, Colloids Surf., A, 2000, 171, 105 CrossRef CAS.
- T. Gilányi, R. Mészáros and I. Varga, Langmuir, 2000, 16, 3200 CrossRef CAS.
- B. D. Casson and C. D. Bain, J. Phys. Chem. B, 1998, 102, 7434 CrossRef CAS.
- B. D. Casson and C. D. Bain, J. Phys. Chem. B, 1999, 103, 4678 CrossRef CAS.
- D. Bonn and D. Ross, Rep. Prog. Phys., 2001, 64, 1085 CrossRef CAS.
- M. Aratono, H. Kawagoe, T. Toyomasu, N. Ikeda, T. Takiue and H. Matsubara, Langmuir, 2001, 17, 7344 CrossRef CAS.
- H. Matsubara, N. Ikeda, T. Takiue, M. Aratono and C. D. Bain, Langmuir, 2003, 19, 2249 CrossRef CAS.
- F. Brochard-Wyart, J. M. Di Meglio, D. Quéré and P. G. De Gennes, Langmuir, 1991, 7, 335 CrossRef CAS.
- C. E. McKenna, M. M. Knock and C. D. Bain, Langmuir, 2000, 16, 5853 CrossRef CAS.
- J. C. Earnshaw and C. J. Hughes, Phys. Rev. A: At., Mol., Opt. Phys., 1992, 46, R4494 CrossRef CAS.
- X. Z. Wu, E. B. Sirota, S. K. Sinha, B. M. Ocko and M. Deutsch, Phys. Rev. Lett., 1993, 70, 958 CrossRef CAS.
- B. M. Ocko, X. Z. Wu, E. B. Sirota, S. K. Sinha, O. Gang and M. Deutsch, Phys. Rev. E: Stat. Phys., Plasmas, Fluids, Relat. Interdiscip. Top., 1997, 55, 3164 CrossRef CAS.
- A. M. Tikhonov, D. M. Mitrinovic, M. Li, Z. Huang and M. L. Schlossman, J. Phys. Chem. B, 2000, 104, 6336 CrossRef CAS.
- Q. Lei and C. D. Bain, Phys. Rev. Lett., 2004, 92, 176103 CrossRef.
- F. Vidal and A. Tadjeddine, Rep. Prog. Phys., 2005, 68, 1095 CrossRef CAS.
- G. L. Richmond, Chem. Rev., 2002, 102, 2693 CrossRef CAS.
- D. A. Beattie and C. D. Bain, in Handbook of Vibrational Spectroscopy, ed. J. M. Chalmers and P. R. Griffiths, Wiley, Chichester, 2002, vol. 1, p. 801 Search PubMed.
- P. B. Miranda and Y. R. Shen, J. Phys. Chem. B, 1999, 103, 3292 CrossRef CAS.
- C. D. Bain, J. Chem. Soc., Faraday Trans., 1995, 91, 1281 RSC.
- Y. R. Shen, Nature, 1989, 333, 519 CrossRef.
- C. D. Bain, in Modern Characterisation Methods of Surfactant Systems, ed. B. P. Binks, Marcel Dekker, Inc., New York, 1999, p. 335 Search PubMed.
- P. Drude, Ann. Phys. Chem., 1891, 43, 126 Search PubMed.
- M. Born and E. Wolf, Principles of Optics, Cambridge University Press, Cambridge, 6th edn., 1980 Search PubMed.
- D. Beaglehole, J. Phys., 1983, 44, C10–147 Search PubMed.
- H. Bercegol, F. Gallet, D. Langevin and J. Meunier, J. Phys., 1989, 50, 2227 Search PubMed.
- D. L. Dorset, H. L. Strauss and R. G. Snyder, J. Phys. Chem., 1991, 95, 938 CrossRef CAS.
- G. R. Bell, C. D. Bain and R. N. Ward, J. Chem. Soc., Faraday Trans., 1996, 92, 515 RSC.
- C. D. Bain, P. B. Davies, T. H. Ong, R. N. Ward and M. A. Brown, Langmuir, 1991, 7, 1563 CrossRef CAS.
- R. A. Macphail, H. L. Strauss, R. G. Snyder and C. A. Elliger, J. Phys. Chem., 1984, 88, 334 CrossRef CAS.
- R. N. Ward, D. C. Duffy, P. B. Davies and C. D. Bain, J. Phys. Chem., 1994, 98, 8536 CrossRef CAS.
- R. N. Ward, PhD Thesis, Cambridge University, 1993 Search PubMed.
- J. P. Rieu, J. F. Legrand, A. Renault, B. Berge, B. M. Ocko, X. Z. Wu and M. Deutsch, J. Phys. II, 1995, 5, 607 CrossRef CAS.
- J. R. Lu, R. K. Thomas, R. Aveyard, B. P. Binks, P. Cooper, P. D. I. Fletcher, A. Sokolowski and J. Penfold, J. Phys. Chem., 1992, 96, 10971 CrossRef CAS.
- J. R. Lu, R. K. Thomas, B. P. Binks, P. D. I. Fletcher and J. Penfold, J. Phys. Chem., 1995, 99, 4113 CrossRef CAS.
- G. A. Sefler, Q. Du, P. B. Miranda and Y. R. Shen, Chem. Phys. Lett., 1995, 235, 347 CrossRef CAS.
- G. R. Bell, S. Manning-Benson and C. D. Bain, J. Phys. Chem. B, 1998, 102, 218 CrossRef CAS.
- B. D. Casson and C. D. Bain, Langmuir, 1997, 13, 5465 CrossRef CAS.
- J. Meunier, J. Phys., 1987, 48, 1819 Search PubMed.
- R. Aveyard, P. Cooper and P. D. I. Fletcher, J. Chem. Soc., Faraday Trans., 1990, 86, 3623 RSC.
- S. Manning-Benson, S. R. W. Parker, C. D. Bain and J. Penfold, Langmuir, 1998, 14, 990 CrossRef.
- C. R. C. Handbook of Chemistry and Physics, ed. D. R. Lide, CRC Press, Boca Raton, 74th edn., 1993 Search PubMed.
- P. W. Atkins, Physical Chemistry, Oxford University Press, Oxford, 5th edn., 1994 Search PubMed.
- P. W. Atkins, Physical Chemistry, Oxford University Press, Oxford, 3rd edn., 1986 Search PubMed.
- J. R. Lu, M. Hromadova, E. Simister, R. K. Thomas and J. Penfold, Physica B, 1994, 198, 120 CrossRef CAS.
- J. R. Lu, M. Hromadova, E. A. Simister, R. K. Thomas and J. Penfold, J. Phys. Chem., 1994, 98, 11519 CrossRef CAS.
- J. R. Lu, Z. X. Li, J. Smallwood, R. K. Thomas and J. Penfold, J. Phys. Chem., 1995, 99, 8233 CrossRef CAS.
- D. J. Lyttle, J. R. Lu, T. J. Su and R. K. Thomas, Langmuir, 1995, 11, 1001 CrossRef CAS.
- S. R. Goates, D. A. Schofield and C. D. Bain, Langmuir, 1998, 15, 1400.
- A. Renault, J. F. Legrand, G. Goldmann and B. Berge, J. Phys. II, 1993, 3, 761 CrossRef CAS.
- R. Aveyard, B. P. Binks, P. Cooper and P. D. I. Fletcher, Prog. Colloid Polym. Sci., 1990, 81, 36 CAS.
- R. Aveyard, B. P. Binks, P. Cooper and P. D. I. Fletcher, Adv. Colloid Interface Sci., 1990, 33, 59 CrossRef CAS.
- D. M. Small, The Physical Chemistry of Lipids: From Alkanes to Phospholipids, ed. D. J. Hanahan, Plenum Press, New York, 1986 Search PubMed.
- A. V. Tkachenko and Y. Rabin, Phys. Rev. Lett., 1996, 76, 2527 CrossRef CAS.
- A. Tkachenko and Y. Rabin, Phys. Rev. Lett., 1997, 79, 532 CrossRef CAS.
- A. J. Colussi, M. R. Hoffmann and Y. Tang, Langmuir, 2000, 16, 5213 CrossRef CAS.
- C. Merkl, T. Pfohl and H. Riegler, Phys. Rev. Lett., 1997, 79, 4625 CrossRef CAS.
- U. G. Volkmann, M. Pino, L. A. Altamirano, H. Taub and F. Y. Hansen, J. Chem. Phys., 2002, 116, 2107 CrossRef CAS.
- H. Mo, H. Taub, U. G. Volkmann, M. Pino, S. N. Ehrlich, F. Y. Hansen, E. Lu and P. Miceli, Chem. Phys. Lett., 2003, 377, 99 CrossRef CAS.
Footnotes |
| † Current address: Department of Chemistry, Zhejiang University, Hangzhou. 310027 P. R. China |
| ‡ Current address: Department of Chemistry, University of Durham, South Road, Durham, UK DH1 3LE. E-mail: c.d.bain@durham.ac.uk |
| This journal is © The Royal Society of Chemistry 2006 |