Organocatalytic asymmetric allylic carbon–carbon bond formation†
Thomas B.
Poulsen
,
Mark
Bell
and
Karl Anker
Jørgensen
*
Danish National Research Foundation: Center for Catalysis, Department of Chemistry, Aarhus University, DK-8000, Aarhus C, Denmark. E-mail: kaj@chem.au.dk; Fax: +45 8619 6199; Tel: +45 8942 3910
First published on 23rd November 2005
Abstract
Organocatalytic allylic C–C bond-forming addition of activated alkylidenes to alkyl and aryl nitroalkenes has been achieved with high diastereo- and enantioselectivity. Chiral tertiary amine catalysts are used to give allyl intermediates which exhibit γ-selectivity in the C–C bond forming step. The reactions proceed with up to >99 : 1 syn : anti ratio for both the alkyl- and aryl nitroalkenes with up 96% and 98% ee, respectively. The products of this conjugate addition are transformed into a range of intermediates, such as optically active conjugated dienes and 1-substituted tetralones, which are difficult to access via alternative methods.
Introduction
One of the fundamental reactions in organic chemistry is that between active methylene compounds and carbonyl compounds to give unsaturated (Knoevenagel) condensation products.1 Since the first reports by Knoevenagel2 and the classic papers on the efficient synthesis and reactivity of such systems3 (e.g. alkylidene malonates, cyanoacetates, and malononitriles), attention from the synthetic community has been continuous.4 Activated alkylidene structures are common intermediates in complex molecule synthesis, where their electrophilic character has been especially exploited.5As part of our recent research on organocatalyzed asymmetric reactions,6 we have introduced a new concept exploiting the latent nucleophilic reactivity of activated alkylidenes for performing allylic aminations with excellent regio- and stereocontrol.7 This concept relies on the deprotonation of an allylic hydrogen by a chiral base.
Scheme 1 shows the activated alkylidene systems (1), which undergo a stereoselective electrophilic addition leading to a substitution of the allylic C–H bond with the electrophile (E) via a chiral ion-pair intermediate.
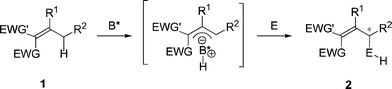 | ||
| Scheme 1 Organocatalytic asymmetric electrophilic addition to allylic C–H bonds. | ||
Asymmetric functionalization of the allylic position plays an important role in organic chemistry due to the great utility of the optically active allylic compounds formed e.g. as intermediates in total synthesis.8 Catalysis using chiral metal complexes, to give π-allyl-metal intermediates, which undergo nucleophilic addition, is a well developed method for asymmetric allylic functionalization.9
The organocatalyzed asymmetric C–C bond formation by addition of carbon-electrophiles to the allylic position of activated alkylidenes has not yet been investigated in great detail. A recent letter10 presenting the asymmetric direct vinylogues Michael reaction of activated alkenes with aromatic nitroalkenes prompted us to present a full account of our contribution towards organocatalyzed asymmetric C–C bond formation by addition of carbon-electrophiles to the allylic position of activated alkylidenes.
Results and discussion
Our studies commenced by examining the feasibility of the reaction between the alkylidene malononitrile 3a derived from 1-tetralone and Michael-acceptor trans-β-nitrostyrene 4a in the presence of different cinchona alkaloid derivatives under various reaction conditions [eqn (1), Table 1].11 The reaction was found to take place smoothly, with near quantitative conversion to the γ−substituted product as well as traces of side products. To our surprise, the 1H NMR spectrum of the reaction mixture showed only one diastereoisomer. This turned out to be the case for all screening experiments.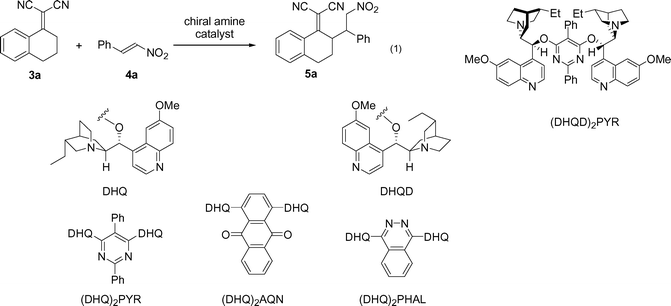
| Entry | Cat./mol% | Temp./°C | Solvent (0.25 M) | Time/h | Conv. (%)b | dr (syn/anti)b | ee (%)c |
|---|---|---|---|---|---|---|---|
| a Reaction performed at a 0.15 mmol scale (3a) with 1.5 eq. of 4a. b Determined by 1H NMR analysis of the crude mixture. c Of the syn-isomer, determined by CSP-HPLC analysis (see Experimental section for details). d Reaction mixture is heterogenous. TBME = tert-butyl methyl ether. 1,2-DCE = 1,2-dichloroethane. | |||||||
| 1 | (DHQ)2AQN (10) | −15 | CH2Cl2 | 16 | >95 | >99:1 | +50 |
| 2 | (DHQ)2PHAL (10) | −15 | CH2Cl2 | 24 | 95 | >99:1 | +61 |
| 3 | (DHQ)2PYR (10) | −15 | CH2Cl2 | 16 | >95 | >99:1 | +76 |
| 4 | (DHQD)2PYR (10) | −15 | CH2Cl2 | 14 | >95 | >99:1 | −86 |
| 5 | (DHQD)2PYR (10) | −15 | Toluene | 14 | 95 | >99:1 | −88 |
| 6 | (DHQD)2PYR (10) | −15 | 1,2-DCE | 20 | >95 | >99:1 | −88 |
| 7 | (DHQD)2PYR (10) | −40 | CH2Cl2 | 40 | >95 | >99:1 | −92 |
| 8 | (DHQD)2PYR (10) | −40 | THF | 20 | 95 | >99:1 | −86 |
| 9 | (DHQD)2PYR (10) | −40 | THF/Tol 1:4 | 20 | 68 | >99:1 | −90 |
| 10 | (DHQD)2PYR (10) | −40 | TBME | 20 | 11d | >99:1 | −90 |
| 11 | (DHQD)2PYR (10) | −40 | EtOAc | 20 | 70 | >99:1 | −95 |
| 12 | (DHQD)2PYR (10) | −40 | acetone | 25 | >95 | >99:1 | −95 |
A screening of reaction conditions in CH2Cl2 showed that (DHQD)2PYR was the most effective catalyst at −15 °C giving 95% conversion with 86% ee (entry 4). The quasienantiomer (DHQ)2PYR gave the opposite configuration as expected with 76% ee (compare entries 3 and 4). Lower temperatures slightly improved the enantiocontrol (86% ee at −15 °C and 92% ee at −40 °C, entries 4 and 7). Poor conversion in the case of TBME as the solvent is attributed to low solubility of the starting materials. Surprisingly, acetone was identified as the best solvent for this reaction (95% conversion, >99:1 dr and 95% ee, entry 12), which is unusual for chiral ion-pair based reactions, and improves the enantioselectivity of the reaction compared to CH2Cl2. It should be noted that the reaction also proceeds in EtOAc with excellent enantioselectivity (entry 11). The use of 1.5 equiv. of the nitroalkene was found to be beneficial to the reaction rate.
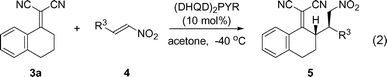
The scope of the asymmetric allylic C–C bond-forming reaction was probed using a range of nitroalkenes 4 reacting with alkylidene malononitriles 3a [eqn (2), Table 2].
| Entry | R3 | Yieldb(%) | drc (syn/anti) | eed(%) |
|---|---|---|---|---|
| a Reaction carried out with 0.25 mmol of 3a and 1.5 eq. of 4, values in parenthesis obtained with the quasienantiomer (DHQ)2PYR in acetone/THF (3:1). b Isolated yield. c Determined by 1H NMR. d Of the syn-isomer determined by CSP-HPLC (see Experimental section). e Reaction carried out in acetone/EtOAc (1:1, 0.25 M). | ||||
| 1 | C6H5 | 5a-98(95) | >99:1( >99:1) | 95(89) |
| 2e | 2-naphthyl | 5b-98 | >99:1 | 92 |
| 3 | 2-thienyl | 5c-93 | >99:1 | 97 |
| 4 | 2-furyl | 5d-99 | 13:1 | 94 |
| 5 | 4-MeO–C6H4 | 5e-97 | >99:1 | 98 |
| 6 | 4-NO2–C6H4 | 5f-99(92) | >99:1( >99:1) | 95(90) |
| 7 | 4-Br–C6H4 | 5g-96 | >99:1 | 96 |
| 8 | 2-Cl–C6H4 | 5h-97 | >99:1 | 53 |
| 9 | 3-NO2–C6H4 | 5i-92 | >99:1 | 91 |
| 10 | c-hexyl | 5j-82 | >99:1 | 96 |
| 11 | n-pentyl | 5k-99 | 19:1 | 94 |
Aryl and heteroaryl nitroalkenes reacted cleanly to give the desired products in 92–99% yield with 92–98% ee (Table 2, entries 1–7, 9) with the exception of 2-Cl-trans-β-nitrostyrene (97% yield and 53% ee, entry 8). This lower selectivity is possibly due to the closeness of the ortho-substituent to the new C–C bond. Aromatic substrates with a para- or meta-substituent reacted in a comparable way to the benchmark reaction regardless of the electron density of the aryl ring. The alkyl substituted nitroalkenes also reacted smoothly to give high yields and enantioselectivities (82 and 99% yield, 96 and 94% ee, entries 10 and 11). Only in two cases did we observe the formation of the anti-diastereomer, although in very small amounts (entry 4, 13:1 and entry 11, 19:1). Our experiments so far indicate, that the excellent diastereomeric ratios observed are due to kinetic control and, therefore, not due to base-mediated equilibration of the γ-stereocenter.
The generality of the allylic functionalization as demonstrated in Table 2 prompted us to investigate the reaction for a series of alkylidene malononitriles 3b–i with trans-β-nitrostyrene 4a. The results are presented in Table 3.
| Entry | Substrate | Product | Yield (%)b | dr (syn/anti)c | ee (%)d | |
|---|---|---|---|---|---|---|
| a Reaction carried out with 0.25 mmol of 3 and 1.5 eq. of 4a at −40 °C in acetone (0.25 M) in the presence of 10 mol% (DHQD)2PYR, value in parenthesis obtained with the quasienantiomer (DHQ)2PYR in acetone/THF (3:1). b Isolated yield. c Determined by 1H NMR. d Of the syn-isomer determined by CSP-HPLC (see Experimental section). e Reaction carried out in EtOAc (0.25 M). f No conversion observed, even at higher temperatures. | ||||||
| 1e |
|
3b |
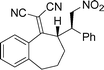
|
5l-96 | 15:1 | 92 |
| 2 |
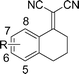
|
7-MeO-3c |
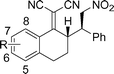
|
5m-96(94) | >99:1( >99:1) | 96(86) |
| 3 | 6-MeO-3d | 5n-99 | >99:1 | 94 | ||
| 4 | 5-MeO-3e | 5o-90 | >99:1 | 93 | ||
| 5 | 6,7-MeO-3f | 5p-99 | >99:1 | 80 | ||
| 6 | 5,7-Me-3g | 5q-95 | >99:1 | 91 | ||
| 7 | 7-NO2-3h | —f | — | — | ||
| 8 |
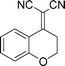
|
3i |
|
5r-95 | >99:1 | 74 |
Increasing the ring-size to seven carbon atoms (3b) afforded similar results to the six-membered system with the enantioselectivity remaining high at 92% ee and a small decrease in diastereoselectivity (15:1, Table 3, entry 1). Many important bioactive compounds contain a methoxy-substituted aromatic ring fused with a cyclohexane ring. We were therefore pleased that placing a methoxy group in the 5, 6, or 7 position gave high enantioselectivity (93, 96 and 94% ee), yield (90, 96 and 99%) and diastereocontrol (>99:1 in all cases) (Table 3, entries 2–4). An alkylidene possessing the ubiquitous 6,7-dimethoxy motif (3f) reacted cleanly to give good, if slightly lower enantiocontrol (99% yield and 80% ee, entry 5). The 5,7-dimethyl substituted compound (3g) reacted with high yield and stereocontrol (95%, 91% ee, entry 6). Surprisingly the introduction of a nitro-group in the 7-position of the alkylidene (3h) completely retarded the reaction even at higher temperatures. Finally the 4-chromanone derived alkylidene (3i) gave 95% yield and 75% ee (entry 8).
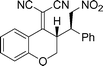
The absolute configuration of the products was established to be (2S,3R) by examination of an X-ray crystal structure obtained from the 4-Br substituted nitrostyrene (5g) (Table 4, Fig. 1).12,13
| 5g | 6 | |
|---|---|---|
| Formula, weight | C21H16BrN3O2 | C21H17N3O2 |
| Crystal system | orthorhombic | monoclinic |
| a, Å | 6.7062 (5) | 8.990 (1) |
| b, Å | 12.4969 (10) | 23.048 (2) |
| c, Å | 22.780 (2) | 15.980 (1) |
| α, deg | 90 | 90 |
| β, deg | 90 | 96.130 (3) |
| γ, deg | 90 | 90 |
| Volume, Å3 | 1909.1 (3) | 3292.1 (4) |
| Temperature | 150 (1) | 100 (1) |
| Space group | P2(1)2(1)2(1) | P2(1)/c |
| Z, Z′ | 4, 4 | 8, 2 |
| µ | 2.174 | 0.091 |
| N meas | 38312 | 65363 |
| N unique | 5814 | 4696 |
| R int | 0.043 | 0.081 |
| R(F), all | 0.061 | 0.107 |
| R w(F2), all | 0.064 | 0.148 |
| Flack | −0.01 (1) | N/A |
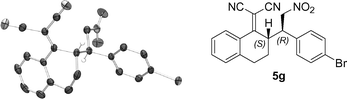 | ||
| Fig. 1 X-ray crystal structure of (2S,3R)-5g (most hydrogen atoms have been omitted for clarity). | ||
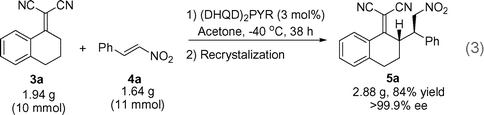
To show the ease with which the present reaction allows access to larger quantities of enantiopure material, we subjected 10 mmol of alkylidene 3a and 1.1 eq. nitroalkene 4a to the standard reaction conditions in the presence of 3 mol% of (DHQD)2PYR [eqn (3)]. After the nucleophile had been consumed as judged by TLC-analysis the product was filtered through a short pad of silica to remove the catalyst. Recrystalization from EtOH afforded the Michael adduct as a single stereoisomer in 84% yield.
During the course of our initial studies we sometimes noted the formation of intensely coloured orange/red reaction mixtures, especially when the catalytic reactions were carried out at higher temperatures. The origin of this phenomenon was identified to be the formation of an interesting, highly conjugated three ring system. Optically active diene 6 (structure confirmed by X-ray analysis,12Scheme 2) arises from an intramolecular cyclisation between the nitroalkane and one of the cyano groups followed by tautomerization. Previous reports on similar ring-closures performed under harsher conditions14 yielded the fully aromatized compounds. Different bases were evaluated for this transformation (Scheme 2) and quinidine was found to give the highest isolated yield.15 A chiral base is obviously not necessary to effect this transformation, however, the efficiency of quinidine compared to achiral bases tested combined with its low cost renders this the best alternative. In fact, the high loadings necessary of e.g. Et3N to afford even low isolated yields of diene 6 leads to an erosion of the optical purity (>99.9% ee to 93% ee). This observation might be ascribed to racemization through a retro-Michael mechanism, being increasingly favored under high base concentrations.
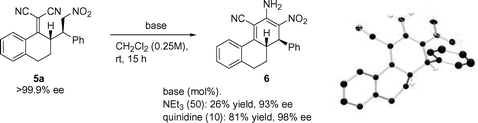 | ||
| Scheme 2 Ring-closing reaction of 5a to afford optically active diene 6 under different conditions. To the right, X-ray crystal structure of 6 (most hydrogen atoms have been omitted for clarity). | ||
As mentioned in the introduction, activated alkylidene systems are related to their parent carbonyl compounds through Knoevenagel condensation with malononitrile. Obviously, the reverse transformation—i.e. the formation of the carbonyl compound from the Knoevenagel condensation product—does not have much synthetic relevance, unless the formation of the activated alkylidene system allows for chemical transformations not possible with the parent carbonyl compound. The similarity between the CO and the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C(CN)2 functionality has been noted by several groups.16 The similarity, however, is of course only qualitative, and in fact very fundamental differences in reactivity do exist. As we have demonstrated here and previously,7 the formation of an activated alkylidene system permits deprotonation in the α-position of the group (the γ-position of the alkylidene system), thereby enabling chiral base-catalyzed asymmetric reactions to be performed. To the best of our knowledge, similar transformations cannot be carried out on the parent carbonyl compound. Thus, the C(CN)2 group can be regarded as a transient activating group in a synthetic sequence towards the catalytic asymmetric functionalization of the carbonyl compound. In fact, the justification of this indirect α-functionalization of carbonyl compounds is increased, when efficient direct approaches to the same transformation are not available—as is indeed currently the case for substituted 1-tetralones (Scheme 3).17
C(CN)2 functionality has been noted by several groups.16 The similarity, however, is of course only qualitative, and in fact very fundamental differences in reactivity do exist. As we have demonstrated here and previously,7 the formation of an activated alkylidene system permits deprotonation in the α-position of the group (the γ-position of the alkylidene system), thereby enabling chiral base-catalyzed asymmetric reactions to be performed. To the best of our knowledge, similar transformations cannot be carried out on the parent carbonyl compound. Thus, the C(CN)2 group can be regarded as a transient activating group in a synthetic sequence towards the catalytic asymmetric functionalization of the carbonyl compound. In fact, the justification of this indirect α-functionalization of carbonyl compounds is increased, when efficient direct approaches to the same transformation are not available—as is indeed currently the case for substituted 1-tetralones (Scheme 3).17
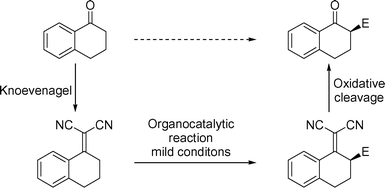 | ||
| Scheme 3 Indirect catalytic asymmetric functionalization of 1-tetralone, via carbonyl activation, chiral base chemistry, and oxidative cleavage. | ||
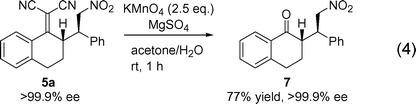
The desired oxidative cleavage could be carried out using potassium permanganate in a mixture of acetone and water at ambient temperature [eqn (4)]. After column chromatography, optically active substituted 1-tetralone 7 was obtained as a single stereoisomer in 77% yield.
Conclusions
In conclusion, we have presented a direct organocatalytic enantioselective C–C bond forming reaction in the allylic position of activated alkylidene systems. The reaction is broad in scope and takes place with excellent regio- and diastereoselectivity, and with good to excellent enantioselectivity for both aryl and alkyl nitroalkenes. Furthermore, we have demonstrated the synthetic utility of the optically active products by performing reactions exploiting the electron-deficient alkylidene double bond. Thus, oxidative cleavage of the alkylidene moiety allows for the preparation of optically active substituted 1-tetralones.Experimental
General Methods
NMR spectra were acquired on a Varian AS 400 spectrometer, running at 400 and 100 MHz for 1H and 13C, respectively. Chemical shifts (δ) are reported in ppm relative to residual solvent signals. 13C NMR spectra were acquired on a broad band decoupled mode. Mass spectra were recorded on a micromass LCT spectrometer using electrospray (ES+) ionization techniques. Flash column chromatography (FC) was carried out using the FlashMaster II from Jones Chromatography with columns containing silica gel. Analytical thin layer chromatography (TLC) was performed using pre-coated aluminium-backed plates (Merck Kieselgel 60 F254) and visualized by ultraviolet irradiation or KMnO4 dip. Optical rotations were measured on a Perkin-Elmer 241 polarimeter. The enantiomeric excess (ee) of the products was determined by chiral stationary phase HPLC (Daicel Chiralpak AS/AD or Daicel Chiralcel OD columns).Materials
Analytical grade solvents were used as received. For flash chromatography (FC) silica gel was purchased from Iatron Laboratories Inc. (Iatrobeads 6RS-8060) or from Fluka (Silica gel 60, 230–400 mesh). All catalysts are commercially available and were used as received. Alkylidene malononitriles 3a and 3c-j were prepared from the corresponding commercially available ketone and malononitrile by Al2O3-mediated Knoevenagel condensation.183b was prepared by condensing commercially available 1-benzosuberone with malononitrile in the presence of catalytic amounts of triethylamine.19 Nitroalkenes 4a,c-i were obtained from commercial suppliers and used as received. Naphthyl-substituted nitroalkene 4b was prepared by Henry-reaction,20 followed by elimination.21 Alkyl-substituted nitroalkenes 4j-k were prepared according to a literature procedure.21syn-diastereomer. 1H NMR (CDCl3) δ 7.49 (t, J 6.5 Hz, 1H), 7.40 (t, J 7.6 Hz, 1H), 7.23–7.36 (m, 5H), 7.14 (d, J 6.3 Hz, 2H), 4.56 (dd, J 12.4, 10.8 Hz, 1H), 4.39 (dd, J 12.4, 8.6 Hz, 1H), 3.60 (dt, J 11.6, 4.1 Hz, 1H), 3.30 (td, J 10.8, 3.7 Hz, 1H), 2.71–2.87 (m, 2H), 1.61–1.83 (m, 4H). 13C NMR (CDCl3) δ 183.6, 138.8, 135.5, 133.2, 132.0, 130.8, 129.4 (2C), 128.6, 128.4, 128.0 (2C), 127.4, 111.5 (2C), 88.2, 78.7, 46.5, 44.8, 35.8, 33.2, 21,7. HRMS calc.: C22H19N3NaO2 380.1375; found: 380.1376. [α]D20 − 202 (c = 0.25, CH2Cl2, 92% ee). After separation of the diastereomers by FC (Et2O/hexane 0 : 100 to 50 : 50), the ee of the syn-diastereomer was determined by HPLC using a Chiralpak AS column [hexane/i-PrOH (85 : 15)]; flow rate 1.0 mL min−1; τmajor = 14.9 min, τminor = 13.8 min (92% ee).
Acknowledgements
This work was made possible by a grant from The Danish National Research Foundation and EU-HMPT-CT-2001-00317. We are grateful to Dr. Jacob Overgaard for X-ray crystallographic analysis and to Karen Skov for performing HRMS-analysis.References
- For reviews see: (a) L. Tietze, in Comprehensive Organic Synthesis, Vol. 2, ed. B. M. Trost and I. Fleming, Pergamon Press, New York, 1991, 341 Search PubMed; (b) R. L. Reeves, in The Chemistry of the Carbonyl Group, Vol. 1, ed. S. Patai, Wiley-Interscience, New York, 1966, 567 Search PubMed.
- (a) E. Knoevenagel, Chem. Ber., 1894, 27, 2345; (b) E. Knoevenagel, Chem. Ber., 1896, 29, 172.
- See e.g.: (a) A. C. Cope, J. Am. Chem. Soc., 1937, 59, 2327–2330 CrossRef CAS; (b) A. C. Cope and K. E. Hoyle, J. Am. Chem. Soc., 1941, 63, 733–736 CrossRef CAS; (c) F. S. Prout, J. Org. Chem., 1953, 18, 928–933 CrossRef CAS.
- For investigations on the regioselectivity in alkylations of alkylidene malononitriles, see: (a) H. Karlsen, P. H. Sånge, L. K. Sunsby, L. C. Hagen, P. Kolsaker and C. Rømming, J. Chem. Soc., Perkin Trans. 1, 2001, 497–507 RSC; For investigations on the structure of the isopropylidenemalononitrile dimer see: (b) J. K. Williams, J. Org. Chem., 1964, 28, 1054–1059.
- For examples see e.g.: (a) E. Fillion and D. Fishlock, J. Am. Chem. Soc., 2005, 127, 13144–13145 CrossRef CAS; (b) L. F. Tietze and Y. Zhou, Angew. Chem., Int. Ed., 1999, 38, 2045–2047 CrossRef CAS.
- General reviews on organocatalysis: (a) P. I. Dalko and L. Moisan, Angew. Chem., Int. Ed., 2004, 43, 5138–5175 CrossRef CAS; (b) ed. K. N. Houk and B. List, Acc. Chem. Res., 2004, 37(8) special issue Search PubMed; (c) E. R. Jarvo and S. J. Miller, Tetrahedron, 2002, 58, 2481–2495 CrossRef; (d) P. I. Dalko and L. Moisan, Angew. Chem., Int. Ed., 2001, 40, 3726–3748 CrossRef CAS; For recent examples of cinchona alkaloid catalyzed asymmetric reactions see e.g.: (e) M. A. Calter, R. M. Phillips and C. Flaschenriem, J. Am. Chem. Soc., 2005, 127, 14566–14567 CrossRef CAS; (f) G. Bartoli, M. Bosco, A. Carlone, M. Locatelli, P. Melchiorre and L. Sambri, Angew. Chem., Int. Ed., 2005, 44, 6219–6222 CrossRef CAS; (g) T. B. Poulsen, C. Alemparte, S. Saaby, M. Bella and K. A. Jørgensen, Angew. Chem., Int. Ed., 2005, 44, 2896–2899 CrossRef CAS; (h) S. Lou, B. M. Taoka, A. Ting and S. E. Schaus, J. Am. Chem. Soc., 2005, 127, 11256 CrossRef CAS; (i) S. Saaby, M. Bella and K. A. Jørgensen, J. Am. Chem. Soc., 2004, 126, 8120–8121 CrossRef CAS; (j) M. Bella and K. A. Jørgensen, J. Am. Chem. Soc., 2004, 126, 5672–5673 CrossRef CAS ; see also refs 11d–e,j–k.
- T. B. Poulsen, C. Alemparte and K. A. Jørgensen, J. Am. Chem. Soc., 2005, 127, 6964–6965 CrossRef CAS.
- (a) B. M. Trost, J. Org. Chem., 2004, 69, 5813–5837 CrossRef CAS; (b) B. M. Trost and M. L. Crawley, Chem. Rev., 2003, 103, 2921–2944 CrossRef CAS.
- B. M. Trost and D. L. Van Vranken, Chem. Rev., 1996, 96, 395–422 CrossRef CAS.
- D. Xue, Y.-C. Chen, Q.-W. Wang, L.-F. Cun, J. Zhu and J.-G. Deng, Org. Lett., 2005, 7, 5293–5296 CrossRef CAS.
- For recent examples of catalytic asymmetric additions to conjugated nitroalkenes, see: (a) R. P. Herrera, V. Sgarzani, L. Bernadi and A. Ricci, Angew. Chem., Int. Ed., 2005, 44, 6576–6579 CrossRef CAS; (b) D. A. Evans and D. Seidel, J. Am. Chem. Soc., 2005, 127, 9958–9959 CrossRef CAS; (c) Y. Hayashi, H. Gotoh, T. Hayashi and M. Shoji, Angew. Chem., Int. Ed., 2005, 44, 4212–4215 CrossRef CAS; (d) H. Li, Y. Wang, L. Tang, F. Wu, X. Liu, C. Guo, B. M. Foxman and L. Deng, Angew. Chem., Int. Ed., 2005, 44, 105–108 CrossRef CAS; (e) H. Li, Y. Wang, L. Tang and L. Deng, J. Am. Chem. Soc., 2004, 126, 9906–9907 CrossRef CAS; (f) T. Ishii, S. Fujioka, Y. Sekiguchi and H. Kotsuki, J. Am. Chem. Soc., 2004, 126, 9558–9559 CrossRef CAS; (g) T. Okino, Y. Hoashi, T. Furukawa, X. Xu and Y. Takemoto, J. Am. Chem. Soc., 2005, 127, 119–125 CrossRef CAS; (h) T. Okino, Y. Hoashi and Y. Takemoto, J. Am. Chem. Soc., 2003, 125, 12672–12673 CrossRef CAS; (i) A. J. A. Cobb, D. A. D. Lonbottom, M. Shaw and S. Ley, Chem. Commun., 2004, 1808 RSC; (j) J. Wang, H. Li, W. Duan, L. Zu and W. Wang, Org. Lett., 2005, 7, 4713–4716 CrossRef CAS; (k) S. H. McCooey and S. J. Connon, Angew. Chem., Int. Ed., 2005, 44, 6367–6370 CrossRef CAS ; for reviews see:; (l) O. M. Berner, L. Tedeschi and D. Enders, Eur. J. Org. Chem., 2002, 1877–1894 CrossRef CAS; (m) N. Kranse and A. Hoffmann-Röder, Synthesis, 2001, 171 CrossRef CAS.
- CCDC 285437 and 285436 contains the supplementary crystallographic data for this paper.† These data can be obtained free of charge viahttp://www.ccdc.cam.ac.uk/conts/retrieving.html (or from the Cambridge Crystallographic Data Centre, 12, Union Road, Cambridge CB2 1EZ, UK; fax: (+44) 1223-336-033; or E-mail: deposit@ccdc.cam.ac.uk).
- By analogy with HPLC traces, NMR spectra, and optical rotations the same configuration for the Michael adducts 5a–f,h–s could be inferred.
- F. M. A. El-Taweel, M. A. Sofan, T. M. Abu El-Maati and A. A. Elagamey, Boll. Chim. Farm., 2001, 140, 306–310 Search PubMed.
- It should be noted that the (DHQD)2PYR cannot be used in a domino-type reaction (initial addition to the nitroalkene followed by ring-closure) due to low yields in the cyclization step.
- See e.g.: (a) K. Wallenfels, K. Friedrich, J. Rieser, W. Ertel and H. K. Thieme, Angew. Chem., Int. Ed., 1976, 15, 261–270 CrossRef; (b) H. Karlsen, P. Kolsaker, C. Rømming and E. Uggerud, J. Chem. Soc., Perkin Trans. 2, 2002, 404–409 RSC.
- Although highly successful direct asymmetric functionalizations of ketones have been carried using enamine-catalysis (see e.g.refs 11i,f), we are currently unaware of reports describing such reactions for substituted tetralones and similar compounds.
- R. E. Sammelson and M. J. Allen, Synthesis, 2005, 543–546 CrossRef CAS.
- A. De, S. Bhattacharya, S. S. Jash, S. Mukherjee, U. Saha and P. K. Sen, J. Heterocycl. Chem., 1992, 29, 1213–1217 Search PubMed.
- D. E. Worrall and A. Tatilbaum, J. Am. Chem. Soc., 1942, 64, 1739–1740 CrossRef CAS.
- S. E. Denmark and L. R. Marcin, J. Org. Chem., 1992, 58, 3850–3856.
- At this temperature the compound had changed colour from pale yellow to red/brown which might indicate some decomposition.
Footnote |
| † CCDC reference numbers [285436 and 285437]. For crystallographic data in CIF or other electronic format see DOI: 10.1039/b514564c |
| This journal is © The Royal Society of Chemistry 2006 |