Highly efficient methods for the one-pot synthesis of β-substituted enones
William J.
Kerr
*,
Colin M.
Pearson
and
Graeme J.
Thurston
WestCHEM, Department of Pure and Applied Chemistry, University of Strathclyde, 295 Cathedral Street, Glasgow, Scotland, UK G1 1XL. E-mail: w.kerr@strath.ac.uk; Fax: +44 141 548 4246; Tel: +44 141 548 2959
First published on 25th November 2005
Abstract
A mild and practically-convenient one-pot procedure for the direct β-substitution of enones has been developed using a conjugate addition–oxidation strategy with a full range of copper-based reagents and N-tert-butylphenylsulfinimidoyl chloride; alkyl- and aryl-substituted enones are delivered in good to excellent yields.
β-Functionalised enones are of elevated importance in organic chemistry and have wide preparative utility in transformations such as Michael addition processes and Diels–Alder reactions. Having stated this, synthetic methods for the production of this moiety are somewhat limited. Although effective preparative strategies exist, in general, most techniques involve multistage processes, harsh reaction conditions, or the use of expensive transition metals. In this regard, perhaps the most widely used method for the formation of β-substituted enones is based on the conjugate addition of an alkyl or aryl cuprate to an enone, followed by the introduction of a selenyl species to provide an α-arylselenyl ketone, which can then be oxidised and eliminated.1 Despite the efficacy of this strategy, the toxicity and two-step nature of the process somewhat detract from its synthetic utility. The analogous technique with a substituted sulfide2 also has severe limitations with regard to functional group compatibility, given the relatively harsh reaction conditions required for the oxidation/elimination. Another applicable method for enone functionalisation is the use of β-halo enones which, following 1,4-addition, readily eliminate to generate the substituted enone product.3 Although valuable, the difficulty in accessing β-halo enones and the formation of disubstituted by-products, limit the utility of this methodology severely. More recently, Nicolaou4 and co-workers have reported on the use of IBX to generate β-substituted enones in an efficient manner, from β-substituted trimethylsilyl enol ethers, themselves generated by conjugate addition to enones. Although this methodology is very mild and efficient, it again involves a multistep process.
In 2000, Mukaiyama and co-workers reported on the use of the versatile reagent, N-tert-butylphenylsulfinimidoyl chloride (1),5 as an oxidant which mediated the oxidation (dehydrogenation) of a series of organic compounds.6,7 In particular, this reagent can be employed in the oxidation of alcohols, amines, and hydroxylamines,7 and in the dehydrogenation of ketones to enones under very mild, low temperature conditions.6b,8 In attempts to further widen the applicability of this sulfinimidoyl species, and given the commercial availability9 and relative ease of preparation of 1 on a large scale,5b we believed that this reagent could be strategically utilised in a novel fashion to directly access β-substituted enones. More specifically and as illustrated in Scheme 1, we envisaged that, following conjugate addition of an organocuprate to an α,β-unsaturated enone, N-tert-butylphenylsulfinimidoyl chloride (1) could be applied as an electrophilic quench to generate β-substituted enones directly from unsubstituted enones in a one-pot, two-step procedure. Accordingly, this strategy was embarked upon. However, following completion of our studies and during the preparation of our manuscript for publication, Matsuo and Aizawa divulged their findings using a very similar approach.10 Based on this, we now communicate the preparative outcomes from our endeavours in this area, whilst highlighting the differences and potentially wider practical utility of our techniques over the recently published procedures.
 | ||
| Scheme 1 | ||
Based on the utility and synthetic complementarity of the range of known organocuprate reagents and protocols,11 we felt that any procedure that was developed as part of this programme should be compatible with as wide a spectrum of the available copper-based methods as possible. Nonetheless, to initiate our studies in this area, we investigated the reaction of a simple Gilman cuprate,12 lithium di-n-butylcuprate, with cyclohex-2-enone, followed by application of 1 as the electrophilic quench. As shown in Table 1, Entry 1, using 2.5 equivalents of N-tert-butylphenylsulfinimidoyl chloride (1), the desired one-pot transformation was found to be achievable, albeit in a rather low 39% yield. In order to optimise the efficiency of this process, the number of equivalents of 1 was progressively increased. Gratifyingly, an improved yield was obtained following each increase in the quantity of the sulfinimidoyl chloride employed. Indeed, using 5 equivalents of 1 produced a good yield of 82% of the desired β-substituted enone product (Table 1, Entry 4). Importantly, at this stage, we felt that to enhance the reactivity of the intermediate enolate ion, formed following conjugate addition, tuning of the polarity of the reaction medium could be beneficial. Accordingly, the oxidant 1 was added in the more polar THF. When this practical amendment was made, the two-step, one-pot addition–oxidative quench was achieved in an excellent and near quantitative 98% yield (Table 1, Entry 5). Consequently, following this brief programme of optimisation, by employing readily prepared and handled simple cuprate reagents, followed by sulfinimidoyl chloride (1), a practically-convenient protocol had been established for the direct preparation of β-substituted enones from the equivalent unsubstituted starting materials. Importantly, this outcome contrasts with the work of Matsuo and Aizawa, who found that use of a simple Gilman cuprate, followed by the addition of 1 in ether (cf. in THF within our optimised system), only produced a low yield of the desired substituted enone product from cyclohex-2-enone (Me2CuLi, 3 equiv. 1; 27% yield).10
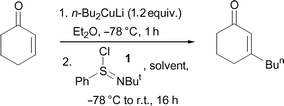
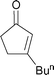
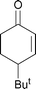
Following these successful initial optimisation studies, the generality and applicability of our developed method with alternative α,β-unsaturated substrates was investigated using a Gilman cuprate.† As Table 2 illustrates, this technique provides good to excellent yields of β-substituted enones from all of the unsaturated starting materials employed. In this regard, enones of varied ring size (5–7) were applied without difficulty. Furthermore, a six-membered ring substrate with a sterically demanding t-butyl group present in the 4-position also reacted very efficiently (Table 2, Entry 4). In addition, this method is not restricted to cyclic substrates, as can be seen from Entry 5, with a good 83% yield being obtained from the acyclic substrate shown. Again, this shows some advantages over the recently published protocols, where the optimised conditions (with higher-order cuprates) delivered only low to moderate yields of substituted acyclic enones.10









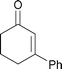
Having demonstrated the use of various α,β-unsaturated substrates, our attention turned to the delivery of alternative suitable lithium dialkylcuprates. Gratifyingly, the introduction of alternative alkyl groups was readily achieved, again in excellent yields. As illustrated in Table 3, delivery of methyl and n-octyl groups provided the product enones in 86 and 80% yield, respectively, with the efficiency of the former process contrasting with the 27% yield obtained with the same cuprate reagent and 1 under the recently published conditions.10 In addition and pleasingly, the conjugate delivery of a phenyl group was also highly effective (Table 3, Entry 4) demonstrating that this methodology is not solely limited to the introduction of alkyl groups.
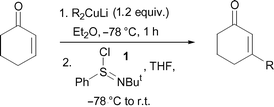



Having established successful protocols with pre-formed Gilman reagents, we then further expanded our study and initiated investigations into alternative copper-mediated methods for 1,4-attack on enones. In this regard, we firstly turned to copper iodide-catalysed conjugate additions with Grignard reagents.14 As shown in Table 4, the delivery of β-functionalised enones was, again, possible using this method, with primary, secondary, and tertiary alkyl groups all being delivered successfully. Whilst these copper-catalysed Grignard processes were marginally less efficient than the methods described above, good yields (73–74%) of the functionalised enones were still obtained using this practically-convenient protocol.




It is widely recognised that the requirement for two equivalents of the nucleophilic component within dialkyl- or diarylcuprates lowers their overall effectiveness as reagents in organic synthesis, especially if the transferring alkyl or aryl unit is synthetically valuable. Despite the general effectiveness of the protocols developed here, the cuprates (and the copper-catalysed Grignard processes) employed, in conjunction with the sulfinimidoyl chloride 1, do require excesses of the nucleophilic agent. Based on this, we proceeded to investigate a number of alternative and potentially more preparatively economical organocuprate reagents, where only one equivalent of the transferring unit is employed. In this regard, use of a mono-organocuprate in dimethyl sulfide15 proceeded effectively to deliver a 74% yield of the desired β-substituted enone product (Table 5, Entry 1). Indeed, as shown by Entry 2 in Table 5, for the delivery of such a simple alkyl group in our addition–oxidation procedure, the sulfide additive was not required. Finally in this series, the popular thienylcuprates developed by Lipshutz,16 in which the 2-thienyl group is used as a non-transferable ligand within mixed higher-order cyanocuprates, were applied to our system with 1. Pleasingly, this also proved highly effective and produced a very good 76% yield of the desired enone product (Table 5, Entry 3). Interestingly, Matsuo and Aizawa have reported that, when using their established techniques, with ether as solvent for both the cuprate and sulfinimidoyl chloride, use of Lipshutz-type higher-order cuprates bearing either a 2-thienyl or an N-imidazolyl group only gave very low to moderate yields of the α,β-unsaturated ketone product. In contrast and to complement the methods reported here, this previously published study shows how higher order dialkyl (or diaryl) cyanocuprates can be employed successfully in this addition–oxidation process, albeit with excesses of the transferring unit being required.10

In summary, we have developed a mild and practically-convenient one-pot conjugate addition–oxidation protocol, to directly produce β-substituted enones in a highly efficient manner. This methodology has proven to be effective with both cyclic and acyclic enones. Furthermore, the application of a series of individual organocuprate reagents has also been successfully demonstrated, to install primary, secondary, and tertiary alkyl units, as well as aryl moieties. Given all of these features, coupled with the ease of preparation and commercial availability of the oxidant, and linked with the simple experimental procedures developed, we feel that these techniques will be of value to the wider scientific community.
We thank the EPSRC for funding (G.J.T.), the C. K. Marr Trust for a Postgraduate Scholarship (C.M.P.), Pfizer Global Research and Development, Sandwich for generous funding of our research, and the EPSRC Mass Spectrometry Service, University of Wales, Swansea for analyses.
References
- (a) H. J. Reich, J. M. Renga and I. L. Reich, J. Org. Chem., 1974, 39, 2133–2135 CrossRef CAS; (b) H. J. Reich, J. M. Renga and I. L. Reich, J. Am. Chem. Soc., 1975, 102, 5434–5447 CrossRef.
- B. M. Trost, Chem. Rev., 1978, 78, 363–382 CrossRef CAS.
- (a) E. Piers and I. Nagakura, J. Org. Chem., 1975, 40, 2694–2696 CrossRef CAS; (b) E. Piers, K. F. Cheng and I. Nagakura, Can. J. Chem., 1982, 60, 1256–1263 CAS; (c) E. Piers, J. R. Grierson, C. K. Lau and I. Nagakura, Can. J. Chem., 1982, 60, 210–223 CAS.
- K. C. Nicolaou, D. L. F. Gray, T. Montagnon and S. T. Harrison, Angew. Chem., Int. Ed., 2002, 41, 996–1000 CrossRef CAS.
- For the original preparation of N-tert-butylphenylsulfinimidoyl chloride, see: (a) L. N. Markovskii, T. N. Dubinina, E. S. Levchenko and A. V. Kirsanov, J. Org. Chem. USSR, 1973, 9, 1435–1439; for a more recent and practically-utilisable preparation, see: (b) J. Matsuo, D. Iida, K. Tatani and T. Mukaiyama, Bull. Chem. Soc. Jpn., 2002, 75, 223–234 CrossRef CAS.
- (a) T. Mukaiyama, J. Matsuo and M. Yanagisawa, Chem. Lett., 2000, 1072–1073 CrossRef CAS; (b) T. Mukaiyama, J. Matsuo and H. Kitagawa, Chem. Lett., 2000, 1250–1251 CAS.
- For reviews, see: (a) T. Mukaiyama, Angew. Chem., Int. Ed., 2004, 43, 5590–5614 CrossRef CAS; (b) J. Matsuo, J. Synth. Org. Chem., Jpn., 2004, 62, 574–583 CAS.
- J. Matsuo and Y. Aizawa, Tetrahedron Lett., 2005, 46, 407–410 CrossRef CAS.
- TCI Organic Chemicals, TCI Europe, B-2070 Zwijndrecht, Belgium.
- J. Matsuo and Y. Aizawa, Chem. Commun., 2005, 2399–2401 RSC.
- (a) J. A. Kozlowski, Comprehensive Organic Synthesis, ed. B. M. Trost and I. Fleming, Permagon, Oxford, 1991, vol. 4, pp. 169–198 Search PubMed; (b) Organocopper Reagents, ed. R. J. K. Taylor, Oxford University Press, Oxford, 1994 Search PubMed; (c) B. H. Lipshutz, R. S. Wilhelm and J. A. Kozlowski, Tetrahedron, 1984, 40, 5005–5038 CrossRef CAS.
- H. Gilman, R. G. Jones and L. A. Woods, J. Org. Chem., 1952, 17, 1630–1634 CrossRef CAS.
- (a) G. Bartoli, E. Marcantoni, M. Petrini and L. Sambri, Chem. Eur. J., 1996, 2, 913–918 CrossRef CAS; (b) R. K. Dieter, L. A. Silks, J. R. Fishpaugh and M. E. Kastner, J. Am. Chem. Soc., 1985, 107, 4679–4692 CrossRef CAS; (c) L. M. Jackman and S. Sternhell, Application of Nuclear Magnetic Resonance Spectroscopy in Organic Chemistry, 2nd edn, Pergamon, London, 1969, pp. 221–224 Search PubMed.
- For a review, see: E. Endrik, Tetrahedron, 1984, 40, 641–657 Search PubMed.
- S. H. Bertz and G. Dabbagh, Tetrahedron, 1989, 45, 425–434 CrossRef CAS.
- (a) B. H. Lipshutz, J. A. Kozlowski, D. A. Parker, S. L. Nguyen and K. E. McCarthy, J. Organomet. Chem., 1985, 285, 437–447 CrossRef CAS; (b) B. H. Lipshutz, D. A. Parker, J. A. Kozlowski and S. L. Nguyen, Tetrahedron Lett., 1984, 25, 5959–5962 CrossRef CAS; (c) B. H. Lipshutz, M. Koerner and D. A. Parker, Tetrahedron Lett., 1987, 28, 945–948 CrossRef CAS.
- N. L. Hungerford and W. Kitching, J. Chem. Soc., Perkin Trans. 1, 1998, 1839–1858 RSC.
Footnote |
† Representative experimental procedure: to a slurry of CuI (229 mg, 1.2 mmol) in Et2O (10 ml) cooled to −78 °C, was added n-BuLi (2.5 M, 0.96 ml, 2.4 mmol) dropwise. After stirring for 1 h, cyclohexanone (96.1 mg, 1 mmol) in Et2O (2 ml) was carefully added. After stirring for a further 1 h, N-tert-butylphenylsulfinimidoyl chloride (1) (1077 mg, 5 mmol) was added rapidly as a solution in THF (3 ml). The reaction was then allowed to slowly warm to room temperature and stirring was continued for 16 h. The reaction was then quenched with 2 M HCl (30 ml) and extracted with EtOAc (3 × 20 ml). The combined organics were then washed with brine (30 ml) and dried over MgSO4. The crude residue was then carefully purified by silica chromatography eluting with ether–petroleum ether mixtures. The product, 3-n-butylcyclohex-2-en-1-one was obtained as a pale yellow oil (150 mg, 98%), with analysis identical to that described in the literature;17 IR (liq. film) 1670 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 1624 (CC) cm−1; δH (400 MHz, CDCl3): 5.87 (s, 1H), 2.34–2.37 (m, 2H), 2.27–2.30 (m, 2H), 2.17–2.23 (m, 2H), 1.95–2.02 (m, 2H), 1.19–1.51 (m, 4H), 0.92 (t, J = 7.2 Hz, 3H). All other compounds exhibited satisfactory spectral and analytical data. O), 1624 (CC) cm−1; δH (400 MHz, CDCl3): 5.87 (s, 1H), 2.34–2.37 (m, 2H), 2.27–2.30 (m, 2H), 2.17–2.23 (m, 2H), 1.95–2.02 (m, 2H), 1.19–1.51 (m, 4H), 0.92 (t, J = 7.2 Hz, 3H). All other compounds exhibited satisfactory spectral and analytical data. |
| This journal is © The Royal Society of Chemistry 2006 |