A new approach to combretastatin D2†
David
Cousin
a,
John
Mann
*a,
Mark
Nieuwenhuyzen
a and
Hendrik
van den Berg
b
aSchool of Chemistry, Queen′s University Belfast, Stranmillis Rd., Belfast, UK BT9 5AG
bDepartment of Oncology, Queen's University Belfast, Belfast City Hospital, Lisburn Rd., Belfast, UK BT9 7AB
First published on 25th November 2005
Abstract
A concise and convergent route to combretastatin D2 is described together with some preliminary biological data.
Introduction
Shrubs and trees of the Combretaceae family are much used in Africa and India as a source of medicinal products, and the constituent combretastatins have been extensively studied as potential drugs.1 Of these, combretastatin A4 (1a) and its phosphate ester (1b) have shown particular promise as potential anti-cancer agents. The latter is currently undergoing phase II clinical trials as an inhibitor of angiogenesis.2All of the combretastatins are acyclic with the exception of combretastatins D1 (2) and D2 (3) and the recently described combretastatins D3 (4) and D4 (5).3 Several syntheses of D1 and D2 have been described4 but none of the routes was efficient, and only one cytotoxicity study has been reported (using mouse leukaemia P388) which showed modest activity (ED50 3–5 µg ml−1) for these compounds.5 Our interest in the design and synthesis of angiogenesis inhibitors prompted us to design a new, convergent route to this class of compounds with a view to further study of the biological properties of both the natural products and their analogues.
Results and discussion
Our synthetic route is shown in Scheme 1 and uses a copper-catalysed coupling of the phenol (6) and boronic acid (7) to provide the key ether linkage of combretastatin D2. The phenol (6) was synthesised from 3-hydroxy-4-methoxybenzaldehyde by means of a Wittig reaction with carbomethoxymethylidene triphenylphosphorane, with subsequent catalytic transfer hydrogenation of the resultant alkene (85% overall). In a parallel synthesis, the ethylene ketal of 4-bromobenzaldehyde was converted into the aryl lithium and then reacted with triisopropyl borate followed by acid hydrolysis (70–75% overall). The coupling was accomplished using a modification of the method of Evans et al.6 using copper(II) acetate to provide the required diaryl ether (8). This reaction was somewhat capricious with yields ranging from 32–81% but could be optimised to around 70% on the multigram scale. Transesterification using allyl alcohol and dibutyltin oxide7 produced the allyl ester (9) in excellent yield (88–95%), and this was reduced to the alcohol (10) using NaBH4 and then converted into the bromide (11) using CBr4 and Ph3P (overall yield 60%). Ozonolysis followed by cleavage of the ozonide (Me2S) yielded aldehyde (12) (ca. 60–70% crude) which could be converted into the phosphonium salt (13) over a period of 2 days (quantitative yield). This participated in an intramolecular Wittig reaction following treatment with potassium carbonate in dichloromethane containing 18-crown-6. The yield of the methyl ether of combretastatin D2 (14) was only 30% and has not yet been optimised, although a large number of other bases, solvents and reaction conditions have been investigated (Table 1). Stronger bases than potassium carbonate invariably caused hydrolysis of the ester group. Since compound (14) has been converted into combretastatin D2 by Boger et al. (using BI3 and dimethylaniline in benzene),4 this constitutes a formal total synthesis of the natural product. However, our approach is both shorter and more efficient than the other published routes, and should allow access to a range of analogues for biological evaluation.| X | Solvent | Base | C/mM | T/°C | Yield (for 3 steps) |
|---|---|---|---|---|---|
| a After isomerisation on silica gel. b After irradiation. | |||||
O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) P(OMe)2 P(OMe)2 |
DME | tBuOK | 2 | 0 to 20 | Decomp. |
| OP(OMe)2 |
DME | NaH | 2 | 0 to 20 | Decomp. |
| Ph3P+, Cl− | DMF | tBuOK | 2 | 0 to 20 | <10% |
| Ph3P+, Br− | DCM | K2CO3/18-C-6 | 2 | 20 | 31%a |
| Ph3P+, Br− | DCM | K2CO3/18-C-6 | 5 | 20 | 29%b |
| Ph3P+, Br− | DCM | K2CO3/18-C-6 | 10 | 20 | 26%b |
| Ph3P+, Br− | DCM | DBU | 2 | 0 to 20 | <10%a |
| Ph3P+, Br− | THF–DCM | NaHMDS | 2.5 | 0 to 20 | 15%a |
| Ph3P+, Br− | THF–DCM | Amberlite | 2.5 | 20 | 17%a |
| Ph3P+, Br− | DCM–H2O | Sat. K2CO3 | 10 | Reflux | 0% |
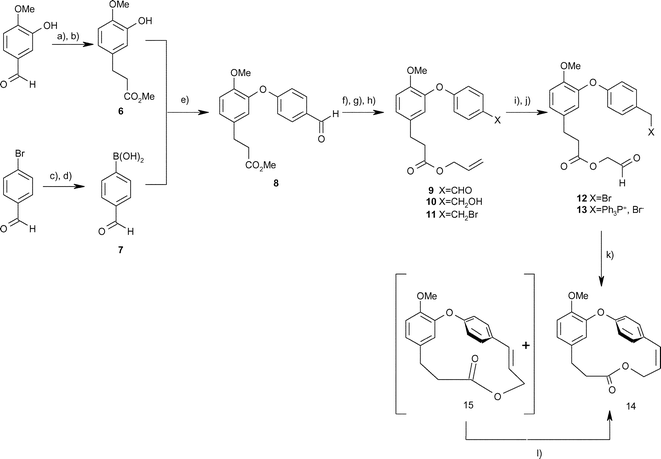 | ||
| Scheme 1 Synthesis of combretastatin D2 methyl ether via intramolecular Wittig reaction. (a) Ph3PCHCO2Me, DCM, r.t., overnight, 95%, (b) HCO2NH4, 10% Pd/C, MeOH, reflux 3 h, 91%, (c) ethylene glycol, PhCH3, pTsOH, 18 h, 75–89%, (d) i) nBuLi, THF, B(OiPr)3, −78 °C to r.t. overnight, ii) 3 N HCl, THF, 2 h, r.t., 70–75% overall, (e) Et3N, Cu(OAc)2, DCM, 4A molecular sieves, r.t., 18 h, 55–70%, (f) allyl alcohol, Bu2SnO, reflux, 20 h, 88–95%, (g) NaBH4, MeOH, r.t., 3 h, 65%, (h) CBr4, PPh3, DCM, r.t, 3 h, 85–90%, (i) O3, DCM, −78 °C, then Me2S, DCM, −78 °C to r.t., 4 h, (j) PPh3, MeCN, r.t., 2 d, (k) K2CO3–18-C-6, DCM, slow addition, 20 °C, overnight, 26–31% from 11, (l) light, CCl4–CHCl3, overnight, quantitative. | ||
Interestingly, in the product mixture from the Wittig reaction the trans-alkene (15) could be seen, though this isomerised to the cis-alkene during silica chromatography (isomerisation was also effected using light). This isomer has never been reported before and both alkenes exhibited interesting 1H NMR signals for the aryl hydrogen β to the methoxy group. For the cis-alkene this resonated at 5.11 ppm while for the trans-isomer it appeared at 4.59 ppm, providing evidence in both cases of significant diamagnetic shielding by the aromatic ring current (Figs. 1 and 2). An X-ray crystal structure of compound (14) confirmed the relationship of the β-hydrogen to the aromatic ring (Fig. 3)‡.
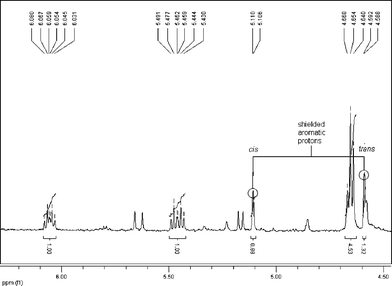 | ||
| Fig. 1 NMR spectrum of the cis–trans mixture obtained after attempted isolation of the trans-isomer on neutralized silica gel. | ||
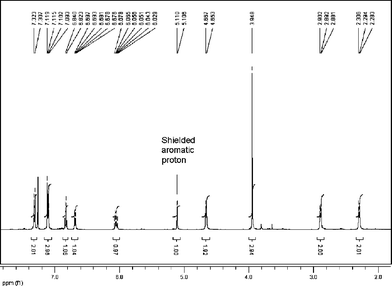 | ||
| Fig. 2 NMR spectrum of combretastatin D2 methyl ether. | ||
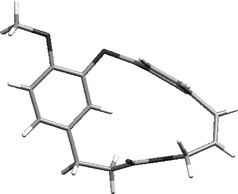 | ||
| Fig. 3 X-Ray structure of combretastatin D2 methyl ether. | ||
An alternative approach to (14) is shown in Scheme 2 and involved an attempted ring-closing metathesis reaction. The aldehydo-ester (8) was converted into the styrene (16) via a Wittig reaction (80–87%), and transesterification as before with allyl alcohol produced the diene (17) (85–95%). Attempted RCM reactions with Grubbs’ catalyst I under a variety of conditions (Table 2) provided no products, while reaction with Grubbs’ catalyst II yielded the dimeric product (18). The best conditions employed 20% of catalysts in toluene at 80 °C with a 75% conversion and 40% yield of the dimer. To date, other metathesis catalysts (e.g. Schrock catalyst) have not been tried.
| Solvent (%cat.) | C/mM | T/°C | t | Conversion (%)a |
|---|---|---|---|---|
| a Conversion determined by analysis of the NMR spectrum of the crude mixture. b Isolated yield. | ||||
| DCM (I, 6%) | 5 | 50 | 20 h | 0 |
| DCM (I, 10% + 50% Ti(OiPr)4) | 5 | 50 | 3 d | 80% (decomp.) |
| Toluene (I, 20% + 30% Ti(OiPr)4) | 5 | 80 | 3 d | 45% (decomp.) |
| DCM (II, 5%) | 5 | 50 | 2 d | 0 |
| DCE (II, 10%) | 5 | 90 | 2 d | <10 |
| Toluene (II, 10%) | 5 | 80 | 3 d | 60 |
| Toluene (II, 20%) | 5 | 80 | 2 d | 75 (40% yield)b |
| Toluene (II, 10%) | 0.5 | 110 | 1 h | 0 |
| Toluene (II, 10%) | 2 | 80 | 2 d | <15 |
| Toluene (II, 10%) | 7.5 | 80 | 2 d | 37 |
| Toluene (II, 10%) | 10 | 80 | 2 d | 40 |
 | ||
| Scheme 2 Attempted synthesis of combretastatin D2 methyl ether via ring-closing metathesis. a) Ph3P+CH3, Br−, NaHMDS, THF, 0 °C to r.t., 20 h, 80–87%, b) allyl alcohol, Bu2SnO, reflux, 20 h, 85–95%, c) Grubbs’ catalyst II, toluene, 80 °C, 30–75% conversion, 30–40% yield. | ||
Biological evaluation
Compound (14) was evaluated for antiproliferative activity against MCF-7 human breast carcinoma, RKO human colon carcinoma and CRL 1730 human umbilical endothelial cells. It exhibited activity in all the cell lines at around 5–10 µM (IC50 values, see Figs. 4–6). No comparable data is available for (14) or combretastatin D2 using these cell lines. The other products produced in this work have not yet been evaluated.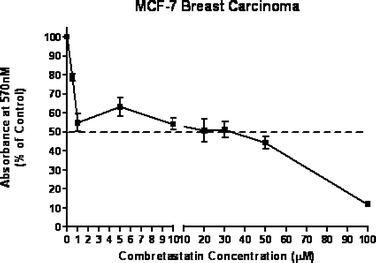 | ||
| Fig. 4 Antiproliferative activity of combretastatin D2 methyl ether against MCF-7 breast cancer cells. | ||
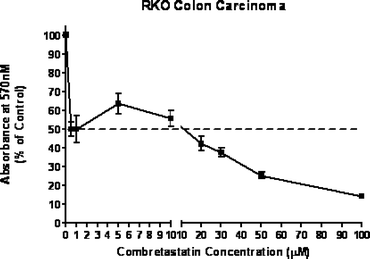 | ||
| Fig. 5 Antiproliferative activity of combretastatin D2 methyl ether against RKO colon cancer cells. | ||
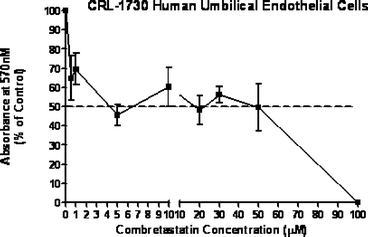 | ||
| Fig. 6 Antiproliferative activity of combretastatin D2 methyl ether against human endothelial cells. | ||
Experimental
In vitro drug sensitivity as determined by MTT dye-exclusion assay
In vitro drug sensitivity was determined as previously described.8 Combretastatin was dissolved in dimethyl sulfoxide (DMSO) to yield a stock solution of 10 mM. The fraction of viable cells remaining after drug treatment was determined by the ability of the cells to metabolise the water-soluble tetrazolium salt, 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide (MTT), into a water insoluble formazan precipitate. Exponentially growing cells (MCF-7 human breast carcinoma, RKO human colon carcinoma and CRL 1730 human umbilical endothelial cells) were seeded 24 hours prior to treatment into sterile flat-bottomed 96 well plates, at varied seeding densities determined by their growth characteristics. Cells were exposed to combretastatin (0.5–100 µM) for 96 hours. MTT (50 µl of a 2 mg ml−1 solution) was then added to each well. The plates were incubated for a further four hours and then the medium and any unconverted MTT was aspirated from the wells. The remaining formazan precipitate was dissolved in 100 µl DMSO and the absorbance read at 570 nm using a Molecular Devices plate reader. IC50 values (compound concentration that produces a 50% reduction in the growth of the cells) were determined from plots of absorbance vs. drug concentration.General experimental procedures
All solvents were dried before use. Acetonitrile, dichloromethane and methanol were either distilled from calcium hydride under nitrogen or argon or used after drying on a high pressure alumina column device (MBRAUN). Tetrahydrofuran was either dried by distillation in the presence of sodium and benzophenone or used after drying on a high pressure alumina column device (MBRAUN). Dry DMF was purchased from Aldrich. Thin layer chromatography was used to monitor reactions using Polygram® SIL G/UV254 precoated plastic sheets with a 0.2 mm layer of silica gel containing fluorescent indicator UV254. Plates were visualised using a 254 nm UV lamp and potassium permanganate stain. Flash column chromatography was carried out using Sorbsil® (or Fluorochem®) C60 silica gel (40–60 mesh) with the eluent or the gradient of eluents reported. NMR spectra were recorded using Bruker Avance300 or Bruker DRX500 spectrometers. Samples were dissolved in CDCl3 with tetramethylsilane as a reference. IR spectra were recorded using a Perkin-Elmer RXI FT-IR system spectrometer. Mass spectra were recorded on a VG Autospec spectrometer and were carried out by ASEP (Queen's University of Belfast). Elemental analyses were carried out by ASEP (Queen's University of Belfast). Melting points were recorded on a Stuart melting point apparatus and are uncorrected.X-Ray crystallography
X-Ray crystallographic data for combretastatin D2 methyl ether were collected using a Bruker SMART diffractometer with graphite monochromated Mo–Kα radiation. Data were collected at low temperature, ca. 153 K. Omega/phi scans were employed for data collection and absorption, Lorentz and polarisation corrections were applied.The structure was solved by direct methods and ordered non-hydrogen atoms were refined with anisotropic atomic displacement parameters. Hydrogen-atom positions were added, and idealised positions and a riding model with fixed thermal parameters (Uij = 1.2Ueq for the atom to which they are bonded (1.5 for CH3)), was used for subsequent refinements. The function minimised for wR2 was Σ[w(|Fo|2 − |Fc|2)] with reflection weights w−1 = [σ2 |Fo|2 + (g1P)2 + g2P] where P = [max |Fo|2 + 2|Fc|2]/3 for all F2 and the function minimised for R1 was Σ[w(|Fo| − |Fc|)]. The SAINT9 and SHELXTL10 packages were used for data collection, reduction, structure solution and refinement. Additional material available from the Cambridge Structural Database includes atomic co-ordinates, thermal parameters, remaining bond lengths and angles, and structure factors (CCDC no. 283472)‡.
O), 1511 (CC), 1267 (C–O–C), 1162 (C–O–C); mp: 74–76 °C (lit.11 72.4 °C); 1H NMR δH (CDCl3): 3.68 (0.82H, s, OMe-cis), 3.79 (3H, s, OMe-trans), 3.91 (0.82H, s, OMe-cis), 3.92 (3H, s, OMe-trans), 5.63 (0.28H, s, OH-cis), 5.72 (1H, s, OH), 5.26–5.32 (d, 2H, H3′, Jtrans = 18 Hz), 5.79–5.86 (1.61H, dm, H5′-trans
+ Ar-cis
+ H2-cis, Jo = 9 Hz), 7.01–7.04 (1H, dd, H6′, J = 2.1 Hz, Jo = 9 Hz), 7.13–7.14 (1H, d, H2′, Jm = 2.1), 7.22–7.25 (dd, Ar-cis), 7.32 (0.28, d, Jm = 1.8), 7.57–7.63 (1H, d, H2, Jtrans = 18 Hz); 13C NMR δC (CDCl3): 168 (CO), 149 (C4′), 146 (C3), 145 (C3′), 129 (C1′), 122 (C6′), 116 (C2), 113 (C2′), 111 (C5′), 56 (OMe), 52 (CO2Me); m/z (EIMS): 208 (M+, 100), 193 (13), 177 (52), 149 (11), 133 (20), 117 (11), 105 (12), 89 (17), 78 (14); mass found 208.073877, C11H12O4 requires 208.073559; found: C: 63.98, H: 5.66, C11H12O4 requires C: 63.45, H: 5.81, O: 30.74%.
O), 1506 (Ar), 1127 (C–O–C); mp: 96–97 °C (lit.12: 94–95 °C); 1H NMR δH (CDCl3): 2.57–2.62 (t, 2H, H3, J = 8.1 Hz), 2.83–2.89 (t, 2H, H2, J = 8.1), 3.67 (s, 3H, OMe), 3.86 (s, 3H, OMe), 5.58 (s, 1H, OH), 6.66–6.69 (dd, 1H, H6′, Jm = 1.9 Jo = 8.2), 6.76–6.78 (m, 2H, H2′ and H5′); 13C NMR δC (CDCl3): 174 (CO), 146 (C4′), 145 (C3′), 134 (C1′), 120 (C6′), 115 (C5′), 111 (C2′), 56 (OMe), 52 (OMe), 36 (C2), 31 (C3); m/z (EIMS): 211 (M+
+ H+, 11), 210 (M+, 63), 179 (9), 150 (34), 137 (100), 135 (20), 122 (9), 107 (14), 94 (9), 91 (14), 79 (11), 77 (13), 65 (10); mass found 210.088452, C11H14O4 requires 210.089209; found C: 63.09, H: 6.62, C11H14O4 requires C: 62.85, H: 6.71, O: 30.44%.
O), 137 (C4), 135 (C3 and C5), 129 (C2 and C6), 116 (C1); m/z (EIMS): 150 (M+, 25), 149 (M+
− H+, 64), 121 (M+
− H+
− CO, 100), 105 (M+
− B(OH)2, 16); mass found: 150.048187, C7H7BO3 requires 150.048824; found C: 55.41, H: 4.68, C7H7BO3 requires C: 56.07, H: 4.71, B: 7.21, O: 32.01%.
O), 164 (C1″), 150 (C3′), 143 (C4′), 134 (C1′), 132 (C3″ and C5″), 131 (C4″), 126 (C5′), 123 (C6′), 117 (C2″ and C6″), 113 (C2′), 61 (OMe), 52 (OMe), 36 (C2), 30 (C3); m/z (CIMS): 333 (M+
+ H+
+ NH4+), 332 (M+
+ NH4+), 315 (M+
+ H+), 314 (M+), 241, 228, 210; mass found: 314.116638, C18H18O5 requires 314.115424; found C: 68.47, H: 5.83, C18H18O5 requires C: 68.78, H: 5.77, O: 25.45%.
C), 1512 (Ar), 1273–1230–1126 (C–O–C); 1H NMR δH (CDCl3): 2.59–2.64 (t, 2H, H3, J = 8.0 Hz), 2.85–2.94 (t, 2H, H2, J = 7.9 Hz), 3.77 (s, 3H, OMe), 4.55–4.60 (d, 2H, CH2CC, J = 4.6 Hz), 5.19–5.30 (m, 2H, CCH2), 5.83–5.87 (ddt, 1H, CHC, Jcis = 11.0 Hz, Jtrans = 17.6 Hz, J = 4.6 Hz), 6.94–7.06 (m, 5H, H6′, H2″, H6″, H2′ and H5′), 7.80–7.84 (dd, 2H, H3″ and H5″), 9.93 (CHO); 13C NMR δC (CDCl3): 191 (CHO), 174 (CO), 164 (C1″), 150 (C3′), 144 (C4′), 134 (C1′ and CHC), 132 (C3″ and C5″), 131 (C4″), 126 (C5′), 123 (C6′), 119 (CCH2), 117 (C2″ and C6″), 113 (C2′), 66 (CH2CC), 56 (OMe), 36 (C2), 30 (C3); m/z (CIMS): 359 (M+
+ H+
+ NH4+, 22), 358 (M+
+ NH4+, 100), 342 (27), 341 (M+
+ H+, 96); mass (calculated for M + H+) found: 341.140213, C20 H21 O5 requires 341.138899.
C), 1507 (Ar), 1270–1225–1126 (C–O–C); 1H NMR δH (CDCl3): 2.60–2.65 (t, 2H, H3, J = 7.7 Hz), 2.87–2.92 (t, 2H, H2, J = 7.7 Hz), 3.84 (s, 3H, OMe), 4.57–4.59 (d, 2H, CH2CC, J = 5.7 Hz), 4.67 (s, 2H, CH2OH), 5.22–5.33 (m, 2H, CCH2), 5.84–5.97 (ddt, 1H, CHC, Jcis = 11 Hz, Jtrans = 17 Hz, J = 5.7 Hz), 6.84 (d, 2H, H2′, Jm = 1.7 Hz), 6.93–6.99 (m, 4H, H6′, H5′, H2″, H6″), 7.31–7.34 (d, 2H, H3″ and H5″, Jo = 8.4 Hz); 13C NMR δC (CDCl3): 173 (CO), 158 (C1″), 150 (C3′), 145 (C4′), 135 (C1′), 134 (C4″), 133 (CHC), 129 (C3″ and C5″), 125 (C5′), 121 (C6′), 119 (CCH2), 118 (C2″ and C6″), 113 (C2′), 66 (CH2OH), 65 (CH2CC), 56 (OMe), 36 (C2), 30 (C3); m/z (EIMS): 342 (M+, 100), 256 (15), 243 (M+
− CH2CHCH2
− CO2, 58), 239 (39), 197 (11), 153 (14), 137 (26), 134 (17), 121 (13), 105 (17), 77 (25); mass found: 342.146004, C20H22O5 requires 342.146724; found C: 69.56, H: 6.50, C20H22O5 requires C: 70.16, H: 6.48, O: 23.36%.
C), 1506 (Ar), 1272–1226–1168–1126 (C–O–C); 1H NMR δH (CDCl3): 2.61–2.67 (t, 2H, H3, J = 7.7 Hz), 2.89–2.94 (t, 2H, H2, J = 7.7 Hz), 3.83 (s, 3H, OMe), 4.53 (s, 2H, CH2Br), 4.58–4.60 (d, 2H, CH2CC, J = 5.8 Hz), 5.23–5.33 (m, 2H, CCH2), 5.84–5.97 (ddt, 1H, CHC, Jcis = 11.0 Hz, Jtrans = 17.0 Hz, J = 5.7 Hz), 6.88–7.04 (m, 5H, H2′, H6′, H5′, H2″, H6″), 7.33–7.36 (d, 2H, H3″ and H5″, Jo = 8.54 Hz); 13C NMR δC (CDCl3): 173 (CO), 159 (C1″), 150 (C3′), 145 (C4′), 134 (C1′), 133 (C4″), 132 (CHC), 131 (C3″ and C5″), 125 (C5′), 122 (C6′), 119 (CCH2), 117 (C2″ and C6″), 114 (C2′), 66 (CH2CC), 57 (OMe), 36 (C2), 34 (C3), 30 (CH2Br); m/z (EIMS): 406 (M+(81Br), 8), 404 (M+(79Br), 8), 336 (M+(81Br) − CO2CH2CHCH2, 9), 334 (M+(79Br) − CO2CH2CHCH2, 9), 325 (M+
− Br, 76), 299 (26), 253 (98), 251 (M+
− Br − CO2CH2CHCH2, 100), 249 (36), 211 (14), 172 (26), 149 (18), 91 (44), 79 (38); mass found: 404.060319, C20H21BrO4 requires 404.062320.
O), 159 (C1″), 151 (C3′), 145 (C4′), 134 (C1′), 132 (C4″), 131 (C3″ and C5″), 125 (C5′), 122 (C6′), 117 (C2″ and C6″), 113 (C2′), 69 (CH2CHO), 56 (OMe), 36 (C2), 34 (CH2Br), 30 (C3); m/z (EIMS): 407 (M+(81Br), 22), 406 (M+(81Br) − H+, 98), 405 (M+(79Br), 22), 404 (M+(79Br) − H+, 98), 391 (39), 380 (M+(81Br) − H+
− CO, 46), 378 (M+(79Br) − H+
− CO, 46), 370 (37), 366 (M+(81Br) − CH2CHO, 43), 364 (M+(79Br) − CH2CHO), 360 (58), 333 (10), 325 (14), 299 (14), 241 (13), 185 (17), 167 (16), 149 (54), 78 (58); mass found: 405.033638, C19H19BrO5 requires 405.033760.
O), 159 (C1″), 150 (C3′), 145 (C4′), 135, (s, P+Ph3p), 134.8–134.9 (d, P+Ph3o, 2JPC = 9.7 Hz), 134 (C1′), 132 (C4″), 132 (d, C3″ and C5″, 3JPC = 9.84 Hz), 130.5–130.6 (d, P+Ph3m, 3JPC = 12.6 Hz), 128 (P+Ph3i, 1JPC = 34 Hz), 126 (C6′), 117 (C2″ and C6″), 117 (C2′), 114 (C5′), 69 (CH2CHO), 56 (OMe), 36 (C3), 30.5–30.9 (d, CH2P+, 1JPC = 50.3 Hz), 30 (C3); m/z (FAB+): 607 (M+
+ H2O, 17), 589 (M+, 52), 561 (31), 547 (33), 503 (14), 455 (15), 369 (56), 293 (28), 279 (100), 262 (52), 183 (33), 154 (49), 136 (41); mass found: 589.214401, C37H34O5P+ requires 589.214388.
C), 1507 (Ar), 1270–1225–1126 (C–O–C); 1H NMR δH (CDCl3): 2.62–2.67 (t, 2H, H3, J = 7.8 Hz), 2.89–2.94 (t, 2H, H2, J = 7.8 Hz), 3.83 (s, 3H, OMe), 4.58–4.60 (d, 2H, CH2CC, J = 5.8 Hz), 4.61 (s, 2H, CH2Cl), 5.23–5.33 (m, 2H, CCH2), 5.85–5.98 (ddt, 1H, CHC, Jcis = 10.5 Hz, Jtrans = 16.5 Hz, J = 5.8 Hz), 6.88–7.04 (m, 5H, H2′, H6′, H5′, H2″, H6″), 7.32–7.35 (d, 2H, H3″ and H5″, Jo = 7.34 Hz); 13C NMR δC (CDCl3): 173 (CO), 159 (C1″), 150 (C3′), 145 (C4′), 134 (C1′), 133 (C4″), 132 (CHC), 130 (C3″ and C5″), 125 (C5′), 122 (C6′), 119 (CCH2), 117 (C2″ and C6″), 113 (C2′), 66 (CH2CC), 56 (OMe), 46 (CH2Cl), 36 (C2), 30 (C3); m/z (EIMS): 362 (M+(37Cl), 36), 360 (M+(35Cl), 100), 325 (M+, 35), 319 (M+, 15), 261 (40), 241 (26), 136 (32), 215 (27), 153 (32), 137 (43), 113 (20), 91 (22); mass found: 360.112236, C20H21ClO4 requires 360.112837; found C: 65.58, H: 5.72, C20H21ClO4 requires C: 66.57, H: 5.87, O: 17.73, Cl: 9.83%.
O), 1506.5 (Ar), 1270.8–1227.3–1171.0–1127.2 (C–O–C, PO and P–O); 1H NMR δH (CDCl3): 2.60–2.65 (t, 2H, H3, J = 7.8 Hz), 2.87–2.92 (t, 2H, H2, J = 7.8 Hz), 3.12–3.19 (d, 2H, CH2P, 2JPH = 21.3 Hz), 3.68–3.72 (d, 6H, P(OMe)2, 3JPH = 10.8 Hz), 3.82 (s, 3H, OMe), 4.57–4.58 (d, 2H, CH2CC, J = 5.6 Hz), 5.22–5.32 (m, 2H, CCH2), 5.83–5.96 (ddt, 1H, CHC, Jcis = 10.7 Hz, Jtrans = 17 Hz, J = 5.6 Hz), 6.84 (d, 1H, H2′, Jm = 1.7 Hz), 6.88–7.00 (m, 4H, H6′, H5′, H2″, H6″), 7.22–7.26 (dd, 2H, H3″ and H5″, Jo = 8.54 Hz, Jm = 2.20 Hz); 13C NMR δC (CDCl3): 173 (CO), 158 (C1″), 150 (C3′), 145 (C4′), 134 (C1′), 133 (CHC), 131 (d, C3″ and C5″, 3JPC = 6.6 Hz), 130 (C4″), 125 (C5′), 122 (C6′), 119 (CCH2), 118 (d, C2″ and C6″, 4JPC = 2.8 Hz), 113 (C2′), 66 (CH2CC), 56 (OMe), 53 (P(OMe)2, 2JPC = 6.9 Hz), 36 (C2), 32–33 (CH2P, 1JPC = 139 Hz), 30 (C3); m/z (EIMS): 434 (M+, 8), 360 (10), 348 (6), 325 (7), 261 (8), 239 (18), 211 (15), 134 (15), 109 (100), 89 (27), 78 (33); mass found: 434.149195, C22H27O7P requires: 434.149442.
O), 1507.8 (Ar), 1271–1226–1171–1127 (C–O–C); 1H NMR δH (CDCl3): 2.70–2.76 (t, 2H, H3, J = 7.6 Hz), 2.88–2.95 (t, 2H, H2, J = 7.6 Hz), 3.12–3.19 (d, 2H, CH2P, 2JPH = 21.3 Hz), 3.68–3.71 (d, 6H, P(OMe)2, 3JPH = 10.8 Hz), 3.82 (s, 3H, OMe), 4.66 (s, 2H, CH2CHO), 6.85–7.00 (m, 5H, H2′, H6′, H5′, H2″, H6″), 7.22–7.24 (dd, 2H, H3″ and H5″, Jo = 8.54 Hz, Jm = 2.20 Hz), 9.56 (CHO); 13C NMR δC (CDCl3): 196 (CHO), 172 (CO), 158 (C1″), 150 (C3′), 145 (C4′), 134 (C1′), 131 (d, C3″ and C5″, 3JPC = 6.6 Hz), 130 (C4″), 125 (C5′), 122 (C6′), 118 (d, C2″ and C6″, 4JPC = 3.0 Hz), 113 (C2′), 69 (CH2CHO), 56 (OMe), 53 (P(OMe)2, 2JPC = 6.9 Hz), 36 (C2), 32–33 (CH2P, 1JPC = 139 Hz), 30 (C3); m/z (EIMS): 436 (M+, 10), 408 (27), 394 (28), 348 (46), 334 (23), 327 (17), 299 (46), 285 (62), 261 (30), 239 (47), 216 (100), 211 (75); mass found: 436.129509, C21H25O8P requires 436.128707.
Procedure with tBuOK–DMF. Potassium t-butoxide was added at 0 °C to a dilute solution of the phosphonium salt (13) in DMF. The resulting yellow solution was allowed to warm to room temperature and was stirred overnight. The reaction was quenched with saturated NH4Cl and the aqueous layer was extracted with EtOAc. The combined organic extracts were washed several times with brine, then dried with MgSO4.
General procedure for intramolecular Wittig reactions with K2CO3–18-C-6. A slurry of potassium carbonate (4 eq.) and 18-crown-6 (4 eq.) in DCM (15 mL mmol−1) was stirred at room temperature for 3 h before the slow addition of a solution of the phosphonium salt (13) via a syringe pump (3 mL h−1). The resulting yellow solution was stirred overnight and the mixture was concentrated under vacuum. The reaction was quenched with saturated NH4Cl, and the aqueous layer was extracted with DCM. The combined organic extracts were washed with brine, then dried with MgSO4. The crude product was purified by flash chromatography (EtOAc–hexane, 2 : 8).
(The same procedure was used for the intramolecular Wittig reactions with the other bases listed in Table 1.)
Combretastatin D2 methyl ether data:
white powder; Rf: 0.43 (EtOAc–hexane, 2 : 8); νmax/cm−1: 2925 (ArH), 1732 (CO), 1586, 1519, 1502, 1265–1219–1149–1128 (C–O–C); mp: 130–131 °C (lit.4: 130–132), 1H NMR δH (CDCl3): 2.28–2.30 (t, 2H, H16, J = 5.4 Hz), 2.88–2.90 (t, 2H, H15, J = 5.4 Hz), 3.95 (s, 3H, OMe), 4.65–4.67 (d, 2H, H2, J = 6.68 Hz), 5.12 (d, 1H, H20, J = 1.99 Hz), 6.03–6.08 (dt, 1H, H3, J1 = 11.1 Hz, J2 = 6.68 Hz), 6.67–6.69 (dd, 1H, H13, J1 = 8.2 Hz, J2 = 1.99 Hz), 6.82–6.84 (d, 1H, H12, J = 8.2 Hz), 7.09–7.12 (m, 3H, H4 H7 H19), 7.31–7.32 (d, 2H, H6 H18, J = 8.2 Hz); 13C NMR δC (CDCl3): 173 (C17), 156 (C8), 151 (C10), 146 (C11), 138 (C3), 135 (C4), 133 (C5), 129 (C6/C14), 125 (C18), 121 (C7/C19), 122 (C13), 113 (C12), 112 (C20), 59 (C2), 56 (OMe), 31 (C16), 27 (C15); m/z (EIMS): 310 (M+, 100), 253 (16), 239 (35), 183 (37), 149 (39), 123 (44), 115 (36), 112 (21), 97 (23), 91 (20), 83 (26), 71 (37), 57 (65); mass found: 310.120979, C19H18O4 requires 310.120509.
C), 1506 (Ar), 1227–1270–1127 (C–O–C); 1H NMR δH (CDCl3): 2.47–2.54 (t, 2H, H3, J = 7.5 Hz), 2.76–2.81 (t, 2H, H2, J = 7.5 Hz), 3.57 (s, 3H, OMe), 3.74 (s, 3H, OMe), 5.07–5.11 (d, 1H, CCH2, Jcis = 10.9 Hz), 5.55–5.60 (d, 1H, CCH2, Jtrans = 17.6 Hz), 6.55–6.65 (dd, 1H, CHC, Jcis = 10.9 Hz, Jtrans = 17.6 Hz), 6.74–6.91 (m, 5H, H6′, H5′, H2′, H3″ and H5″), 7.26–7.28 (dd, 2H, H2″ and H6″, Jm = 1.9 Hz, Jo = 6.8 Hz); 13C NMR δC (CDCl3): 172 (CO), 157 (C1″), 149 (C3′), 144 (C4′), 135 (ArCC), 132 (C1′), 131 (C4″), 127 (C3″ and C5″), 123 (C5′), 120 (C6′), 116 (C2″ and C6″), 112 (ArCHCH2), 111 (C2′) 55 (OMe), 51 (CO2Me), 35 (C2), 29 (C3); m/z (CIMS): 313 (M+
+ H+), 281 (M+
+ H+
− OMe); mass (calculated for M + H+) found: 313.143349, C19H21O4 requires 313.143984.
C), 1506 (Ar), 1227–1270–1127 (C–O–C); 1H NMR δH (CDCl3): 2.50–2.55 (t, 2H, H3, J = 7.9 Hz), 2.77–2.82 (t, 2H, H2, J = 7.9 Hz), 3.73 (s, 3H, OMe), 4.46–4.48 (d, 2H, CH2–CC, J = 4.6 Hz), 5.07–5.11 (d, 1H, ArCCH2, Jcis = 11.0 Hz), 5.15–5.22 (m, 2H, CHCH2), 5.53–5.59 (d, 1H, ArCCH2, Jtrans = 17.6 Hz), 5.79–5.85 (ddt, 1H, CHC, Jcis = 11.0 Hz, Jtrans = 17.6 Hz, J = 4.6 Hz), 6.55–6.65 (dd, 1H, ArCHC, Jcis 10.9 Hz, Jtrans = 17.6 Hz), 6.75–6.91 (m, 5H, H2′, H5′, H6′, H2″ and H6″), 7.20–7.28 (dd, 2H, H3″ and H5″, Jm = 2.0 Hz, Jo = 6.9 Hz); 13C NMR δC (CDCl3): 171 (CO), 156 (C1″), 149 (C3′), 144 (C4′), 135 (ArCC), 132 (C1′), 131 (C4″ and CHC), 126 (C3″ and C5″), 123 (C5′), 120 (C6′), 117 (CCH2), 116 (C2″ and C6″), 112 (ArCHCH2), 111 (C2′), 64 (CH2CC), 55 (OMe), 35 (C2), 29 (C3); m/z (CIMS): 339 (M+
+ H+); mass (calculated for M + H+) found: 339.160606, C21H23O4 requires: 339.159634; found C: 74.12, H: 7.02, C21H22O4 requires: C: 74.54, H: 6.55, O: 18.91%.
C), 1515.6 (Ar), 1506.4 (Ar), 1269.0–1230.2–1123.0 (C–O–C); mp: 216–217 °C; 1H NMR δH (CDCl3): 2.49–2.54 (t, 2H, H16, J = 8.8 Hz), 2.78–2.84 (t, 2H, H15, J = 8.8 Hz), 3.88 (s, 3H, OMe), 4.65–4.67 (dd, 2H, H2, J1 = 6.9 Hz, J2 = 0.9 Hz), 6.07–6.17 (dt, 1H, H3, Jtrans = 15.9 Hz), 6.58–6.61 (d, 1H, H4, J = 15.9 Hz), 6.59 (d, 1H, H20, Jm = 1.8 Hz), 6.89–6.91 (dd, 1H, H13, Jo = 8.3 Hz, Jm = 1.8 Hz), 6.92–6.93 (d, 1H, H12, Jo = 8.3 Hz), 6.94–6.97 (dd, 2H, H7, H19, Jo = 8.7 Hz, Jm = 2.0 Hz), 7.30–7.32 (dd, 2H, H6 and H18, Jo = 8.7 Hz, Jm = 2.0 Hz); 13C NMR δC (CDCl3): 29.3 (C15), 35.3 (C16), 55.1 (OMe), 64.3 (C2), 112 (C13), 117 (C20), 118 (C7 and C19), 121 (C3), 123 (C12), 127 (C6 and C18), 130 (C5), 132 (C14), 134 (C4), 145 (C11), 148 (C10), 156 (C8), 171 (C17); m/z (FAB): 621 (M+, 36), 391 (6), 311 (23), 307 (16), 251 (34), 195 (17), 176 (14), 154 (100), 136 (85), 131 (46), 121 (33); mass found: 620.241592, C38H36O8 requires 620.241019.
References
- A. Cirla and J. Mann, Combretastatins: from natural products to drug discovery, Nat. Prod. Rep., 2003, 20, 558–564 RSC.
- P. E. Thorpe, D. J. Chaplin and D. C. Blakey, The First International Conference on Vascular Targeting: Meeting Overview, Cancer Res., 2003, 63, 1144–1147 CAS.
- N. Vongvanich, P. Kittakoop, P. Charoenchai, S. Intamas, K. Danwisetkanjana and Y. Thebtaranonth, Planta Med., 2005, 71, 191 CrossRef CAS.
- V. H. Deshpande and N. J. Gokhale, Tetrahedron Lett., 1992, 33, 4213 CrossRef CAS; S. D. Rychnovski and K. H. Hwang, J. Org. Chem., 1994, 59, 5414 CrossRef CAS; E. A. Couladouros and I. C. Soufli, Tetrahedron Lett., 1995, 36, 9369 CrossRef CAS; D. L. Boger, S. M. Sakya and D. Yohannes, J. Org. Chem., 1991, 56, 4204 CrossRef CAS; E. A. Couladouros, I. C. Soufli, V. Moutsos and R. K. Chadha, Chem.–Eur. J., 1998, 4(1), 33 CrossRef CAS; K. K. Gangakhedkar, Synth. Commun., 1996, 26, 1887 CrossRef CAS.
- G. R. Pettit, S. B. Singh and M. L. Niven, J. Am. Chem. Soc., 1988, 110, 8539 CrossRef CAS; S. B. Singh and G. R. Pettit, J. Org. Chem., 1990, 55, 2797 CrossRef CAS.
- D. A. Evans, J. L. Katz and T. R. West, Tetrahedron Lett., 1998, 39, 2937 CrossRef CAS; M. T. Chan, K. L. Monaco, R. P. Wang and M. P. Winters, Tetrahedron Lett., 1998, 39, 2933 CrossRef CAS.
- P. Baumhof, R. Mazitschek and A. Giannis, Angew. Chem., Int. Ed., 2001, 40, 3672 CrossRef CAS.
- A. Seaton, C. Higgins, J. Mann, A. Baron, C. Bailly, S. Neidle and H. van den Berg, Eur. J. Cancer, 2003, 39, 2548–2555 CrossRef CAS.
- SMART Software Reference Manual, version 5.054, Bruker Analytical X-Ray Systems Inc., Madison, WI, 1998 Search PubMed.
- G. M. Sheldrick, SHELXTL, An Integrated System for Data Collection, Processing, Structure Solution and Refinement, Bruker Analytical X-Ray Systems Inc., Madison, WI, 1998 Search PubMed.
- J. Lee and J. Lee, Synth. Commun., 1999, 29, 4127 CrossRef CAS.
- F. R. Blase and K. Banerjee, Synth. Commun., 1995, 25, 3187 CrossRef CAS.
- I. N. Zhmurova, V. G. Yurchenko, R. I. Yurchenko and T. V. Savenko, J. Gen. Chem. USSR (Engl. Transl.), 1977, 47, 2015–2019; I. N. Zhmurova, V. G. Yurchenko, R. I. Yurchenko and T. V. Savenko, Zh. Obshch. Khim., 1977, 47, 2207–2212 CAS.
- H. R. Snyder, A. J. Reedy and W. J. Lennarz, J. Am. Chem. Soc., 1958, 80, 835 CrossRef CAS.
- K. C. Park, K. Yoshino and H. Tomiyasu, Synthesis, 1999, 12, 2041 CrossRef.
Footnotes |
| † This paper is dedicated to Professor Steve Ley on the occasion of his 60th birthday. |
| ‡ Crystal data for C19H18O4: the crystals are small and show very weak diffraction and hence a low ratio of observed reflections (approx. 25%). However, we were able to establish the atomic connectivity from this crystal. M = 310.33, orthorhombic, space group Pbca, a = 13.47(5) Å, b = 10.86(4) Å, c = 21.40(10) Å, U = 3132(22) Å3, Z = 8, µ = 0.092 mm−1. A total of 5614 reflections were measured for the angle range 2 < 2θ < 45 and 2022 independent reflections were used in the refinement. The final parameters were wR2 = 0.2935 and R1 = 0.1041 [I > 2σI]. CCDC reference number 283472. For crystallographic data in CIF or other electronic format see DOI: 10.1039/b513515j |
| This journal is © The Royal Society of Chemistry 2006 |