Rapid and efficient synthesis of poly-substituted quinolines assisted by p-toluene sulphonic acid under solvent-free conditions: comparative study of microwave irradiation versus conventional heating
Cheng-Sheng
Jia
a,
Ze
Zhang
a,
Shu-Jiang
Tu
b and
Guan-Wu
Wang
*a
aHefei National Laboratory for Physical Sciences at Microscale and Department of Chemistry, University of Science and Technology of China, Hefei, Anhui 230026, P. R. China. E-mail: gwang@ustc.edu.cn; Fax: +86-551-360-7864
bDepartment of Chemistry and Key Laboratory of Biotechnology for Medicinal Plants, Xuzhou Normal University, Xuzhou, Jiangsu 221116, P. R. China
First published on 18th November 2005
Abstract
A rapid and efficient method for the preparation of various poly-substituted quinolines has been developed through the Friedländer condensation of 2-aminoarylketone or 2-aminoarylaldehyde with carbonyl compounds in the presence of p-toluene sulphonic acid, which was achieved by both microwave irradiation and conventional heating under solvent-free conditions.
Introduction
It is well known that the quinoline ring system is an important structural unit widely existing in alkaloids, therapeutics and synthetic analogues with interesting biological activities.1 A large variety of quinoline derivatives have been used as antimalarial, anti-inflammatory agents, antiasthmatic, antibacterial, antihypertensive and tyrokinase PDGF-RTK inhibiting agents.2,3 Furthermore, poly-substituted quinolines have been found to undergo hierarchical self-assembly into a variety of nano- and mesostructures with enhanced electronic and photonic functions.4 In view of these points, a great deal of effort has been drawn to develop new and efficient synthetic routes to quinoline derivatives in both synthetic organic and medicinal chemistry. Up to now, versatile methods have been developed for the preparation of these kinds of derivatives, such as Skraup, Doebner-von Miller, Friedländer, Combes methods and so on.5–7 Among these methods, Friedländer annulation,7c,d an acid- or base-catalyzed condensation followed by a cyclodehydration between an aromatic 2-aminoaldehyde or ketone and a carbonyl compound containing a reactive α-methylene group, is one of the most simple and straightforward approaches for the synthesis of poly-substituted quinolines. But even with this classical method, it is still not fully satisfactory with regard to relatively low yield, drastic reaction conditions, generality and operational complexity due to the occurrence of several side reactions. Furthermore, under base-catalyzed conditions, o-aminobenzophenone fails to react with simple ketones such as cyclohexanone and β-ketoesters.8 Therefore, a simple, general and efficient procedure is still in demand for the synthesis of these important heterocyclic compounds.On the other hand, for the stringent and growing environmental regulations, organic chemists are requested to develop environmentally benign synthetic methodologies. One of the most promising approaches is to perform organic reactions under solvent-free conditions, including using microwave irradiation and conventional heating techniques.9
To the best of our knowledge, p-toluene sulphonic acid (p-TsOH) is a convenient, easily available and cheap reagent, which has been used as a catalyst under microwave irradiation leading to a variety of heterocyclic compounds.10 Although p-TsOH has been employed in the Friedländer reaction, it is limited to a substantial extent due to the use of an organic solvent such as toluene and a very low yield.11a Recently, p-TsOH has been used to catalyze the solvent-free Friedländer condensation of 2-aminonicotinaldehyde with acetoacetanilide or benzoylacetanilide by hand grinding, but the utilized carbonyl substrates were just amides.11b As a continuation of our interest in solvent-free organic reactions,12 herein we report a solvent-free methodology for the p-TsOH-assisted synthesis of a large variety of poly-substituted quinolines by microwave irradiation and conventional heating with improved efficiencies and cost effectiveness. Concretely speaking, treatment of 2-aminoarylketone or 2-aminoarylaldehyde with various carbonyl compounds in the presence of p-TsOH promoted by microwave irradiation or conventional heating results in the formation of the corresponding quinoline derivatives with good to excellent yields.
Results and discussion
2-Amino-4′-fluorobenzophenone (1a) has been used as the starting material to synthesize quinoline derivatives, which can functionalize as HMG-CoA reductase inhibitors.11a,13 We therefore first chose 1a and searched for the optimized conditions for its reaction with ethyl acetoacetate (2a) affording quinoline 3a under microwave (SANYO EM-350S microwave oven) conditions (Scheme 1).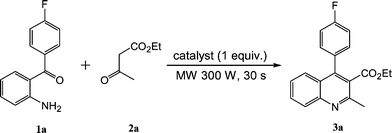 | ||
| Scheme 1 | ||
First, we employed various catalysts to determine which one has the most effective catalytic activity. The results of these comparative experiments are summarized in Table 1.
| Entry | Catalyst | Yield (%) |
|---|---|---|
| 1 | — | Trace |
| 2 | CuSO4·5H2O | 35 |
| 3 | CuCl | 50 |
| 4 | CuCl2·2H2O | 48 |
| 5 | ZnCl2 | 68 |
| 6 | NiCl2·6H2O | 10 |
| 7 | FeSO4·7H2O | Trace |
| 8 | FeCl3·6H2O | 82 |
| 9 | BiCl3 | 86 |
| 10 | AlCl3 | 35 |
| 11 | SnCl4·5H2O | 86 |
| 12 | H3PO4 | 71 |
| 13 | CH3CO2H | 18 |
| 14 | CF3CO2H | 58 |
| 15 | p-TsOH | 92 |
From the above results it can be seen that all the catalysts except FeSO4·7H2O can promote the reaction to a certain extent. Although FeCl3·6H2O (entry 8), BiCl3 (entry 9) and SnCl4·5H2O (entry 11) exhibited good catalytic activity, the water-insoluble solids formed from these catalysts when treated with an aqueous solution of NaOH make the work-up procedure more complicated. It is obvious that p-TsOH (entry 15) demonstrated superior catalytic activity and was the best catalyst among those examined. In order to further evaluate the influence of p-TsOH, this reaction was carried out using different amount of p-TsOH under microwave irradiation and conventional heating conditions. The results are listed in Table 2.
| Entry | p-TsOH/equiv. | Microwave irradiation | Conventional heating | ||
|---|---|---|---|---|---|
| Time/s | Yield (%) | Time/min | Yield (%) | ||
| 1 | 0 | 75 | Trace | 5 | Trace |
| 2 | 0.1 | 75 | 15 | 5 | 50 |
| 3 | 0.2 | 75 | 50 | 5 | 55 |
| 4 | 0.3 | 60 | 80 | 5 | 67 |
| 5 | 0.4 | 45 | 84 | 3 | 82 |
| 6 | 0.5 | 45 | 88 | 3 | 90 |
| 7 | 1.0 | 30 | 92 | 3 | 95 |
| 8 | 2.0 | 30 | 86 | 3 | 94 |
From Table 2 it can be seen that the reaction hardly proceeded in the absence of p-TsOH. The increase in the amount of p-TsOH afforded higher yield. When 1.0 equivalent of p-TsOH was used, the yield was significantly increased up to 92% under microwave irradiation and 95% under conventional heating, respectively (entry 7). However, an excess of p-TsOH did not help to increase the yield. On the contrary, more than one equivalent of p-TsOH resulted in a slightly lower yield (entry 8).
These optimization results prompted us to select 1.0 equivalent of p-TsOH as a catalyst for further study; that is, we used 1.0 equivalent of p-TsOH to catalyze the library synthesis of quinoline 3 from 1a or 2-aminoacetophenone (1b) or 2-amino-3,5-dibromobenzaldehyde (1c) and various carbonyl compounds (2) under microwave irradiation conditions (Scheme 2).
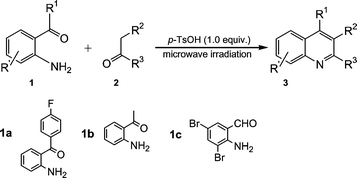 | ||
| Scheme 2 | ||
In a typical case, a molar equivalent of substrates 1, 2 and p-TsOH was mixed and then irradiated in a SANYO EM-350S microwave oven at 300 W or 150 W for a designated time as required to complete the reaction (determined by TLC). The resulting reaction mixture was treated with aqueous NaOH (10%) and the desired product was collected simply by filtration. Under these conditions, the reactions proceed smoothly and efficiently with almost quantitative yield in very short time (not exceeding 60 s). The detailed results for these reactions are collected in Table 3.
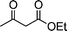
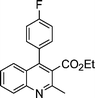
| Substrate 1 | Substrate 2 | Quinoline 3 | Time/s | Yieldb (%) | Mp (lit.)/°C | ||
|---|---|---|---|---|---|---|---|
| a Products 3a–3p were obtained at 300 W and products 3q–3v were obtained at 150 W (the employed microwave output and reaction time for each product are the optimized conditions from several comparative entries). b Yields refers to those of pure isolated products, which were fully characterized by spectral data. c Product 3c has been characterized11a by spectral data but no melting point. | |||||||
| 1a |
|
2a |
|
3a | 30 | 92 | 118–120 (120–12113a) |
| 1a |
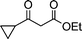
|
2b |
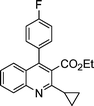
|
3b | 45 | 91 | 74–76 (73.5–75.513c) |
| 1a |
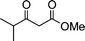
|
2c |
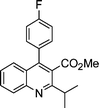
|
3c | 30 | 94 | 114–116c |
| 1a |
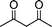
|
2d |
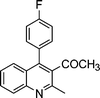
|
3d | 30 | 93 | 140–142 |
| 1a |
|
2e |
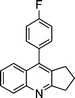
|
3e | 30 | 93 | 139–141 |
| 1a |
|
2f |
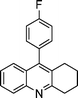
|
3f | 30 | 92 | 172–174 |
| 1a |
|
2g |
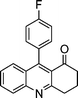
|
3g | 60 | 93 | 170–172 |
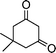
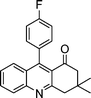
| 1a |
|
2h |
|
3h | 60 | 94 | 211–213 |
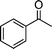
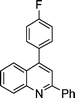
| 1a |
|
2i |
|
3i | 60 | 88 | 82–84 |
| 1a |
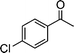
|
2j |
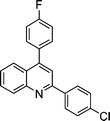
|
3j | 60 | 89 | 98–100 |
| 1b |
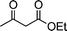
|
2a |
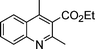
|
3k | 15 | 91 | Oil (oil14) |
| 1b |
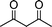
|
2d |
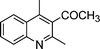
|
3l | 15 | 92 | Oil (oil14) |
| 1b |
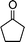
|
2e |
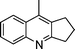
|
3m | 15 | 87 | 58–60 (6014) |
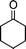
| 1b |
|
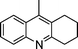
2f |
|
3n | 15 | 85 | 75–77 (7814) |
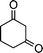
| 1b |
|
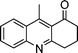
2g |
|
3o | 15 | 95 | 65–66 (6815) |
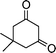
| 1b |
|
2h |
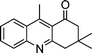
|
3p | 15 | 95 | 105–106 |
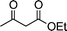
| 1c |
|
2a |
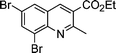
|
3q | 60 | 92 | 116–118 |
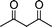
| 1c |
|
2d |
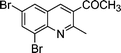
|
3r | 60 | 96 | 162–164 |
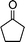
| 1c |
|
2e |
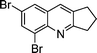
|
3s | 60 | 91 | 110–112 |
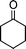
| 1c |
|
2f |
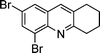
|
3t | 60 | 93 | 106–108 (105–10616) |
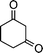
| 1c |
|
2g |
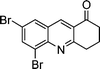
|
3u | 60 | 92 | 176–178 |
| 1c |
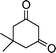
|
2h |
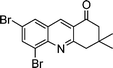
|
3v | 60 | 96 | 164–166 |
From the above results, it can be seen that the generality of this facile condensation was established in the presence of p-TsOH under solvent-free conditions to furnish the corresponding poly-substituted quinolines. By comparison, substrate 1c demonstrates a superior reaction activity to 1a and 1b, in which the employed microwave output is only 150 W.
Because the foregoing optimization work has demonstrated that this kind of reaction can also be efficiently promoted by conventional heating under neat conditions, we tried to obtain all the above products under solvent-free conditions (Scheme 3). As expected, the desired poly-substituted quinolines 3 were also obtained with almost quantitative yields, as shown in Table 4.
| Entry | Quinoline 3 | Time/min | Yield (%) |
|---|---|---|---|
| 1 | 3a | 3 | 90 |
| 2 | 3b | 3 | 93 |
| 3 | 3c | 3 | 94 |
| 4 | 3d | 3 | 94 |
| 5 | 3e | 3 | 95 |
| 6 | 3f | 3 | 92 |
| 7 | 3g | 5 | 95 |
| 8 | 3h | 5 | 93 |
| 9 | 3i | 10 | 90 |
| 10 | 3j | 10 | 91 |
| 11 | 3k | 5 | 90 |
| 12 | 3l | 5 | 93 |
| 13 | 3m | 5 | 89 |
| 14 | 3n | 5 | 86 |
| 15 | 3o | 5 | 96 |
| 16 | 3p | 5 | 94 |
| 17 | 3q | 3 | 94 |
| 18 | 3r | 3 | 95 |
| 19 | 3s | 3 | 92 |
| 20 | 3t | 3 | 95 |
| 21 | 3u | 4 | 94 |
| 22 | 3v | 4 | 95 |
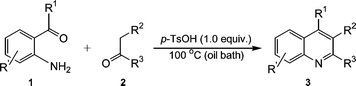 | ||
| Scheme 3 | ||
It should be noted that p-TsOH has recently been used to catalyze solvent-free Friedländer condensation of 2-aminonicotinaldehyde with acetoacetanilide or benzoylacetanilide by hand grinding.11b We have examined the solvent-free reactions of substrates 1a–c with 2a–h under similar conditions, and found that these reactions could hardly proceed by hand grinding. For example, the reaction of 1a with 2a, 2d, 2f or 2h in the presence of p-TsOH in the solid state at room temperature by hand grinding for 30 min gave none of, or only trace amounts of products. Therefore, heating is required for the success of the neat reactions.
In order to draw a direct comparison between microwave irradiation and conductive heating (100 °C), a few selected reactions were carried out at an identical temperature in a monomodal Emrys™ Creator microwave synthesizer. The results are summarized in Table 5.
| Substrate 1 | Substrate 2 | Quinoline 3 | Time/min | Temp/°C | Power/W | Yield (%) |
|---|---|---|---|---|---|---|
| 1a | 2a | 3a | 0.5 | 100 | 300 | 94 |
| 1a | 2h | 3h | 3 | 100 | 300 | 96 |
| 1b | 2a | 3k | 1 | 100 | 300 | 92 |
| 1b | 2h | 3p | 3 | 100 | 300 | 95 |
| 1c | 2a | 3q | 1 | 100 | 150 | 94 |
| 1c | 2h | 3v | 4 | 100 | 150 | 96 |
By comparison of the data in Tables 4 and 5, it can be seen that the microwave irradiation protocol is generally faster than conventional heating at the same temperature to reach a comparable yield. For the microwave-assisted reactions in a SANYO E-350S microwave oven (Table 3), the reaction time required could be even shorter than that in a monomodal Emrys™ Creator. The possible reason is that the temperature in a SANYO E-350S microwave oven was uncontrolled and likely reached at a temperature higher than 100 °C. Furthermore, the test tubes were uncapped and the water formed could evaporate from the reaction mixture at a higher temperature, thus providing extra driving force for the reactions. One can see from the results in Tables 3 and 5 that the good yields in a commercially available and cheap SANYO E-350S domestic microwave oven can be reproduced in a monomodal Emrys™ Creator and thus can be transferred to a more modern microwave synthesizer.
Conclusion
In summary, we have developed a rapid and efficient version of the Friedländer annulation for the synthesis of various poly-substituted quinolines assisted by p-TsOH. All the reactions were conducted under completely solvent-free conditions using either microwave irradiation or a conventional heating technique. In both methods, the process is environmentally benign and the experimental procedure is very simple. Our protocol can be applied to a wide range of substrates, tolerating the presence of halogen, ketone and ester groups. These methods not only afford significant improvements in reaction rates and yields but also present a more straightforward and easy work-up procedure, thus providing a very efficient alternative to traditional processes.Experimental
General
Melting points were determined on an XT-4 apparatus and were uncorrected. IR spectra were taken on a Bruker Vector-22 spectrometer in KBr pellets and reported in cm−1. 1H NMR spectra were recorded at 300 MHz on a Bruker Avance-300 spectrometer in CDCl3 with chemical shifts (δ) given in ppm relative to TMS as an internal standard. 13C NMR spectra were recorded on a Bruker Avance-300 (75.5 MHz) spectrometer with complete proton decoupling; chemical shifts are reported in ppm relative to the solvent resonance as the internal standard (CDCl3, δ 77.16 ppm). All intensities in the 13C NMR spectral data are 1C except where indicated. High-resolution mass spectra (HRMS) were obtained on a Micromass GCT mass spectrometer with a positive EI mode.General procedure for the synthesis of compound 3 with microwave irradiation in a SANYO EM-350S microwave oven
2-Aminoarylketone or 2-aminoarylaldehyde 1 (1 mmol) and carbonyl compound 2 (1 mmol) were mixed with the given amount of p-TsOH or other catalyst and introduced into a test tube (10 mL). Then the mixture was subjected to microwave irradiation at an output of 300 W or 150 W for a given time. When it was cooled to room temperature, water (3 mL) was added to the reaction mixture. The resulting suspension was neutralized with 0.4 mL of 10% NaOH. Then the mixture was stirred for 5 min and the solid was collected by Büchner filtration, washed with H2O (6 mL × 3) and dried in a desiccator to give the product as a white or slightly yellow powder. For the oil or low-melting-point products, the purification procedure was different. The neutralized mixture was extracted with ethyl acetate (5 mL × 2). The organic layer was separated out and dried over anhydrous MgSO4. Evaporation of the solvent afforded the crude product as a slightly yellow oil, which was further purified by column chromatography when necessary.General procedure for the synthesis of compound 3 with conventional heating
A mixture containing 2-aminoarylketone or 2-aminoarylaldehyde 1 (1 mmol), carbonyl compound 2 (1 mmol) and p-TsOH (1 mmol) was introduced into a test tube (10 mL) and stirred at 100 °C (oil bath temperature) for the designated time. When the reaction was completed (monitored by TLC), the subsequent work-up procedure was the same as that of the above microwave irradiation conditions.General procedure for the synthesis of compound 3 with microwave irradiation in a monomodal Emrys™ Creator microwave synthesizer
All reactions were performed in a monomodal Emrys™ Creator from Personal Chemistry, Uppsala, Sweden. Typically, in a 10 mL Emrys™ reaction vial, 2-aminoarylketone or 2-aminoarylaldehyde 1 (1 mmol), carbonyl compound 2 (1 mmol) and p-TsOH (1 mmol) were mixed and then capped. The mixture was irradiated at 300 W or 150 W and at 100 °C for a given time. The subsequent work-up procedure was the same as that in a SANYO EM-350S microwave oven.Acknowledgements
We are grateful for financial support from the National Science Fund for Distinguished Young Scholars (20125205), Fund for Innovative Research Groups of National Science Foundation of China (20321101) and Anhui Provincial Bureau of Human Resources (2001Z019).References
- (a) R. D. Larsen, E. G. Corley, A. O. King, J. D. Carrol, P. Davis, T. R. Verhoeven, P. J. Reider, M. Labelle, J. Y. Gauthier, Y.-B. Xiang and R. J. Zamboni, J. Org. Chem., 1996, 61, 3398 CrossRef CAS; (b) J. P. Michael, Nat. Prod. Rep., 1997, 14, 605 RSC; (c) G. Roma, M. D. Braccio, G. Grossi, F. Mattioli and M. Ghia, Eur. J. Med. Chem., 2000, 35, 1021 CrossRef CAS; (d) Y.-L. Chen, K.-C. Fang, J.-Y. Sheu, S.-L. Hsu and C.-C. Tzeng, J. Med. Chem., 2001, 44, 2374 CrossRef CAS.
- (a) B. Kalluraya and S. Sreenivasa, Farmaco, 1998, 53, 399 CrossRef CAS; (b) D. Dubé, M. Blouin, C. Brideau, C.-C. Chan, S. Desmarais, D. Ethier, J.-P. Falgueyret, R. W. Friesen, M. Girard, Y. Girard, J. Guay, D. Riendeau, P. Tagari and R. N. Young, Bioorg. Med. Chem. Lett., 1998, 8, 1255 CrossRef CAS.
- M. P. Maguire, K. R. Sheets, K. McVety, A. P. Spada and A. Zilberstein, J. Med. Chem., 1994, 37, 2129 CrossRef CAS.
- (a) A. K. Agarwal and S. A. Jenekhe, Macromolecules, 1991, 24, 6806 CrossRef CAS; (b) X. Zhang, A. S. Shetty and S. A. Jenekhe, Macromolecules, 1999, 32, 7422 CrossRef CAS; (c) X. Zhang and S. A. Jenekhe, Macromolecules, 2000, 33, 2069 CrossRef CAS; (d) S. A. Jenekhe, L. Lu and M. M. Alam, Macromolecules, 2001, 34, 7315 CrossRef CAS.
- (a) M. Abass, Heterocycles, 2005, 65, 901 CrossRef CAS; (b) V. V. Kouznetsov, L. Y. V. Méndez and C. M. M. Gómez, Curr. Org. Chem., 2005, 9, 141 CrossRef CAS.
- (a) G. Jones, in Comprehensive Heterocyclic Chemistry II, eds. A. R. Katritzky, C. W. Rees and E. F. V. Scriven, Pergamon, New York, 1996, vol. 5, p. 167 Search PubMed; (b) C. S. Cho, B. H. Oh, J. S. Kim, T.-J. Kim and S. C. Shim, Chem. Commun., 2000, 1885 RSC and references therein; (c) B. Jiang and Y.-G. Si, J. Org. Chem., 2002, 67, 9449 CrossRef CAS.
- (a) H. Skraup, Ber., 1880, 13, 2086 Search PubMed; (b) R. H. F. Mansake and M. Kulka, Org. React., 1953, 7, 59; (c) P. Friedländer, Ber., 1882, 15, 2572 Search PubMed; (d) C.-C. Cheng and S.-J. Yan, Org. React., 1982, 28, 37 CAS; (e) R. J. Linderman and K. S. Kirollos, Tetrahedron Lett., 1990, 31, 2689 CrossRef CAS; (f) C. S. Cho, J. S. Kim, B. H. Oh, T.-J. Kim, S. C. Shim and N. S. Yoon, Tetrahedron, 2000, 56, 7747 CrossRef CAS; (g) M. Arisawa, C. Theeraladanon, A. Nishida and M. Nakagawa, Tetrahedron Lett., 2001, 42, 8029 CrossRef CAS; (h) M.-E. Theoclitou and L. A. Robinson, Tetrahedron Lett., 2002, 43, 3907 CrossRef CAS; (i) S. Dumouchel, F. Mongin, F. Trécourt and G. Quéguiner, Tetrahedron Lett., 2003, 44, 2033 CrossRef CAS.
- E. A. Fehnel, J. Heterocycl. Chem., 1967, 4, 565 Search PubMed.
- (a) S. Deshayes, M. Liagre, A. Loupy, J. L. Luche and A. Petit, Tetrahedron, 1999, 55, 10851 CrossRef CAS; (b) A. Loupy, Top. Curr. Chem., 1999, 206, 153; (c) K. Tanaka and F. Toda, Chem. Rev., 2000, 100, 1025 CrossRef CAS; (d) P. Lidström, J. Tierney, B. Wathey and J. Westman, Tetrahedron, 2001, 57, 9225 CrossRef CAS; (e) M. Nüchter, B. Ondruschka, W. Bonrath and A. Gum, Green Chem., 2004, 6, 128 RSC; (f) Y. Xu and Q.-X. Guo, Heterocycles, 2004, 63, 903 CrossRef CAS.
- (a) B. Pério, M.-J. Dozias, P. Jacquault and J. Hamelin, Tetrahedron Lett., 1997, 38, 7867 CrossRef CAS; (b) P. Goncalo, C. Roussel, J. M. Mélot and J. Vébrel, J. Chem. Soc., Perkin Trans. 2, 1999, 2111 RSC; (c) S. Paul, M. Gupta, R. Gupta and A. Loupy, Tetrahedron Lett., 2001, 42, 3827 CrossRef CAS.
- (a) D. R. Sliskovic, J. A. Picard, W. H. Roark, B. D. Roth, E. Ferguson, B. R. Krause, R. S. Newton, C. Sekerke and M. K. Shaw, J. Med. Chem., 1991, 34, 367 CrossRef CAS; (b) K. Mogilaiah and G. R. Sudhakar, Indian J. Chem., Sect. B: Org. Chem. Incl. Med. Chem., 2003, 42B, 1170 CAS.
- (a) Z. Zhang, Y.-W. Dong, G.-W. Wang and K. Komatsu, Synlett, 2004, 61; (b) Z. Zhang, Y.-W. Dong, G.-W. Wang and K. Komatsu, Chem. Lett., 2004, 33, 168 CrossRef CAS; (c) Z. Zhang, G.-W. Wang, C.-B. Miao, Y.-W. Dong and Y.-B. Shen, Chem. Commun., 2004, 1832 RSC; (d) Z. Zhang, J. Gao, J.-J. Xia and G.-W. Wang, Org. Biomol. Chem., 2005, 3, 1617 RSC.
- (a) Y. Fujikawa, M. Suzuki, H. Iwasaki, M. Sakashita and M. Kitahara, Eur. Pat. Appl., EP 0304063, 1989; Y. Fujikawa, M. Suzuki, H. Iwasaki, M. Sakashita and M. Kitahara, Chem. Abstr., 1989, 111, 134010x Search PubMed; (b) N. Miyachi, Y. Yanagawa, H. Iwasaki, Y. Ohara and T. Hiyama, Tetrahedron Lett., 1993, 34, 8267 CrossRef CAS; (c) M. Suzuki, H. Iwasaki, Y. Fujikawa, M. Kitahara, M. Sakashita and R. Sakoda, Bioorg. Med. Chem., 2001, 9, 2727 CrossRef CAS.
- S. S. Palimkar, S. A. Siddiqui, T. Daniel, R. J. Lahoti and K. V. Srinivasan, J. Org. Chem., 2003, 68, 9371 CrossRef CAS.
- G. Kempter and G. Möbius, J. Prakt. Chem., 1966, 34, 298 CrossRef CAS.
- W. Borsche, Justus Liebigs Ann. Chem., 1910, 377, 70 CrossRef.
| This journal is © The Royal Society of Chemistry 2006 |