Stereoselective synthesis of 2,4,5-trisubstituted piperidines by carbonyl ene and Prins cyclisations
Claire A. M.
Cariou
and
John S.
Snaith
*
School of Chemistry, The University of Birmingham, Edgbaston, Birmingham, UK B15 2TT. E-mail: j.s.snaith@bham.ac.uk; Fax: +44 121 414 4403; Tel: +44 121 414 4363
First published on 29th November 2005
Abstract
Cyclisation of aldehydes 3a–e catalysed by concentrated hydrochloric acid affords predominantly the all cis 2,4,5-trisubstituted piperidines 4a–e when the 2-substituent is small, while catalysis by MeAlCl2 in refluxing chloroform gives the trans piperidines 5a–e with diastereomeric ratios of up to 99 : 1.
Piperidines are widely distributed throughout Nature1 and are an important scaffold for drug discovery,2 forming the core of many pharmaceuticals. Methods for their stereocontrolled synthesis are of continuing interest, driven by the wide variety of functionality and substitution patterns present in piperidine targets.3
Intramolecular carbonyl ene reactions present an attractive method for ring closure, leading to the formation of two contiguous stereocentres with an often high degree of stereocontrol.4 We recently published a route to 3,4-disubstituted piperidines which had a carbonyl ene reaction as the key ring-closing step.5 The Brønsted acid-catalysed reaction at low temperatures strongly favoured a cis relationship between the two new stereocentres, while the Lewis acid-catalysed reaction at elevated temperatures gave the corresponding trans product.
We now describe our efforts towards extending this approach to the synthesis of 2,4,5-trisubstituted piperidines, using cyclisation precursors derived from α-amino alcohols. Such trisubstituted piperidines are of particular interest as they form the core of a number of important natural products, including the pseudodistomin family of anti-tumour compounds. These were isolated by Kobayashi6 from a marine tunicate, and have recently been the focus of synthetic attention.7
The cyclisation precursors were readily synthesised from commercially available α-amino alcohols via a procedure involving a one-carbon homologation by cyanide, Scheme 1. Bis tosylation of the α-amino alcohols 1a–e followed by displacement of the O-tosyl group by sodium cyanide in DMF proceeded smoothly and in good yield to afford the N-tosyl-β-amino nitriles 2a–e. These were alkylated with prenyl bromide before being reduced by Dibal-H to the β-amino aldehyde cyclisation precursors 3a–e in excellent overall yield. The β-amino aldehydes could be readily chromatographed and were unchanged on storing for several weeks at −20 °C.
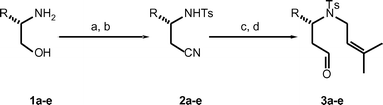 | ||
Scheme 1 Synthesis of cyclisation precursors. (a) TsCl, pyridine, CH2Cl2, 25 °C, 68–85%; (b) NaCN, DMF, 25 °C, 70–90%; (c) BrCH2CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C(CH3)2, Cs2CO3, CH3CN, 25 °C, 82–98%; (d) Dibal-H, CH2Cl2, −78 °C, 83–97%. C(CH3)2, Cs2CO3, CH3CN, 25 °C, 82–98%; (d) Dibal-H, CH2Cl2, −78 °C, 83–97%. | ||
Cyclisation of 3a–e was first studied using our optimised Brønsted acid conditions† of three equivalents of concentrated hydrochloric acid in CH2Cl2 at −78 °C.5 In our earlier work, these conditions were found to favour formation of the kinetic product, in which there is a cis relationship between the hydroxyl and isopropenyl substituents. The results are summarised in Table 1.

In all cases, of the four possible stereoisomers, only two, piperidines 4 and 5, were observed, in excellent combined yields. In the case of β-amino aldehydes with sterically undemanding 2-substituents, entries 1–3, the diastereoselectivity was moderate to good, although it decreased markedly in the case of 3d and 3e with very bulky 2-substituents. Traces (typically <5%) of chloride side-products were often isolated, arising from the addition of HCl across the double bond in 4 or 5. These were generally separable by chromatography, but could also be converted back to the alkenes 4 and 5 by stirring with aqueous ammonia in THF.
The major diastereomer was confirmed as the all cis piperidine 4 by single crystal X-ray analysis of 4c, Fig. 1.‡ Formation of this product can be rationalised by considering two factors. Firstly, there is a strong preference for the 2-substituent to adopt an axial disposition in the chair-like transition state, thus avoiding the pseudo A1,3 strain with the sulfonamide; this stereochemical preference in N-acyl and N-sulfonamido piperidines has been shown to be pronounced in a number of cases.9 The second factor is the kinetic preference for the ene component and the aldehyde to adopt a cis relationship in the cyclisation transition state, as observed in our earlier work.5 This cis relationship is achieved with the aldehyde lying in an axial position in the TS, and the more bulky ene component lying equatorial, Fig. 2. More bulky 2-substituents lead to a lowering of the diastereoselectivity as a result of increased 1,3-diaxial interactions with the aldehyde, forcing the aldehyde into an equatorial position to give 5.
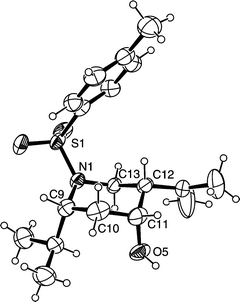 | ||
| Fig. 1 ORTEP8 representation of 4c; ellipsoids drawn at the 30% probability level. | ||
 | ||
| Fig. 2 Conformations leading to major and minor isomers in the Brønsted acid-catalysed reactions. | ||
Turning to the Lewis acid-catalysed reaction, aldehydes 3a–e were treated with one equivalent of methyl aluminium dichloride, which had been found to be the optimal Lewis acid in our earlier studies.5 As in the Brønsted acid-catalysed reactions, only two of the four possible diastereomers were observed (Table 2). The stereoselectivities ranged from good to excellent, with the major diastereomer identified as 5 from a combination of NOE data and 1H NMR coupling constants. Further confirmation came from single crystal X-ray analysis of 5a, Fig. 3.§
| Entry | Aldehyde | R | Temperature | 4 : 5b | Yield (%)c |
|---|---|---|---|---|---|
| a All reactions were performed using 1 equivalent of MeAlCl2. b Ratio determined by 1H NMR or HPLC of crude reaction mixtures. c Isolated yields of major (minor) isomers following chromatography. | |||||
| 1 | 3a | Me | 23 | 12 : 88 | 76 (5) |
| 2 | 3a | Me | 40 | 7 : 93 | 60 (4) |
| 3 | 3a | Me | 60 | 4 : 96 | 71 (4) |
| 4 | 3b | Bn | 23 | 10 : 90 | 61 (10) |
| 5 | 3b | Bn | 40 | 5 : 95 | 64 (5) |
| 6 | 3c | iPr | 40 | 2 : 98 | 82 (2) |
| 7 | 3d | tBu | 60 | 1 : 99 | 88 (1) |
| 8 | 3e | Ph | 60 | 2 : 98 | 80 (2) |
 | ||
| Fig. 3 ORTEP representation of 5a; ellipsoids drawn at 30% probability level. | ||
Under the equilibrating Lewis acidic conditions the thermodynamic product is favoured, in which the 4- and 5-substituents are equatorial, and the 2-substituent is axial to avoid the pseudo A1,3 strain with the sulfonamide. The increased 1,3-diaxial interactions present in the TS leading to 4 results in the equilibration to the thermodynamic product (Fig. 4) being facile even at room temperature, but improved ratios were obtained on heating at 40 or 60 °C (see, for example, entries 1–3).
 | ||
| Fig. 4 Increased 1,3-diaxial interactions in Lewis acid-catalysed reaction favours the equatorial aldehyde. | ||
Removal of the tosyl protecting group from a representative range piperidines was readily effected by stirring with sodium naphthalenide10 for 5 min at −78 °C, Table 3. The crude yields of essentially pure piperidines were near quantitative in most cases, although compounds 4a and 5a in particular were difficult to handle and chromatograph due to their significant polarity and water solubility.
|
|
||||
|---|---|---|---|---|
| Entry | Tosyl piperidine | R | Product | Yield (%) |
| 1 | 4a | Me | 6a | 54 |
| 2 | 5a | Me | 7a | 52 |
| 3 | 4b | Bn | 6b | 61 |
| 4 | 5b | Bn | 7b | 87 |
| 5 | 5c | iPr | 7c | 90 |
| 6 | 5d | tBu | 7d | 99 |
| 7 | 5e | Ph | 7e | 67 |

In summary, we have discovered a highly diastereoselective synthesis of 2,4,5-trisubstituted piperidines from simple acyclic precursors, which should have application to the synthesis of more complex molecules.
We thank the Engineering and Physical Sciences Research Council for the award of a studentship to C. A. M. C.
References
- J. A. Findlay, in The Alkaloids, ed. A. Brossi, Academic Press: London, 1985, vol. 26, p. 89 Search PubMed; A. R. Pinder, Nat. Prod. Rep., 1986, 3, 171 Search PubMed; A. R. Pinder, Nat. Prod. Rep., 1987, 4, 527 RSC; A. R. Pinder, Nat. Prod. Rep., 1989, 6, 67 RSC; A. R. Pinder, Nat. Prod. Rep., 1990, 7, 447 RSC; A. R. Pinder, Nat. Prod. Rep., 1992, 9, 491 RSC; D. O'Hagan, Nat. Prod. Rep., 2000, 17, 435 RSC.
- See for example: P. S. Watson, B. Jiang and B. Scott, Org. Lett., 2000, 2, 3679 Search PubMed and references therein.
- For recent reviews see: S. Laschat and T. Dickner, Synthesis, 2000, 1781 Search PubMed; P. M. Weintraub, J. S. Sabol, J. M. Kane and D. R. Borcherding, Tetrahedron, 2003, 59, 2953 CrossRef CAS; M. G. P. Buffat, Tetrahedron, 2004, 60, 1701 CrossRef CAS.
- For a review see: B. B. Snider, in Comprehensive Organic Synthesis, ed. B. M. Trost and I. Fleming, Pergamon, Oxford, 1991, vol. 2, p. 527 Search PubMed.
- J. T. Williams, P. S. Bahia and J. S. Snaith, Org. Lett., 2002, 4, 3727 CrossRef CAS.
- M. Ishibashi, Y. Ohizumi, T. Sasaki, H. Nakamura, Y. Hirata and J. Kobayashi, J. Org. Chem., 1987, 52, 450 CrossRef CAS; J. Kobayashi, K. Naitoh, Y. Doi, K. Deki and M. Ishibashi, J. Org. Chem., 1995, 60, 6941 CrossRef CAS.
- D. Ma and H. Sun, J. Org. Chem., 2000, 65, 6009 CrossRef CAS; N. Langlois, Org. Lett., 2002, 4, 185 CrossRef CAS; B. M. Trost and D. R. Fandrick, Org. Lett., 2005, 7, 823 CrossRef CAS; F. A. Davis, J. Zhang, Y. Li, H. Xu and C. DeBrosse, J. Org. Chem., 2005, 70, 5413 CrossRef CAS.
- ORTEP-3 for Windows: L. J. Farrugia, J. Appl. Crystallogr., 1997, 30, 565 Search PubMed.
- F. Johnson, Chem. Rev., 1968, 68, 375 CrossRef CAS; L. A. Gandon, A. G. Russell and J. S. Snaith, Org. Biomol. Chem., 2004, 2, 2270 RSC.
- J. C. Adelbrecht, D. Craig, B. W. Dymock and S. Thorimbert, SYNLETT, 2000, 467 CAS.
Footnotes |
| † Brønsted acid-catalysed cyclisation procedure. Preparation of (2S*, 4R*, 5S*)-2-methyl-5-iso-propenyl-1-(p-toluenesulfonyl)piperidin-4-ol 4a. Concentrated HCl (37%, 85 µL) was added to a solution of aldehyde 3a (0.102 g, 0.33 mmol) in dichloromethane (10 mL) at −78 °C. The solution was stirred at −78 °C overnight, after which it was quenched by addition of water (10 mL). The aqueous phase was then extracted with dichloromethane (4 × 10 mL). The combined organic phases were washed with brine (10 mL), dried over MgSO4 and concentrated in vacuo to leave a colourless oil, which was purified by flash column chromatography (silica; ethyl acetate–hexane, 2 : 3, Rf = 0.41) to afford the piperidine 4a (0.07 g, 70%) as a colourless thick oil. [α]27D
−3.6 (c 0.5 in CHCl3); (νmax(CHCl3)/cm−1 3525 (O–H), 2923 (C–H), 1644 (CC aliphatic), 1598 (CC aromatic), 1494 (CC aromatic), 1451 (CC aromatic), 1383 (C–H), 1336 (SO2), 1305 (C–H), 1153 (SO2), 1088 (C–O); δH(300 MHz, CDCl3) 1.21 (3H, d, J 7.0), 1.63–1.67 (2H, envelope), 1.73 (3H, s), 1.80–1.82 (1H, m), 2.12 (1H, broad d, J 11.8), 2.40 (3H, s), 3.30 (1H, t, J 12.7), 3.62 (1H, dd, J 4.1, J 13.2), 3.99 (1H, d, J 2.6), 4.17–4.22 (1H, m), 4.69 (1H, s), 5.00 (1H, s), 7.27 (2H, d, J 8.1), 7.69 (2H, d, J 8.1); δC(75 MHz, CDCl3) 18.9, 21.5, 22.8, 35.7, 37.6, 46.5, 47.4, 64.6, 112.3, 127.0, 129.7, 138.4, 143.0, 144.1; m/z (ES+) 332 (100%, [M + Na]+) [HRMS Found: (M + Na)+ 332.1297. C16H23NNaO3S requires M, 332.1296]. Lewis acid-catalysed cyclisation procedure. Preparation of (2R, 4S, 5S)-2-tert-butyl-5-iso-propenyl-1-(p-toluenesulfonyl)piperidin-4-ol 5d. Methyl aluminium dichloride (1 M solution in hexane, 480 µL, 0.48 mmol) was added to a solution of the aldehyde 3d (0.168 g, 0.48 mmol) in chloroform (20 mL). The solution was stirred overnight at 60 °C, after which it was quenched by addition of water (20 mL). The aqueous phase was then extracted with dichloromethane (4 × 20 mL). The combined organic phases were washed with brine (20 mL), dried over MgSO4 and concentrated in vacuo to leave a colourless oil, that was purified by flash column chromatography (silica; ethyl acetate–petroleum ether, 1 : 2, Rf = 0.22) to afford piperidine 5d as a colourless oil (0.147 g, 88%). [α]19D
−6.0 (c 0.3 in CHCl3); (Found: C, 64.8; H, 8.1; N, 3.8. C19H29NO3S requires C, 64.9; H, 8.3; N, 4.0%); νmax(CHCl3)/cm−1 3498 (O–H), 2964 (C–H), 1646 (CC aliphatic), 1598 (CC aromatic), 1401, 1367 (C–H), 1336 (SO2), 1084 (C–O); δH(300 MHz, CDCl3) 1.05 (9H, s), 1.16–1.27 (1H, m), 1.30–1.38 (1H, m), 1.60 (3H, s), 1.71 (1H, s), 2.13 (1H, dd, J 4.4, J 14.0), 2.42 (3H, s), 3.04 (1H, dd, J 12.3, J 15.4), 3.79 (1H, dd, J 3.7, J 15.4), 3.91 (1H, dt, J 4.7, J 11.0), 3.99 (1H, d, J 8.1), 4.64 (1H, s), 4.89 (1H, s), 7.30 (2H, d, J 8.1), 7.73 (2H, d, J 8.1); δC(75 MHz, CDCl3) 20.3, 21.6, 29.5, 31.7, 36.9, 46.2, 49.9, 61.4, 66.5, 113.9, 127.2, 129.9, 138.4, 142.6, 143.4; m/z (ES+) 374 (100%, [M + Na]+) [HRMS Found: (M + Na)+ 374.1768. C19H29NNaO3S requires M, 374.1766]. Tosyl group removal procedure. Preparation of (2S, 4S, 5S)-2-benzyl-5-iso-propenylpiperidin-4-ol 7b. To a solution of 5b (0.099 g, 0.26 mmol) in tetrahydrofuran (1.5 mL) under nitrogen was added at −78 °C a freshly prepared solution of sodium naphthalenide (1.2 mL of a 1 M solution in tetrahydrofuran, 4.6 eq). After 5 min the reaction was quenched with methanol (0.4 mL), warmed up to room temperature, diluted with water (5 mL) and acidified to pH 1 with aqueous HCl (2 M). The aqueous phase was washed with diethyl ether (3 × 10 mL), basified to pH 9 with aqueous NaOH (2 M) and extracted with ethyl acetate (4 × 10 mL). The combined organic phases were washed with brine (10 mL), dried over MgSO4 and concentrated in vacuo to afford piperidine 7b (0.052 g, 87%) as colourless crystals. Mp 119 °C; [α]23D
−49 (c 0.98 in CHCl3); νmax(CHCl3)/cm−1 3306 (O–H, N–H), 2917 (C–H), 1641 (CC aliphatic), 1602 (CC aromatic), 1493 (CC aromatic), 1455 (CC aromatic); 1090 (C–O); δH(300 MHz, CDCl3) 1.63 (1H, ddd, J 5.1, J 9.9, J 12.9), 1.79 (3H, s), 1.91 (1H, broad s), 1.97 (1H, dt, J 3.7, J 12.9), 2.07–2.15 (1H, m), 2.72 (1H, dd, J 6.4, J 13.4), 2.87–2.94 (3H, envelope), 3.33–3.40 (1H, m), 3.99 (1H, dt, J 4.1, J 9.4), 4.94 (1H, s), 4.98 (1H, s), 7.16–7.33 (5H, m); δC(75 MHz, CDCl3) 21.2, 37.0, 38.8, 43.7, 53.4, 54.4, 66.4, 113.3, 126.4, 128.7, 129.1, 139.6, 144.4; m/z (ES+) 232 (65%, [M + H]+), 214.1 (100, [M − OH]+) [HRMS Found: (M + H)+ 232.1700. C15H22NO requires M, 232.1701]. |
| ‡ Crystal data for 4c. C18H27NO3S, M = 337.47, monoclinic, a = 8.4757(1), b = 19.2080(3), c = 11.8021(2) Å, U = 1902.87(5) Å3, T = 296 K, space group P21, Z = 4, µ(Cu Kα) = 1.617 mm−1, 5917 reflections measured, 5423 unique (Rint = 0.0374) which were used in all calculations. The final wR(F2) was 0.1035 (all data). Crystal data for 4a. C16H23NO3S, M = 309.41, monoclinic, a = 22.4722(3), b = 7.8727(1), c = 19.9610(2) Å, U = 3270.17(7) Å3, T = 296 K, space group C2/c, Z = 8, µ(Cu Kα) = 1.837 mm−1, 2943 reflections measured, 2689 unique (Rint = 0.0392) which were used in all calculations. The final wR(F2) was 0.1150 (all data). CCDC reference number 288320. For crystallographic data in CIF or other electronic format see DOI: 10.1039/b515547a |
| § CCDC reference number 288321. For crystallographic data in CIF or other electronic format see DOI: 10.1039/b515547a |
| This journal is © The Royal Society of Chemistry 2006 |