Mechanism of 1,4,5,8-naphthalene tetracarboxylic acid dianhydride hydrolysis and formation in aqueous solution
T. C.
Barros
a,
I. M.
Cuccovia
*c,
J. P. S.
Farah
b,
J. C.
Masini
b,
H.
Chaimovich
c and
M. J.
Politi
c
aUniversidade Paulista, Instituto de Ciências da Saúde, Bauru, SP, Brazil
bDepartamento de Química Fundamental and Instituto de Química da, Universidade de São Paulo, SP, Brazil
cDepartamento de Bioquímica do, Instituto de Química da, Universidade de São Paulo, SP, Brazil. E-mail: imcuccov@quim.iq.usp.br
First published on 18th November 2005
Abstract
The study of highly conjugated, carbonyl-containing molecules such as 1,4,5,8-naphthalene tetracarboxylic dianhydride, III, is of interest since reactivity differences and transmission of electronic effects through the conjugated framework can be evidenced. The kinetics of hydrolysis of III in aqueous solution were determined from 5 M acid to pH 10. In basic solution hydrolysis of III yields, sequentially, 1,4,5,8-naphthalene diacid monoanhydride, II, and 1,4,5,8-naphthalene tetracarboxylic acid, I. The second order rate constant for alkaline hydrolysis is 200 fold higher for the first ring opening. The water-catalyzed hydrolysis of III yields a pH-dependent mixture of ionic forms of I and II. The rate constant for water-catalyzed hydrolysis of III is 25 fold higher than that for II. In concentrated acid the rates for reaching equilibrium (I, II and III) increase and III is the major product. The pKas of I (3.24, 5.13 and 6.25) and II (3.05, 5.90) were determined by potentiometric, fluorescence and UV spectroscopy titrations and by quantitative fit of the kinetic and equilibrium data. The apparent, pH-dependent, equilibrium constants, KEqII, for anhydride formation between I and II were obtained from the UV spectra. The quantitative fit of kinetic and equilibrium data are consistent with the assumption that anhydride formation only proceeds with the fully protonated species for both I and II and permitted the estimation of the equilibrium constants for anhydride formation, KEqII. The value of KEqII (I ⇌ II) between pH 1 and 6 was ca. 5. Geometry optimization calculations in the gas phase of the reactions of III in alkaline, neutral and acid conditions, at the DFT level of theory, gave electronic distributions that were qualitatively consistent with the experimental results.
1 Introduction
The experimental and theoretical analysis of anhydride formation in intramolecular reactions is a paradigmatic model chemical system, extensively used to analyze ground and transition state contributions to the rates of intramolecular and enzymatic reactions.1Hydrolysis of carboxylic anhydrides in aqueous solution can be spontaneous, acid or base-catalyzed.1–6 Dicarboxylic acids can be in equilibrium with the correspondent anhydrides in aqueous solutions and, in general, the equilibrium is displaced towards the diacid.7,8 The equilibrium constant may vary from 10−7 to 25 depending on structure and the unionized diacid is the kinetically significant species for anhydride formation.7–9
The equilibrium between 1,8-naphthalic anhydride, 1,8-An, and its hydrolysis product 1,8-naphthalene dicarboxylic acid, 1,8-Acid, in aqueous solution has been analyzed previously.10 The pH dependence of hydrolysis kinetics of 1,8-An is complex and indicates an equilibrium reaction between 1,8-An and 1,8-Acid at pHs lower than 5.0. Kinetics and equilibrium results were rationalized by assuming that protonated forms of the 1,8-Acid are in equilibrium with 1,8-An. The values of the equilibrium constants for 1,8-An formation range from 4, between pH 0 and 6, to 100 in concentrated acid. Ab initio calculations for the pathway connecting the undissociated 1,8-Acid to the 1,8-An are consistent with a mechanism involving a rate determining intramolecular proton transfer concerted with oxygen alignment towards the carbonyl center forming a transition state with a neutral and planar intermediate with an sp3 carbon. The second step is dehydration, through another transition state, yielding a complex between water and 1,8-An.10
Tetra-substituted naphthalene acid, namely 1,4,5,8-naphthalene tetracarboxylic acid, I, Scheme 1, and its mono II and dianhydrides, III, are intermediates in the synthesis of polymers and dyes.11
 | ||
| Scheme 1 Reactions involved in the hydrolysis of III to II and I. | ||
Fully protonated I yields 1,4,5,8-naphthalene tetracarboxylic dianhydride, III, upon drying.11 Hydrolysis of III in aqueous base was reported to yield I without the formation of 1,4,5,8-naphthalene diacid monoanhydride, II, as an intermediate, Scheme 1.11 Compound II was obtained by acidification of I with mineral acid at temperatures above 60 °C. Kofman and coworkers prepared I, and its derivatives, II and III, in acid at 90 °C and 140–150 °C, respectively.11 Acid dissociation constants and alkaline hydrolysis in aqueous DMSO were also analyzed.11 Compounds I, II and III showed 4, 3 and 2 dissociable groups, respectively, and the authors rationalized the data by assuming an intermediate, formed after OH− attack at carbonyl groups of III, leads to the appearance of a dissociable group and the opening of the two anhydrides to yield I. The formation of II in aqueous solution upon heating was suggested from NMR spectra and it was proposed that, in moist DMSO, an equilibrium between I and III is possible.12 Crystal structures of compounds I, II and III are known.12–16
1,4,5,8-Naphthalene tetracarboxylic acid, I, Scheme 1, is an interesting model where short-range electronic and steric interactions, as well as protonation states, can modulate chemical reactivity. Intramolecular reactions of I exhibit short range interactions that are typical of those encountered in an enzyme active site.
The detailed pathways of anhydride formation and hydrolysis in the 1,4,5,8-naphthalene tetracarboxylic system have not been analyzed. It seemed unlikely that the hydrolysis of III yielded I without the intervening formation of II, as described previously.11 Here we present kinetic and equilibrium data that contribute to the understanding of the mechanism of formation and hydrolysis of III in aqueous solutions. Cyclization of I to II and further cyclization to III, in acid, were also studied. The pKs of I and II were determined using UV-Vis and fluorescence spectroscopy, potentiometric titration and kinetic data. Cyclization of both I and II depends on the protonation of the carboxylic groups. Geometry optimization of III, selected intermediates and monoanhydrides was performed to define differences in reactivity between the mono and dianhydrides in the acid, water and base-catalyzed reactions.
2 Experimental
2.1 Materials
1,4,5,8-Naphthalene tetracarboxylic dianhydride, III, (Aldrich) was used without further purification. The tetra sodium salt of 1,4,5,8-naphthalene tetracarboxylic acid,17I-Base, (Scheme 1) was prepared by alkaline hydrolysis of III, adding III to yield a final concentration of 0.001 M in aqueous solutions of NaOH and maintaining the sample at 30 °C for 30 min (see results). At the end of the reaction, absorbance measurement at 368 nm guaranteed complete hydrolysis of III to I-Base (see results).1,4,5,8-Naphthalene diacid monoanhydride, II: 12.5 mL of an aqueous solution of I-Base (1 × 10−3 M) in NaOH (0.005 M) was added to a 25 mL volumetric flask containing water and HCl sufficient to give pH 1.0 and 5 × 10−4 M of protonated I-Base, I-H4. The solution was maintained at 30 °C until I-H4 was almost completely cyclized (ca. 80%, see results) to II-H2.
Other reagents (Merck, Aldrich or Sigma) were P.A. or spectroscopic grade and used as supplied. All aqueous solutions and buffers were prepared in freshly glass bi-distilled water. The buffers used were the sodium salts of borate (pH 9.0 to 10.0), phosphate (pH 6.4 to 8.0), acetate (pH 3.8 to 5.6), citrate (pH 2.2 to 6.0), 4-morpholineethanesulfonic acid, MES (pH 5.5 to 6.5), and [2-(hydroxymethyl) aminomethane], TRIS (pH 7.5 to 8.5). Buffer concentrations, shown in the figure legends, varied from 0.01 M to 0.05 M. HCl or HClO4 were used for pHs lower than 2.0, and in the H0 region.18 NaClO4 was obtained by neutralization of concentrated HClO4 with sodium hydroxide.
2.2 Methods
UV-Vis absorption spectra and kinetic measurements were recorded using Hitachi U-2000, Cary 3E, Shimadzu UV-2401 PC or Beckman DU-7 spectrophotometers. Kinetic measurements were done at 30 °C ± 0.1. Fast reactions, above pH 7, were recorded in an Applied Photophysics Model SX-18 MV Stopped Flow system. Steady-state fluorescence spectra were recorded either in SPEX-1681 or Hitachi-F-2000 spectrofluorimeters. The excitation and emission slit widths were set to 1 mm, band-pass of 13 nm. Fluorescence emission spectra obtained with the SPEX fluorometer were corrected using the fabricant software. pHs were measured with a Beckman model Φ71 pH meter. I was titrated using a Radiometer PHM 82 Standard pH Meter equipped with a glass electrode calibrated with standard buffers at 30 °C.pKI-H1 and pKI-H2 of I were determined from measurement of the absorbance (at 250 nm), at several pHs, by adding 40 µL of 1 × 10−3 M of I-Base, prepared at pH 11.5, to 2.0 mL of 0.05 M buffer. The absorbances were measured within intervals of 30 s to minimize cyclization.
All kinetic reactions, titrations and equilibrium measurements were performed at 30 °C. I was also titrated by fluorescence (emission 454 nm, excitation 290 nm).
3 Results and discussion
3.1 Spectra
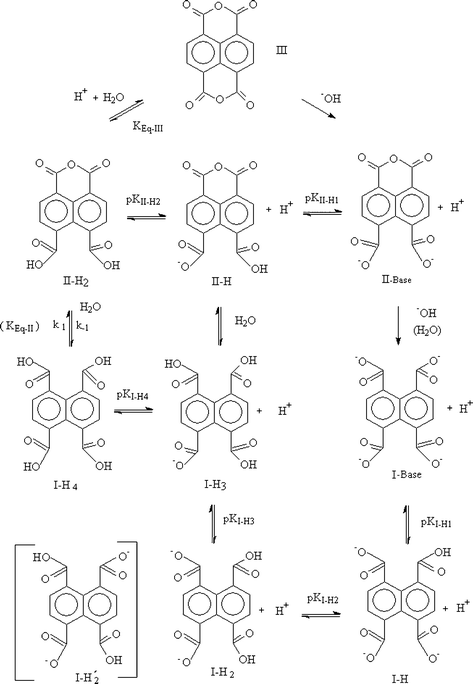
Typical UV-Vis absorption and fluorescence emission spectra of III are presented in Fig. 1. The UV maximum absorbance wavelengths, λmax, and corresponding molar absorptivities, ε, of III in carbon tetrachloride and acetonitrile are collected in Table 1. The value of ε of III in CH3CN (363.5 nm), is 30 618 M−1 cm−1, a typical value of π, π* transitions, indicating a highly conjugated aromatic system. Spectral resolution of III and high ε values were similar to that observed for 1,4,5,8-naphthalene derivatives.21| Solvent | λ 1/nm | ε 1/M−1cm−1 | λ 2/nm | ε 2/M−1 cm−1 | λ 3/nm | ε 3/M−1cm−1 |
|---|---|---|---|---|---|---|
| CH3CN | 329 | 13 214 | 345.5 | 23 410 | 363.5 | 30 618 |
| CCl4 | 329 | 10 189 | 346 | 18 600 | 364 | 25 294 |
| HClO4 0.1 M | 332 | 11 708 | 348 | 19 699 | 368 | 23 737 |
| HClO4 0.1 M + NaClO4 4 M | 333 | 12 215 | 351 | 18 607 | 370 | 17 784 |
| HClO4 12 M | 336 | 11 700 | 354 | 20 751 | 375 | 25 552 |
 | ||
| Fig. 1 (A) Fluorescence excitation (a, b, c) and emission (a′, b′, c′) spectra of III, 1 × 10−5 M, in acetonitrile–water (1:1 v/v) at different times, in minutes: zero (a, a′), 8 (b, b′) and 45 (c, c′). (B) UV-Vis spectra of III, 1 × 10−5 M, in phosphate buffer, 0.02 M, pH 7, as a function of time: (1) t = zero; (2) t = 6 s; (3) t = 12 s; (4) t = 18 s; (5) t = 30 s; (6) t = 54 s; (7) t = 5 min; (8) t = 9 min (9) t = 20 min; (10) t = 40 min; (11) t = 1 h; (12) t = 1.5 h; (13) t = 2 h; (14) t = 3 h; (15) t = 4 h. | ||
Emission and excitation spectra of III in CH3CN showed fine vibrational structure and were symmetric, demonstrating that the ground and excited states have the same geometry (Fig. 1A). Fluorescence quantum yields, Φf, and maximum emission wavelength, λemmax, of III in CH3CN and CCl4 are presented in Table 2. The low Φf value of III in CCl4 can be attributed to a solvent quenching effect.22
The fluorescence spectra (emission and excitation) of III in a mixture of 50% CH3CN–water changed with time (Fig. 1A). This effect is assigned to a hydrolysis reaction which leads to opening of the anhydride moiety and to a decrease in the aromatic conjugation extent and consequently in a decrease in Φf. Using methanol or ethanol as solvents, a time dependent shift was observed in III absorption λmax from 368 nm to 346 nm, also indicative of solvolysis (not shown).
Like in other solvents (Table 1) compound III, as its diimide homologues,21 showed a well-resolved UV-Vis absorbance spectrum in aqueous solution (phosphate buffer 0.02 M, pH 7.0) (Fig. 1B, line 1). The UV-Vis spectrum of III in water exhibits a λmax at 368 nm and a second peak at 348 nm, similar to that obtained for films of III.23 These features show that, as dianhydride or diimide, the chromophoric group remains relatively unsolvated.
Adding III to aqueous buffer (pH 7.0) resulted in a time-dependent spectral change (Fig. 1B). The absorbance at 368 nm (Fig. 1B) shows a rapid decrease, with half-life of ca. 24 s followed by a much slower process with a half-life of ca. 2 h which leads to further decrease in the absorbance at 368 nm and an increase in absorbance at 308 nm. The initial fast decrease in the absorbance at 368 nm is attributed to hydrolysis of III yielding II (Scheme 1). Compound II at pH 7 is almost completely dissociated (see below) and the fully dissociated ionic form of II will be referred as II-Base, (Scheme 1, and see below). Note that, in this initial reaction, an isosbestic point at 338 nm is evident (Fig. 1B). Above 260 nm the UV spectrum of II-Base shows a single λmax at 348 nm (Fig. 1B, line 7). The slower process can be attributed to base catalyzed hydrolysis of II-Base yielding I (Fig. 1B, line 15), which at pH 7.0 is also fully dissociated, I-Base, see below (Scheme 1). The spectrum of I-Base exhibits a λmax at 308 nm. In the alkaline hydrolysis of II → I an isosbestic point at 326 nm is clear.
In the following section a kinetic study of the reactions III → II and II ⇌ I in a wide range of acidities are presented.
3.2 Kinetics
The first order rate constant, kψ, for the reaction III → II was calculated from the absorbance change followed at 368 nm (where III absorbs) using only the fast change process. The variation of the observed rate constants, kψ, for the alkaline hydrolysis of III yielding II-Base with pH is shown in Fig. 2A. The solid line in Fig. 2A was obtained by fitting the data with eqn (1), where kw is the first order rate constant for the water-catalyzed reaction and kOH the second-order rate constant for hydroxide attack (III → II-Base, Scheme 1). The best-fit parameters were: kwIII = 0.005 s−1 and kOHIII = 8.5 × 104 M−1s−1.| kψ = kw + kOH[OH] | (1) |
![(A) pH effect on kψ of hydrolysis of III to II. Inset: pH effect on kψ of hydrolysis of II to I. All buffers used were 0.02 M (see methods). The final concentrations of II and III were 1 × 10−5 M. Lines were calculated using eqn (1). (B) UV-Vis spectra of reaction mixtures of III in equilibrium with II as a function of time, in minutes, in 0.1 M HClO4, pH = 1.0 at: (1) t = zero, (2) t = 0.5; (3) t = 1.5; (4) t = 3; (5) t = 5; (6) t = 9; (7) t = 19; (8) t = 70. The reaction was started with I. (C) Spectra at the end of reaction starting from I-Base, 1 × 10−5 M, in different [HClO4].](/image/article/2006/OB/b512187f/b512187f-f2.gif) | ||
| Fig. 2 (A) pH effect on kψ of hydrolysis of III to II. Inset: pH effect on kψ of hydrolysis of II to I. All buffers used were 0.02 M (see methods). The final concentrations of II and III were 1 × 10−5 M. Lines were calculated using eqn (1). (B) UV-Vis spectra of reaction mixtures of III in equilibrium with II as a function of time, in minutes, in 0.1 M HClO4, pH = 1.0 at: (1) t = zero, (2) t = 0.5; (3) t = 1.5; (4) t = 3; (5) t = 5; (6) t = 9; (7) t = 19; (8) t = 70. The reaction was started with I. (C) Spectra at the end of reaction starting from I-Base, 1 × 10−5 M, in different [HClO4]. | ||
The kψ for the water-catalyzed hydrolysis and hydroxide attack on II-Base yielding I-Base, were obtained starting the reaction by addition of II-Base to solutions of different pHs (insert, Fig. 2A). Using eqn (1) the best-fit parameters were kwII = 2.0 × 10−4 s−1 and kOHII = 4.5 × 102 M−1 s−1. The kwIII/kwII and kOHIII/kOHII ratios (25 and 190, respectively) show the difference in reactivity between the dianhydride and monoanhydride towards water and OH−.
Addition of III to buffers of pHs between 1 and 6, also led to a time dependent changes in λmax from 368 nm to 348 nm and a slower change from 348 to 308 nm (not shown). The fast process can be attributed to hydrolysis of III to II as analyzed above. The slower process, however, is more complex, since the UV spectra of the products correspond to II ⇌ I equilibrium mixture and the concentration of I at equilibrium was pH-dependent, decreasing with acidity (see below).
The time-dependent spectrum of a reaction mixture in 0.1 M HClO4, with III as the initial reagent, is shown in Fig. 2B. Two consecutive isosbestic points can be differentiated: one at 341 nm during the fast first absorbance decrease and another at 320 nm in the slower phase. The existence of two consecutive isosbestic points lends credence to our assumption that the observed kinetics can be best explained by two consecutive reactions also at this pH, i.e.III → II and II ⇌ I. The peak at λmax of III disappears after ca. 10 min (Fig. 2B) therefore, at equilibrium the spectrum corresponds to that of II, with a small contribution of I. It is also clear from the spectra that in the 1–6 pH range the final concentration of III is negligible.
Above 1.0 M acid only the equilibrium between III and II was observed. The final spectrum of the reaction products, using either III or II or I as the initial reagent, shows a mixture of both compounds, with the III/II ratio increasing with [HClO4] (Fig. 2C).
As described above, the rate constant, kψ, for hydrolysis of III yielding II can be obtained following the reaction at 368 nm. The variation of kψ with acidity was studied from pH 6 to H0 = −5 and three different regions are evident (Fig. 3A). Between pH 6 to 0.5 kψ was constant, between pH 0.5 to H0 = −2 kψ decreased with acidity to a minimum and below H0 = −2, kψ increased with acidity.
![(A) Effect of pH on kψ of reactions starting from III to II (○, ●, ▲) and starting from II to III (△). Different symbols correspond to different sets of experiments. Buffers used were 0.02 M. (B) Effect of [NaClO4] on hydrolysis of III in HClO4 0.1 M. (C) Final absorbance, at 368 nm, after attainment of equilibrium, between II and III as a function of H0, starting with [II] = 2.5 × 10−5 M. Different symbols correspond to independent sets of experiments (○, ●, ▲, △).](/image/article/2006/OB/b512187f/b512187f-f3.gif) | ||
| Fig. 3 (A) Effect of pH on kψ of reactions starting from III to II (○, ●, ▲) and starting from II to III (△). Different symbols correspond to different sets of experiments. Buffers used were 0.02 M. (B) Effect of [NaClO4] on hydrolysis of III in HClO4 0.1 M. (C) Final absorbance, at 368 nm, after attainment of equilibrium, between II and III as a function of H0, starting with [II] = 2.5 × 10−5 M. Different symbols correspond to independent sets of experiments (○, ●, ▲, △). | ||
Between pH 0.5 to 6, the reaction must be water catalyzed, since kψ did not vary with acidity and the value was identical to that calculated for the water reaction with eqn (1) for the alkaline region (i.e., kwIII = 0.005 s−1) (Fig. 3A).
From pH 0.5 to H0 = −1.5, kψ decreased with acidity and the product was also II (spectra, not shown). As described previously for the hydrolysis of anhydrides,24 the decrease in kψ from pH 0.5 to −1.5 is due mainly to the increase in ionic strength. The effect of [NaClO4] on the spectrum of IIITable 1, and on kψ was determined in HClO4 at several [NaClO4]. As the concentration of acid increases there is a continuous shift in λmax of III from 368 to 375 nm, due a combination of specific salt (i.e. ClO4−) effects, ionic strength and further protonation of III (Table 1). Addition of NaClO4 (5.0 M) to a solution of III in 0.80 M HClO4 red-shifted the λmax from 368 to 372 nm (Table 1), indicating that part of the spectral changes observed in concentrated acid can be attributed to a medium polarity effect.
The value of kψ of the reaction (III → II) in 0.1 M HClO4 decreased with [NaClO4] from 5 × 10−3 s−1 (no added salt) to 4.3 × 10−4 s−1 (5.0 M NaClO4) (Fig. 3B). This inhibitory salt effect, consistent with the decrease in observed in Fig. 3A in that acidity range, indicates that the minimum in the kψvs. acidity plot results from ionic strength effect on the reaction.
Below H0 −2 the hydrolysis of III is acid catalyzed, kψ increasing with H0 (Fig. 3A). Further protonation of III and/or II can be proposed to rationalize these results. In order to ascertain that in the H0 region II and III are in equilibrium we demonstrated that the value of the rate constants are identical upon starting the reaction with II or III (Fig. 3A).
The change in the spectra at the end of the reactions demonstrates the increase in III/II ratio with acidity (Fig. 2C). Upon increasing HClO4, the spectrum approaches that of III and the absorbance at 368 nm, at the end of the reaction, A∞, increases, indicating that III is the major equilibrium product (Fig. 3C). The equilibrium constant (II ⇌ III) was not calculated because the spectral changes reflect not only alteration in the III/II ratio but also the effects of ionic strength in the λmax and ε of III. It is clear, however, that the equilibrium II ⇌ III is strongly displaced towards the dianhydride.
It has not escaped our attention that the use of the H0 function is subject to debate.25a The HA function may be more appropriate to describe the variation of both rate constant and absorbance with acid concentration in our system.25b,c,d The differences between using several functions to describe the effect of concentrated acid are evident,25e but no difference between HA and H0 is obtained, in our data, in the region where the minimum in the rate constant was obtained (Fig. 3A). We have thus used the H0 function as a descriptive tool rather than as a method to rationalize mechanism or calculate values of equilibrium constants in concentrated acid.
The rate constants for reaching equilibrium in the I ⇌II reaction and the final reaction spectra were determined between pHs 1 and 6. Both rate constants and final spectra were identical upon starting the reaction(s) with either reagent. When using I as a reagent we added an aliquot of I-Base, prepared by complete III hydrolysis at pH 10.2, to solutions of buffers at pHs < 6. A time-dependent change of spectra was obtained with λmax changing from 308 nm to 348 nm, indicating formation of II. Final spectra of these reactions, at several pHs, are presented in Fig. 4A. The absence of a peak at 368 nm indicates no significant formation of III in this pH range. As the acidity increases, the II/I ratio increases, as evidenced by the increase in absorbance at 348 nm (Fig. 4A). The spectra of the final reaction mixture are complex because the carboxylic groups of I and II dissociate at this pH range (Scheme 1, see below) and the spectra, at each pH, reflects also the change in the concentration of ionic species of II and I.
![(A) UV-Vis final spectra of cyclization of I to II at several pH's: (a) pH 2.3; (b) pH 3.0; (c) pH 4.0; (d) pH 4.9; (e) pH 5.2; (f) pH 5.5 and (g) pH 6.5. (B) pH effect on kψ of equilibrium reaction between II and I, starting from I-Base, followed at 348 nm and [I-Base] = 1.05 × 10−5 M. The solid line was obtained using eqn (14) and the different symbols correspond to independent sets of experiments. (C) pH effect on the absorbance at equilibrium between II and I, at 348 nm, starting the reaction from [I-Base] = 8.8 × 10−6 M. The different symbols correspond to independent sets of experiments. The solid line was obtained using eqn (10).](/image/article/2006/OB/b512187f/b512187f-f4.gif) | ||
| Fig. 4 (A) UV-Vis final spectra of cyclization of I to II at several pH's: (a) pH 2.3; (b) pH 3.0; (c) pH 4.0; (d) pH 4.9; (e) pH 5.2; (f) pH 5.5 and (g) pH 6.5. (B) pH effect on kψ of equilibrium reaction between II and I, starting from I-Base, followed at 348 nm and [I-Base] = 1.05 × 10−5 M. The solid line was obtained using eqn (14) and the different symbols correspond to independent sets of experiments. (C) pH effect on the absorbance at equilibrium between II and I, at 348 nm, starting the reaction from [I-Base] = 8.8 × 10−6 M. The different symbols correspond to independent sets of experiments. The solid line was obtained using eqn (10). | ||
The equilibrium constant for II ⇌ I reaction was measured at 348 nm. At this wavelength the ionic species of II have essentially the same absorption coefficient and absorption by I is insignificant (see below, Fig. 5B). Absorbance at the end of the reactions, A∞, at this wavelength was used to calculate the equilibrium constant between (II ⇌ I) at several pHs (see below).
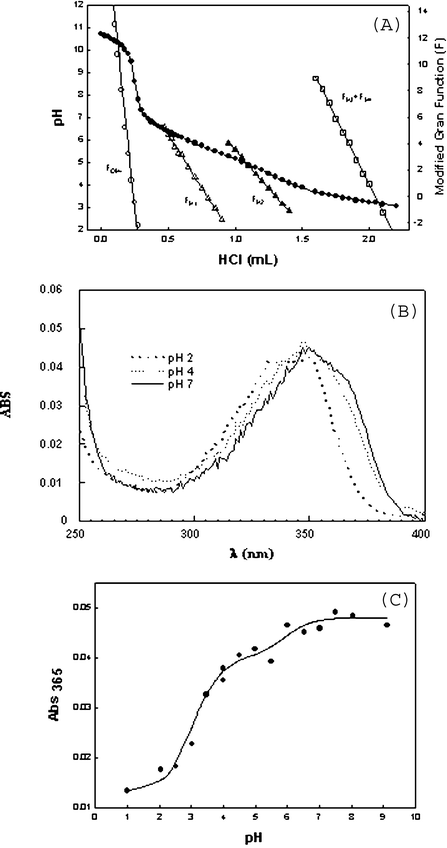 | ||
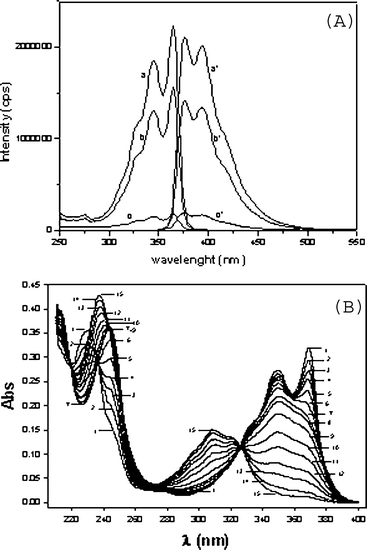
Fig. 5 (A) Experimental titration curve of I (●) linearized by the modified Gran functions (○) FOH− (×103), (△) FH1 (×109), (▲) FH2 (×1010) and (□) FH3 + FH4 (×1012) with FOH−, FH1, FH2 and FH3 + FH4 corresponding to the titration of the excess of OH− and ionizable groups with pKas: 6.25 ± 0.05; 5.13 ± 0.08 and 3.24 ± 0.11, respectively. (B) pH effect on the absorbance spectra of II: (![[dash dash, graph caption]](https://www.rsc.org/images/entities/char_e091.gif) ) pH = 2; (⋯) pH = 4; (—) pH = 7. (C) pH effect on dissociation of II, 2.5 × 10−5 M, at 365 nm. Continuous line was generated using eqn (2). Buffer concentrations were 0.05 M. ) pH = 2; (⋯) pH = 4; (—) pH = 7. (C) pH effect on dissociation of II, 2.5 × 10−5 M, at 365 nm. Continuous line was generated using eqn (2). Buffer concentrations were 0.05 M. | ||
Below pH 6 the kψs for reaching equilibrium (II ⇌ I), followed at 348 nm, increased sigmoidally with pH, reaching a plateau at pH = 1 (Fig. 4B). The A∞ of the reaction mixture plotted against pH increases with acidity to a plateau extending from pH 3 to pH 1 (Fig. 4C). The increase in kψ‘s and A∞ with acidity led us to suggest that the protonated form of I is that involved in the cyclization to II and that formation of II is favored over I at low pH.
In order to determine the dissociation state of the species involved in the reactions of I and II, pKas were determined.
3.3 pKa Determination
The pKas of I and II (Scheme 1) were determined by spectrophotometric and potentiometric methods. Protonation of I-Base, yields species I-H4, I-H3, I-H2, I-H and I-Base and the corresponding pKas are pKI-H4, pKI-H3, pKI-H2, and pKI-H1, respectively. It should be noted that the ionic species I-H2 can correspond to different and indistinguishable isomers (I-H2, I-H2′) as shown in Scheme 1. The ionic forms of II are II-H2, II-H and II-Base and the pKas are pKII-H2 and pKII-H1, respectively.Potentiometric titration allowed the determination of pKas of I (see Methods). Titration data (pH vs. HCl volume) were treated by modified Gran functions (Fig. 5A).19 The calculated pKas of I were 3.24 ± 0.11, 5.13 ± 0.08 and 6.25 ± 0.05. Titration of a fully dissociated 1 mM solution of I with HCl (see Methods) needed 1.96 ± 0.36, 1.23 ± 0.09 and 1.16 ± 0.13 mM acid for full protonation at the above indicated pKa regions. These data indicate a stoichiometry of 2 : 1 : 1, respectively (Table 3). The carboxylate dissociations around pH 3.2 can be attributed to those of the carboxylic groups at opposite sides of the naphthalene ring (pKI-H4 and pKI-H3, Scheme 1). The sensitivity of the titration method and the error in this pH region does not allow differentiation if those pKas are identical or similar. Although IH4 is neutral and IH3 is negatively charged the pKas can be similar. The dissociated carboxylate of IH3 is at the opposite side of the naphthalene ring in relation to the carboxyl group that dissociates to yield IH2.
| Method | pKII-H2 | pKII-H1 | pKI-H4 | pKI-H3 | pKI-H2 | pKI-H1 |
|---|---|---|---|---|---|---|
| Potentiometry | 3.24 ± 0.11 | 3.24 ± 0.11 | 5.13 ± 0.08 | 6.25 ± 0.05 | ||
| Fluorescence | 3.65 | |||||
| Equilibrium | 3.40 | 3.70 | 3.70 | |||
| Kinetic | 3.40 | 3.52 | 3.52 | |||
| UV Spectra | 3.05 | 5.90 |
IH 3 is highly conjugated and, furthermore, H bonding between the dissociated and protonated carboxylates of IH3, at the same side of the ring, may decrease further the effect of the negative charge on the pKa of IH3 yielding IH2. The pKas 5.13 and 6.25 can be assigned to pKI-H2 and pKI-H1 (Scheme 1).
The different ionic species of II and I can also be differentiated spectrophotometrically, allowing faster pKa determinations without significant cyclization.
The UV-Vis spectrum of II changes with pH upon deprotonation (Fig. 5B). The absorbance of II at 365 nm was recorded immediately after injecting an aliquot of II into buffered solutions in order to avoid ring opening. Absorbance vs. pH data are in Fig. 5C and the pKas were calculated using the eqn (2):10
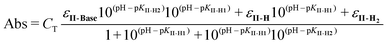 | (2) |
The pH-dependent change of the maximum fluorescence wavelength of I-Base (data not shown) was attributed to changes in the protonation of the carboxylic groups. Only one pKa could be determined by fluorescence, at 454 nm, with a value of 3.65 (Table 3).
Considering the differences in the methods used, the pKas of I obtained by different methods agree very well (Table 3).
3.4 Quantitative analysis of equilibria
Since the absorptivity at 348 nm of II-Base, II-H and II-H2 are similar (Fig. 5B), this wavelength was used to measure the equilibrium constant between II and I, KEq-II. At 348 nm the absorption of I is less than 5% of that of compound II and can be neglected.An initial estimation of KEq-II between I-H4 and II-H2 was obtained from the following experiment: an aliquot of 0.05 mL of III, 1 × 10−3 M in DMSO, was added to 2.5 mL of a 0.02 M phosphate buffer, pH 7.1, and allowed to react for 3 min, yielding II-Base (the half life of this reaction is 24 s). At this time, 0.02 mL of 12 M HClO4 were added to lower the pH to 1.3. At this pH II is in the fully protonated form, II-H2 (Table 3). The absorbance obtained after HClO4 addition was used to calculate εII-H2 and the reaction was allowed to reach equilibrium, where a mixture of II-H2 and I-H2 was obtained. I-H4 does not absorb at this wavelength and, therefore, KEq-II is given by:
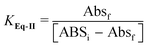 | (3) |
The effect of pH on the [II]/[I] ratio was determined by measuring the A∞ of the reaction as a function of pH. A∞ was measured by adding III in DMSO to buffers with different pHs. III yields II, that subsequently equilibrates with I. The absorbance at equilibrium (II ⇌ I) are in Fig. 4C. The decrease in pH led to the increase of A∞, corresponding to an increase in the II-H2/I-H4ratio. The definitions of the dissociation equilibrium shown in Scheme 1 are in eqn (4)–(7).
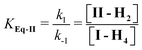 | (4) |
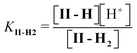 | (5) |
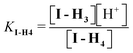 | (6) |
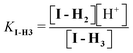 | (7) |
| CT = [I–H4] + [I–H3] + [I–H2] + [II–H2] + [II–H] | (8) |
As commented on above, the absorbance at equilibrium at 348 nm, Abs∞, at each pH, is given by the sum of the absorbance due only to II-H and II-H2 because the ionic forms of I do not contribute to the absorbance at this wavelength:
| Abs∞ = εII-1[II–H] + εII-2[II-H2] | (9) |
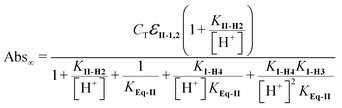 | (10) |
The values of pKI-H4, pKI-H3, pKII-H2 and KEq-II, were obtained by fitting the data in Fig. 4C using eqn (10). The solid line in Fig. 4C was obtained with the following values: CT = 8.8 × 10−6 M, εII-1,2 = 16,715 M−1 cm−1, KII-H2 = 4 × 10−4 (pKII-2 = 3.4), KI-H4 = KI-H3 = 2.0 × 10−4, (pKI-H4 = pKI-H3 = 3.7) (Table 3). The best fit value for KEq-II was 5.0.
3.5 Quantitative analysis of kinetics
From the data presented above it can be concluded that the ionic species involved in the reaction I ⇌ II are I-H4 and II-H2. Between pH 1 and 3, the concentration of II at equilibrium is pH independent (Fig. 4C). Hence, it is reasonable to conclude that equilibrium is established between species I-H4 and II-H2, i.e. between the fully protonated tetra-acid and the di-protonated mono-anhydride.The II/I ratio at equilibrium is negligible above pH 5 (Fig. 4B), supporting the equilibrium data that also indicated that only I-H4 cyclizes. As only I-H4 cyclizes the system can be described as follows:
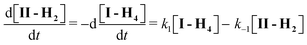 | (11) |
| [I]T = [I-H2] + [I-H3] + [I-H4] | (12) |
| [II]T = [II-H2] + [II-H] | (13) |
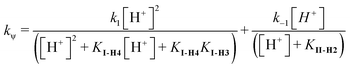 | (14) |
The fit of eqn (14) to the kinetic data in Fig. 4B was obtained using k−1 = 0.000275, k1 = 0.00165 and KII-2 = 4 × 10−4 (pKII-2 = 3.4), KI-H4 = KI-H2 = 3 × 10−4 (pKI-H4 = pKI-H3 = 3.52). The ratio between k1 and k−1 yields the equilibrium constant, KEq-II, eqn (3). The best-fit value of the kinetic data was obtained with KEq-II = k1/k−1 = 6, in good agreement with that obtained from the equilibrium data, i.e., KEq-II = 5.
These results led us to conclude that protonation of the four carboxylic acids of I is necessary for, at least, one ring cyclization leading to II-H2. The value of KEq-II indicates that II is favored over I at low pH, in agreement with the previous data for cyclization of 1,8-naphthalic dicarboxylic acid to the anhydride (KEq = 3 at 30 °C).10
3.6 Structural calculations
Our kinetic and equilibrium data showed unequivocally a major decrease in reactivity upon opening of the first anhydride (i.e., compare the rates of reaction of III to II with those from II to I) from pH 2 to pH 7, i.e. water reaction, and in alkali. Furthermore, it was evident that the monoanhydride II was reversibly transformed into dianhydride III only at high [H+]. It was of interest to obtain independent structural data supporting these experimental observations. For this purpose we calculated the electronic density maps of geometry optimized structures (see Methods) in the gas phase. Calculations, performed exclusively for the first ring opening, were done separately for the acid, water and base-catalyzed reaction manifolds (Schemes 2–4).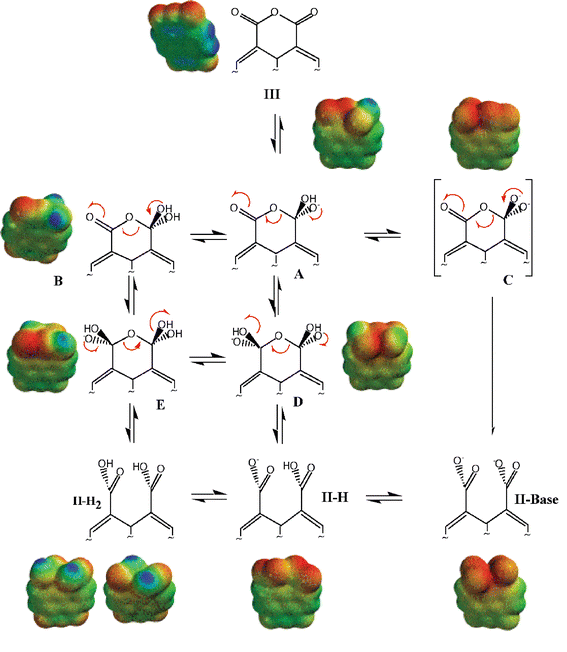 | ||
| Scheme 2 Mechanism of alkaline hydrolysis of III. | ||
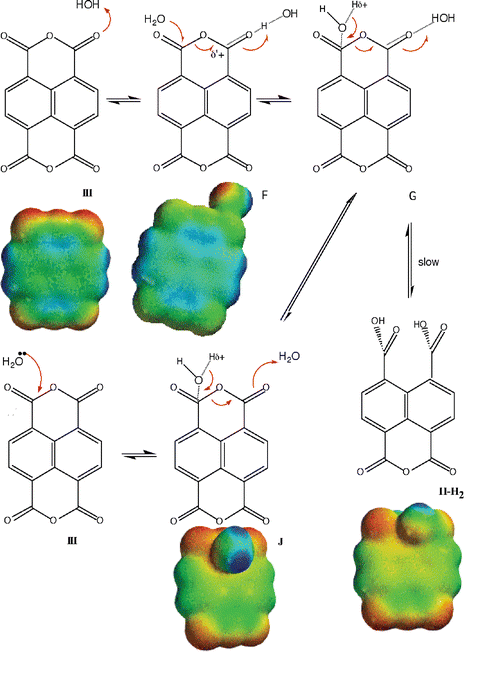 | ||
| Scheme 3 Mechanism of water hydrolysis of III. | ||
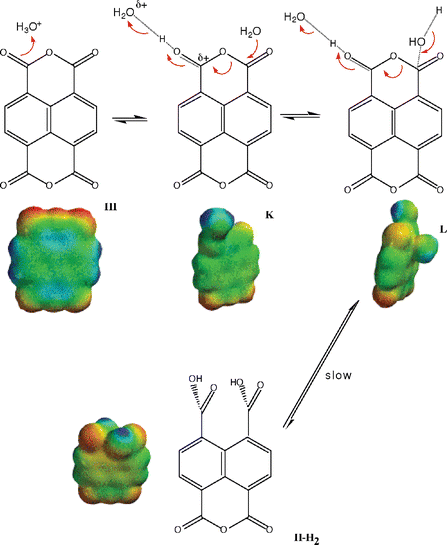 | ||
| Scheme 4 Mechanism of acid hydrolysis of III. | ||
Schemes 2–4 show some model structures possibly involved in the acid, base and water catalysis of hydrolysis of III. The color intensities depict electronic isosurfaces referring to each structure. Relative color intensities, from one structure to another, cannot be compared. Selected values of electrostatic potential at the same of the atoms in the structures shown in Schemes 2–4 are presented in Table 4 for clarity.
| Structures |
V
+ (kcal mol−1)![[hair space]](https://www.rsc.org/images/entities/char_200a.gif) a a |
V
− (kcal mol−1)b |
V+ (kcal mol−1)c |
|---|---|---|---|
| a Potential at carbonyl carbon in the opposite side of reaction center. b Potential at carbonyl oxygen in the opposite side of reaction center. c Potential at carbonyl carbon in the same side of reaction center. | |||
| A | −59 | ||
| B | 9 | ||
| C | −126 | ||
| D | −135 | ||
| E | −62 | ||
| II-H2 | 8 (trans); 8 (cis) | −41 (trans); −41 (cis) | |
| II-H | −56 | ||
| II-Base | −131 | ||
| F | 19 | 18 | |
| J | 14 | −36 | |
| K | 15 | 98 | |
| L | 12 | ||
| III | 17 | −33 | |
Scheme 2 shows the geometry optimization calculations for the presumed intermediates formed upon OH− attack on III. Although the calculated structures in base were not detected here by UV absorption in water, structure B in aprotic DMSO had been observed by IR and 1H NMR spectroscopy.26 In a highly conjugated molecule such as III, hydroxide addition to one of the carbonyl carbons increases the electronic density along the whole structure. This is demonstrated both by the density potential plots in Scheme 2 (compare III with intermediates and products) as well as the values of electrostatic potential (V+) at the carbonyl carbon centered at the opposite side of the reaction (Table 4). Hence we chose to calculate simple model structures, starting material and those most likely to be formed along the reaction pathways, excluding more complex, water bridged models. It should be noted that III, although containing six oxygen atoms and four carbonyls, is poorly water soluble and that the calculated electrostatic potential at the carbonyl carbon of III does not vary significantly after inclusion of a hydrogen bonded water molecule (See Table 4). Our calculations do not allow decisions of preferred pathways but the methodological choice permitted estimation of electronic correlation dependent properties. The calculations, as will be discussed below, were sufficiently accurate to permit better molecular level understanding of part of the experimental findings. Structures shown in Scheme 2, with the exception of C were stationary states, suggesting that a proton loss from A may lead to II-Base with no intervening intermediate. The electronic isosurface shown above C corresponds to that of II-Base. In the first proposed structure formed after OH− attack to one of the carbonyl carbons, (A), we calculated extensive electronic availability along the molecular structure (Scheme 2, Table 4). The redistribution of charge in the molecule is maintained in the final product and while the value of the electrostatic potential at the carbonyl carbon of III was 17 kcal mol−1 that of II-Base was −131 kcal mol−1 (Table 4).
Although our calculations in the gas phase allow for the existence of II-H2, II-H and II-Base, only II-Base is significant in alkali (see above). The increase in negative charge density at the carbonyl carbon of the remaining anhydride is in qualitative agreement with the 200 fold decrease in reactivity observed experimentally when reactions III to II and II to I in alkali are compared (see text).
From pH 2 to 7 the rate of anhydride hydrolysis remains unchanged, suggesting a water-catalyzed hydrolysis (see above). Water attack to the carbonyl carbon can be calculated from a solvated III (F in Scheme 3), similar to a protonated species in acid (K in Scheme 4). A H2O–III complex, hydrogen bonded to a carbonyl oxygen, and a nucleophilic attack by water oxygen to carbonyl carbon, produces structures F and G (Scheme 3). The attempts to calculate structure G (Scheme 3) by correlated DFT level of theory failed, probably due to very weak interactions. A HF/6-31G* calculation did not result in reasonable distances for any significant interactions. Structure F (Scheme 3) presents a weak hydrogen bond with a water molecule, in agreement with the low water solubility of III in that pH range. In fact the small differences observed in the values of the electrostatic potential at the carbonyl carbon in the same side of reaction center of species III (17 kcal mol−1) and J (18 kcal mol−1) (Table 4) also indicate very small water–reagent interaction. In the assumed structure J (Scheme 3) the water O carbonyl C distance is large, and the contribution of this species can be neglected. The calculations shown in Scheme 3 suggest that the preferred pathway for water-catalyzed hydrolysis follows the pathway III ⇌ F ⇌ II-H2. The rate difference between the III to II and II to I water-catalyzed reactions was 20 fold, an order of magnitude lower than that for the corresponding OH− reactions of the same species. The calculated differences in electrostatic potentials also were correspondingly lower (Table 4).
The limited number of structures considered in acid suggests a straightforward protonation followed by water attack (Scheme 4).
4 Conclusions
Product composition and rates for the hydrolysis of III in aqueous solution varied significantly with acidity. In base III hydrolyzed to yield II and I and, at infinite time, the only significant product was I. In moderate acidic solutions the hydrolysis of III gave an I ⇌ II equilibrium mixture. III only equilibrates with II in concentrated acid. pKa determinations using different methods and quantitative analysis of equilibrium and kinetic data permitted the full description of the hydrolysis of III from alkaline solutions to concentrated acid. Carboxylic acid protonation at the same side of the ring was demonstrated to be necessary for anhydride formation. The rates of water or hydroxide attack on the dianhydride III were significantly higher when compared to the monoanhydride II. Geometry optimization calculations clearly indicated that addition of a nucleophile to the dianhydride resulted in a redistribution of the electronic density over the whole molecule and were qualitatively consistent with the observed reactivity differences between III and II. These results represent the first mechanistic description of the hydrolysis of III in aqueous solution over a wide range of conditions and contribute to the understanding of anhydride reactivity in highly conjugated systems.Acknowledgements
Financial support for this work was received from CNPq, FAPESP, Pró-Reitoria de Pesquisa da USP and UNIP.References and notes
- T. C. Bruice and F. C. Lightstone, Acc. Chem. Res., 1999, 32, 127–136 CrossRef CAS.
- A. J. Kirby, Adv. Phys. Org. Chem., 1980, 17, 183–278 CAS.
- W. P. Jencks, Catalysis in Chemistry and Enzymology, Dover, New York, 1987 Search PubMed.
- S. Y. Yunes, J. C. Gesser, H. Chaimovich and F. Nome, J. Phys. Org. Chem., 1997, 10, 461–465 CrossRef CAS.
- T. C. Barros, Formação e Decomposição de Naftalimidas em Solução Aquosa: Dependência Estrutural e Efeito de Micelas, Master Thesis, Chemistry Institute, Biochemical Department, University of São Paulo, USP, São Paulo, Brazil, 1991 Search PubMed.
- M. Bender, Chem. Rev., 1960, 60, 53–113 CrossRef CAS.
- J. A. Knopp, W. S. Linnell and W. C. Child, Jr., J. Phys. Chem., 1962, 66, 1513–1516 CrossRef CAS.
- W. P. Jencks, F. Barley, R. Barnett and M. Gilchrist, J. Am. Chem. Soc., 1966, 88, 4464–4467 CrossRef CAS.
- (a) L. Eberson, Acta Chem. Scand., 1964, 18, 1276–1282 CrossRef CAS; (b) L. Eberson and H. Welinder, J. Am. Chem. Soc., 1972, 93, 5821–5826; (c) L. Higuchi, L. Eberson and J. D. McRae, J. Am. Chem. Soc., 1967, 89, 3001 CrossRef CAS; (d) M. D. Hawkins, J. Chem. Soc., Perkin Trans. 2, 1975, 282 RSC.
- T. C. Barros, S. Yunes, G. Menegon, F. Nome, H. Chaimovich, M. J. Politi, L. G. Dias and I. M. Cuccovia, J. Chem. Soc., Perkin Trans. 2, 2001, 2342–2350 RSC.
- A. G. Kofman, G. V. Chernysh, V. A. Shingalevskii and G. N. Vorozhtsov, Zh. Org. Khim., 1988, 24, 1973–1978 CAS; A. G. Kofman, G. V. Chernysh, V. A. Shingalevskii and G. N. Vorozhtsov, Zh. Org. Khim. (Engl. Transl.), 1989, 1778–1783 Search PubMed.
- L. Born and G. Heywang, Z. Kristallogr., 1990, 190, 147–152 CAS.
- L. J. Fitzgerald, J. Gallucci and R. E. Gerkin, Acta Crystallogr., Sect. C, 1991, 47, 2315–2319 CrossRef.
- L. J. Fitzgerald, J. Gallucci and R. E. Gerkin, Acta Crystallogr., Sect. C, 1992, 48, 460–465 CrossRef.
- L. J. Fitzgerald, J. Gallucci and R. E. Gerkin, Acta Crystallogr., Sect. C, 1992, 48, 1430–1434 CrossRef.
- A. C. Blackburn, L. J. Fitzgerald and R. E. Gerkin, Acta Crystallogr., Sect. C, 1997, 53, 1991–1995 CrossRef.
- The commercial 1,4,5,8-naphthalene tetracarboxylic acid, I, (Aldrich) was not employed here because its absorption spectra in CH3CN corresponds to the 1,4,5,8-naphthalene diacid monoanhydride, II, maximum at 348 nm and not to I, maximum at 308 nm.
- R. H. Boyd, in Solute Solvent Interactions, ed. J. F. Coetzee and C. D. Ritchie, 1969 Search PubMed.
- J. C. Masini, Talanta, 1994, 41, 1383–1389 CrossRef CAS.
- Spartan Version 5.0, Wave Function, Inc., 18401 Von Karman Avenue, Suite 370, Irvine, CA 92612 USA Search PubMed.
- T. C. Barros, S. Brochsztain, V. G. Toscano, P. Berci Filho and M. J. Politi, J. Photochem. Photobiol., A, 1997, 111, 97–104 CrossRef CAS.
- A. Gilbert, and J. Baggott, Essentials of Molecular Photochemistry, CRC Press, London, 1991 Search PubMed.
- A. Jayaraman, M. L. Kaplan and P. H. Schimidt, J. Phys. Chem., 1985, 82, 1682–1687 CrossRef CAS.
- C. A. Bunton, N. A. Fuller, S. G. Perry and I. H. Pitman, J. Chem. Soc., 1962, 4478–4485 RSC.
- (a) R. A. Cox and K. Yates, Can. J. Chem., 1983, 61, 2225–2243 CAS; (b) K. Yates and J. B. Stevens, Can. J. Chem., 1964, 42, 1957–1970 CAS; (c) R. A. Cox, C. R. Smith and K. Yates, Can. J. Chem., 1979, 57, 2952–2959 CAS; (d) R. A. Cox and K. Yates, Can. J. Chem., 1979, 57, 2944–2951 CAS; (e) K. Yates, H. Wai, G. Welch and R. A. McClelland, J. Am. Chem. Soc., 1973, 95, 418–426 CrossRef CAS.
- I. D. Rae, Aust. J. Chem., 1972, 25, 679–681 CAS.
| This journal is © The Royal Society of Chemistry 2006 |