Design and synthesis of iridium(III) azacrown complex: application as a highly sensitive metal cation phosphorescence sensor
Mei-Lin
Ho
a,
Fu-Ming
Hwang
b,
Pei-Nung
Chen
b,
Ya-Hui
Hu
a,
Yi-Ming
Cheng
a,
Kung-Shih
Chen
a,
Gene-Hsiang
Lee
a,
Yun
Chi
*b and
Pi-Tai
Chou
*a
aDepartment of Chemistry, National Taiwan University, Taipei, 106, Taiwan. E-mail: chop@ntu.edu.tw
bDepartment of Chemistry, National Tsing Hua University, Hsinchu, 300, Taiwan. E-mail: ychi@mx.nthu.edu.tw
First published on 18th November 2005
Abstract
A new metal cation probe 1 bearing a central Ir(III) element and 1-aza-15-crown-5-ether substituted pyridyl pyrazolate as the chelate was synthesized. The octahedral molecular structure of 1 was confirmed using single crystal X-ray diffraction analyses. Subsequent photophysical study showed yellow-green emission at ∼560 nm in both fluid solution and solid state at room temperature. Remarkable differentiation in spectral properties upon metal cation (e.g. Ca2+) complexation makes complex 1 a highly sensitive phosphorescence probe.
Introduction
Detection of alkali or alkaline earth ions has great potential for practical applications in areas such as analytical chemistry, environmental chemistry and the biological sciences.1 One method is to use the so-called chemosensor that shows the basic molecular configurations such as chromophore–receptor or chromophore–spacer–receptor. As the receptor can selectively bind with the guest cations, measurable and reversible changes in color and/or luminescence can be detected at the signalling unit (i.e. chromophore) linking to the receptor, which allows the recognition and quantification of the metal cation by conventional spectroscopic methods.2 A common design contains a crown ether to serve as the receptor, together with an organic dye or other metal based fluorophores.3 Recently, extension was made to systems with third-row transition-metal complexes.4 Work by a number of groups, mainly pioneered by Schanze and co-workers,5 has established the importance of utilizing long-lived phosphorescence. Typical signalling involves the switching between non-emissive intra-ligand (3IL) ππ* and metal–to–ligand charge transfer (3MLCT) states,6 so that a positive luminescent response vs. concentration of cation can be obtained. Moreover, it may allow the design of “light-controlled ion switches”, for which the cation ejection from the crown ether can be triggered by the effective reduction of electron donation at the receptor on excitation of the nearby chromophore.In this article, we present a novel system in which an azacrown receptor is attached to the pyridyl pyrazolate chelate of a heteroleptic Ir(III) complex. This design provides three inherent advantages. First, the Ir(III) metal atom forms a highly stable, octahedral coordinated structure and induces strong phosphorescent emission due to the heavy atom effect. Moreover, the ancillary cyclometalated phenyl pyrazole ligands, for which the ππ* energy levels are far higher than those of the respective MLCT and other ligand-centered ππ* excited states,7 enable both the HOMO and LUMO to reside predominantly on the azacrown substituted pyridyl pyrazolate segment. This architecture enhances the effectiveness of this design over others having more delocalized electronic configurations; the latter should be less capable of recognizing the cation due to the spreading of their electronic perturbation over the whole complex. Thirdly, the azacrown fragment is attached to an anionic pyrazolate chelate ligand, forming a neutral Ir(III) metal complex. Such a charge-neutral characteristic is similar to that of the Re(I) and Pt(II) based sensor complexes, but is in sharp contrast to most of the Ru(II) based polypyridyl sensors,8 for which the net cationic charge on the overall metal complex is expected to reside, in part, at the azacrown ether site, giving a much reduced sensitivity in recognizing metal cations.9
Experimental
General information and materials
Elemental analyses and mass spectra (operating in FAB mode) were carried out at the NSC Regional Instrument Centre at the National Chiao Tung University, Taiwan. 1H and 13C NMR spectra were recorded on a Varian Mercury 400 or an Inova-500 MHz instrument; chemical shifts are quoted with respect to an internal standard, Me4Si. All synthetic manipulations were performed under a N2 atmosphere, while solvents were used as received. Synthesis of 1-[4-(1,4,7,10-tetraoxa-13-aza-cyclopentadec-13-yl)-phenyl] ethanone follows the procedures reported by Okahara and co-workers,10 using the starting materials 4-N,N-bis(2-hydroxyethyl)aminoacetophenone and triethylene glycol di(p-toluenesulfonate). Triethylene glycol di(p-toluenesulfonate) was prepared using the literature method,11 while 1-phenyl-3,5-dimethyl pyrazole (pdpz)H was prepared from the condensation of phenyl hydrazine hydrochloride with acetylacetone according to literature procedures.12 Treatment of (pdpz)H with IrCl3·nH2O in refluxing methoxyethanol afforded the chloride bridged dimer [(pdpz)2IrCl]2 in 75% yield;13 it was then used for subsequent reactions without further purification.Synthesis of [3-(4-aza-15-crown-5-phenyl)pyridyl(1,3-dione)]
To a stirred suspension of NaH (0.75 g, 31.3 mmol) in THF (40 mL), was slowly added 15 mL of THF solution of 1-[4-(1,4,7,10-tetraoxa-13-aza-cyclopentadec-13-yl)-phenyl]-ethanone (2.6 g, 7.8 mmol) at room temperature. The solution was heated to reflux, and ethyl picolinate (2.0 mL, 15 mmol) was added over the course of 1 h. After the addition was completed, the temperature was gradually lowered to room temperature. The solution was continuously stirred for another 3 h and then quenched with dilute HCl solution. After the removal of the solvent, the residue was extracted with CH2Cl2 (3 × 60 mL). The extracts were combined, washed twice with water, and dried over anhydrous MgSO4 to afford a yellow solid (2.4 g, 5.4 mmol, 70%).Synthesis of 3-(4-aza-15-crown-5-phenyl) pyridyl pyrazole
A solution of [3-(4-aza-15-crown-5-phenyl) pyridyl(1,3-dione)] (1.7 g, 3.8 mmol) and 98% of hydrazine monohydrate (1.94 mL) in 45 mL of anhydrous ethanol was refluxed for 12 h. Next, the solvent was removed under vacuum, and the residue was dissolved in CH2Cl2, washed twice with water, dried over anhydrous MgSO4, and the solution was concentrated to dryness. The crude product was purified by silica gel column chromatography (ethyl acetate and methanol = 5 : 1, v/v), giving a light yellow powdery material (azppz)H (1.2 g, 2.74 mmol, 71%).Preparation of [(pdpz)2Ir(azppz)] (1)
A mixture of [(pdpz)2IrCl]2 (100 mg, 0.088 mmol), azacrown substituted pyridyl pyrazole (azppzH, 85 mg, 0.19 mmol) and Na2CO3 (93 mg, 0.88 mmol) in 2-methoxyethanol (25 mL) was heated to reflux for 4 h. Excess of water was added after cooling the solution to room temperature. The precipitate was collected by filtration and subjected to silica gel column chromatography using ethyl acetate and methanol (5 : 1) as eluent. Yellow-green crystals of [(pdpz)2Ir(azppz)] (1) were obtained from repeated recrystallization using a mixture of THF and pentane at room temperature (70 mg, 0.07 mmol, 41%).Single crystal X-ray diffraction data were acquired on a Bruker Smart CCD diffractometer using λ(Mo Kα) radiation (λ = 0.71073 Å). Data collection was executed using the SMART program. Cell refinement and data reduction were made with the SAINT program. The structure was determined using the SHELXTL/PC program and refined using full-matrix least squares. All non-hydrogen atoms were refined anisotropically, whereas hydrogen atoms were placed at the calculated positions and included in the final stage of refinements with fixed parameters. Selected crystal data of 1: C58H77IrN8O8, M = 1206.48, triclinic, space group P-1, a = 10.3194(7), b = 16.8549(12), c = 18.3003(13) Å, α = 63.527(2), β = 89.840(2), γ = 76.006(2)°, V = 2744.4(3) Å3, Z = 2, ρcalcd = 1.460 gcm−1, F(000) = 1244, crystal size = 0.35 × 0.17 × 0.08 mm, λ (Mo Kα) = 0.7107 Å, T = 150(1) K, µ = 2.495 mm−1, 12566 reflections collected (Rint = 0.0645), final R1[I > 2((I)] = 0.0558 and wR2(all data) = 0.1406.
Preparation of [(pdpz)2Ir(dappz)] (2)
A mixture of [(pdpz)2IrCl]2 (100 mg, 0.088 mmol), NMe2 substituted pyridyl pyrazole (dappzH, 51 mg, 0.19 mmol) and Na2CO3 (93 mg, 0.88 mmol) in 2-methoxyethanol (25 mL) was heated to reflux for 4 h. Excess of water was added after cooling the solution to room temperature. The precipitate was collected by filtration and subjected to silica gel column chromatography using ethyl acetate and methanol (5 : 1) as the eluent. Yellow-green powders of [(pdpz)2Ir(dappz)] (2) were collected after washing with acetone; yield: 50 mg, 0.063 mmol, 36%.Preparation of [Ph2B(azppz)] (3)
In a 50 mL reaction flask, a mixture of azacrown substituted pyridyl pyrazole (azppzH, 219 mg, 0.5 mmol), 4.4 mL of 2.5 M BPh3, and 20 mL of anhydrous THF solvent was heated to reflux for 24 h. The solution was then concentrated to dryness and an orange-red crystalline solid of 3 was obtained from recrystallization using a mixture of CH2Cl2 and methanol (100 mg, 0.166 mmol, 33%).Measurements
Steady-state absorption and emission spectra were recorded by a Hitachi (U-3310) spectrophotometer and an Edinburgh (FS920) fluorimeter, respectively. Emission quantum yields were measured at excitation wavelength λexc = 380 nm in CH2Cl2 at room temperature. In this approach, coumarin 480 (Exciton, Φr = 0.93 in EtOH) was used as the reference. The association constant Ka of 1 : 1 1/guest complex formation calculated by the UV-Vis absorption method is obtained by the following equation.14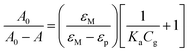 | (1) |
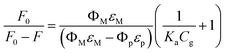 | (2) |
For the phosphorescence lifetime measurements in the microsecond region, a third harmonic of an Nd:YAG laser 355 nm (∼8 ns) was used as an excitation source. Emission decay was detected by a photomultiplier tube and averaged over 500 shots using an oscilloscope. Laser energy was reduced to <1 mJ pulse−1 to prevent possible photochemical decomposition. For the lifetime measurements of <10 ns, the fundamental train of pulses from a Ti-sapphire oscillator (82 MHz, Spectra Physics) was used to produce second harmonics (375 ∼ 425 nm, ∼120 fs) as an excitation light source. The signal was detected by a time-correlated single photon counting system (Edinburgh OB 900-L). The system response time was determined to be ∼30 ps.
Computational methodology
All calculations were performed with the Gaussian03 package.15 Geometrical optimization on the electronic ground state was carried out using the hybrid Hartree–Fock/Density functional theory (HF/DFT) method, B3LYP.16 “Double-ζ” quality basis set consisting of Hay and Wadt's effective core potentials (LANL2DZ)17 was employed for iridium atom and 6-31G* basis18 for H, C, and N atoms. A relativistic effective core potential (ECP) replaced the inner core electrons of Ir(III), leaving the outer core (5s25p6) electrons and the 5d6 valence electrons. Time-dependent DFT (TDDFT) calculations were then performed with the same functional and basis set at the optimized geometry to obtain electronic transition energies. Oscillator strengths were deduced from the dipole transition matrix elements (for singlet states only).Results and discussion
A multi-step synthetic pathway leading to the desired iridium metal complexes is depicted in Scheme 1. First of all, an azacrown substituted pyridyl pyrazole ligand, (azppz)H, was obtained from the condensation reaction of an azacrown substituted acetophenone and ethyl picolinate, followed by treatment with hydrazine monohydrate in ethanol solution (Scheme 1). The subsequent reaction of (azppz)H with the chloride bridged dimer complex [(pdpz)2IrCl]2 and slight excess of Na2CO3 in refluxing methoxyethanol solution afforded the required iridium chelate complex [(pdpz)2Ir(azppz)] (1), where (pdpz)H = 1-phenyl-3,5-dimethyl pyrazole. Moreover, the analogous dimethylamino substituted iridium derivative (2) and a BPh2 substituted complex (3) (see Scheme 1) bearing identical azppz pyrazolate ligand were synthesized from reaction with the boron reagent BPh3, and these then served as the standards for photophysical measurements.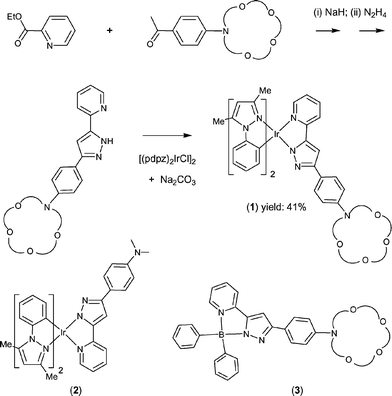 | ||
| Scheme 1 Synthetic scheme of 1 and molecular structures of 2 and 3. | ||
As indicated in Fig. 1, single crystal X-ray structural analysis of 1 shows the expected octahedral geometry around the iridium metal center, along with two N-phenyl pyrazole fragments and one azacrown substituted pyridyl pyrazolate ligand. The cyclometalated N-phenyl pyrazoles adopt an eclipse orientation, while the azacrown pyrazolate chelate is located opposite to the cyclometalated carbon atoms, with the spatial arrangement being akin to those observed for other heteroleptic pyridyl pyrazolate complexes.19 Moreover, the azacrown fragment shows unusually large deformation toward the iridium metal fragment, for which the calculated dihedral angle between the adjacent p-phenyl group and the O4N plane of azacrown is 117.5°.
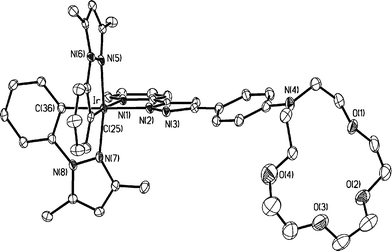 | ||
| Fig. 1 The X-ray crystal structure of 1; selected distances: Ir–C(25) = 1.984(6), Ir–C(36) = 2.020(6), Ir–N(1) = 2.155(5), Ir–N(2) = 2.112(5), Ir–N(5) = 2.023(5) and 2.021(5) Å. | ||
As shown in Fig. 2, ion-free 1 in CH3CN exhibits a 318 nm absorption band, accompanied by a shoulder at ∼375 nm. In comparison, the boron complex 3 bearing the same pyridyl pyrazolate ligand reveals two low-lying absorption bands with peak wavelengths at 315 and 375 nm, the spectral features of which are similar to that of complex 1. Due to lack of both MLCT and the transition associated with cyclometalated phenyl pyrazole ligand in boron complex 3, it is reasonable to assign both 315 and 375 nm bands as the 1ππ* transitions incorporating pyrazolates → pyridyl types of charge transfer. Transitions associated with MLCT in 1 are probably too weak and thus are hidden inside the 1ππ* bands. Further firm support is given in the theoretical approaches (vide infra). Likewise, absorption features associated with the 3ππ* intra-ligand bands and 3MLCT could not be resolved although an effective enhancement of the spin–orbit coupling from Ir is expected.
![Absorption spectrum changes of 1 (2.2 × 10−5 M) upon addition of various concentrations of anhydrous Ca(ClO4)2 in aerated CH3CN solution (a) 0, (b) 1.45 × 10−5, (c) 1.83 × 10−5, (d) 1.84 × 10−5, (e) 2.13 × 10−5, (f) 2.71 × 10−5, (g) 4.39 × 10−5, (h) 8.70 × 10−5, (i) 1.45 × 10−4, (j) 2.03 × 10−4 M. () absorption spectrum of 3 in CH3CN. Insert: the plot of A0/A0
−
A against 1/[Ca2+] at 320 nm.](/image/article/2006/OB/b511943j/b511943j-f2.gif) | ||
Fig. 2 Absorption spectrum changes of 1 (2.2 × 10−5 M) upon addition of various concentrations of anhydrous Ca(ClO4)2 in aerated CH3CN solution (a) 0, (b) 1.45 × 10−5, (c) 1.83 × 10−5, (d) 1.84 × 10−5, (e) 2.13 × 10−5, (f) 2.71 × 10−5, (g) 4.39 × 10−5, (h) 8.70 × 10−5, (i) 1.45 × 10−4, (j) 2.03 × 10−4 M. (![[dash dash, graph caption]](https://www.rsc.org/images/entities/char_e091.gif) ) absorption spectrum of 3 in CH3CN. Insert: the plot of A0/A0
−
A against 1/[Ca2+] at 320 nm. ) absorption spectrum of 3 in CH3CN. Insert: the plot of A0/A0
−
A against 1/[Ca2+] at 320 nm. | ||
The emission spectrum of 1 in CH3CN is depicted in Fig. 3 and the corresponding relaxation dynamics are listed in Table 1. The emission band with a peak wavelength at 560 nm revealed a drastic oxygen quenching effect, the intensity of which decreased from 0.22 in degassed CH3CN, to ∼1.0 × 10−3 upon aeration. Likewise, the corresponding observed lifetime decreased from 8 µs (degassed) to 38 ns (aerated, see Table 1). The ∼1/9 diffusion controlled rate of O2 quenching, in combination with a long radiative decay rate of 3.5 × 104 s−1, leads to an unambiguous conclusion that 1 exhibits predominantly the phosphorescence resulting from the enhancement of Ir(III) spin–orbit coupling.
| PL λmax | Q.Y.a | lifetime (τ)a | K a | |
|---|---|---|---|---|
| a data in parentheses are measured in the degassed solution. | ||||
| 1 | 560 nm | 0.001 (0.22) | 38 ns (8.2 µs) | |
| 1/Ca2+ | 520 nm | 0.012 (0.20) | 42 ns (0.6 µs) | 4.2 × 104 |
| 1/Mg2+ | 518 nm | 0.013 (0.23) | 48 ns (0.8 µs) | 1.6 × 105 |
| 1/Ba2+ | 521 nm | 0.010 (0.20) | 41 ns (0.6 µs) | 5.3 × 103 |
| 1/Na+ | 520 nm | 0.012 (0.18) | 40 ns (0.5 µs) | 4.7 × 103 |
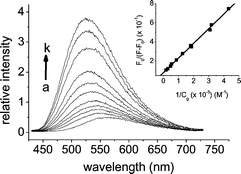 | ||
| Fig. 3 The emission spectrum changes of 1 (2.2 × 10−5 M) upon addition of various concentrations of anhydrous Ca(ClO4)2 in aerated CH3CN solution (a) 0, (b) 3.15 × 10−6, (c) 3.25 × 10−6, (d) 5.40 × 10−6, (e) 5.57 × 10−6, (f) 7.89 × 10−6, (g) 1.16 × 10−5, (h) 2.09 × 10−5, (i) 3.48 × 10−5, (j) 5.8 × 10−5, (k) 1.85 × 10−4 M. λex: 300 nm. Insert: the plot of F0/F − F0 against 1/Cg at 520 nm. | ||
Upon addition of Ca2+, the absorption and emission titration spectra of 1 (2.2 × 10−5 M) in CH3CN are shown in Figs. 2 and 3, respectively. Increasing [Ca2+] leads to a hypsochromic shift of the absorption profile, in which the appearance of an isosbestic point at ∼295 nm verifies a two-species equilibrium. The 1 : 1 1/Ca2+ complexation was supported by a straight-line plot for the ratio of absorbance, A0/(A0 − A), versus 1/[Ca2+] (see the experimental section) throughout the titration, and an association constant Ka of 4.0 × 104 M−1 was thus deduced in CH3CN. Likewise, drastic changes on the Ca2+ phosphorescence titration spectra were also observed. Upon excitation at the isosbestic point of 295 nm, the 560 nm phosphorescence was gradually blue shifted toward 520 nm, accompanied by the increase of the emission intensity. Taking the emission peak intensity of Ca2+-free and Ca2+-added complex 1 to be F0 and F, respectively, a straight line plot of F0/(F − F0) versus 1/[Ca2+] (see the experimental section) at e.g. 520 nm is depicted in the insert of Fig. 3. The deduced Ka value of 4.2 × 104 M−1, within experimental error, is in agreement with that extracted from the absorption titration.
It should be noted that both complexes 2 and 3 showed negligible changes in their absorption and emission spectra upon addition of Ca2+, manifesting the importance of the coexistence of Ir and 1-aza-15-crown-5-ether in complex 1 toward the Ca2+ recognition. The results can be rationalized by weakening the electron donating ability of the aza-nitrogen upon Ca2+/azacrown complex formation and consequently greatly alters the photophysical properties of 1. This viewpoint can be firmly supported by theoretical modelling. Presently, ab initio calculation on 1 is formidable due to its structural complexity. Alternatively, the dimethyl amino analogue 2 was selected, of which the major ligand chromophores remain intact with respect to 1. Furthermore, the protonated form of 2, 2H+, serves as a model for the Ca2+ bonded complex 1. We then applied density functional theory incorporating B3LYP method with 6-31G* basis for non-metal atoms and a relativistic effective core potential for the inner core electrons of Ir(III) metal atom. The resulting frontier orbitals for the low-lying transitions revealed drastic differences between 2 and 2H+. As shown in Fig. 4, the lowest triplet manifold for 2 exhibits predominantly HOMO → LUMO intraligand charge transfer (ILCT) transition, in which HOMO and LUMO are mainly located at the phenyl pyrazolate and pyridyl chromophores, respectively. In sharp contrast, upon protonation (2H+) the transition switches greatly to a ligand–ligand charge transfer (LLCT) incorporating cyclometalated phenyl pyrazole ligand (HOMO) → p-dialkylamino-phenyl pyrazolate (LUMO). Assuming that the differences in photophysical behavior between 2 and 2H+ can be likewise applied to 1 and 1/Ca2+ complex, changes of absorption and emission spectra during Ca2+ titration are thus be rationalized by the swap of lowest transition from ILCT (p-dialkylaminophenyl pyrazolate → pyridine) in 1 to LLCT (cyclometalated phenyl pyrazolate → p-dialkylaminophenyl pyrazolate) in 1/Ca2+ complex.
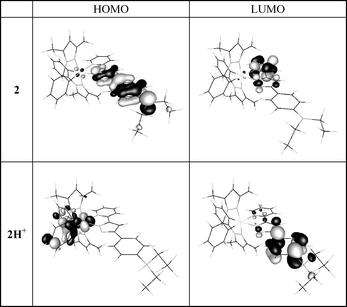 | ||
| Fig. 4 Frontier orbitals of complexes 2 and 2H+, see text for detail description. | ||
Similar titration experiments have been performed for the hard, bivalent metal cation such as Mg2+ and Ba2+ and Ka values of 1.6 × 105 M−1 and 5.3 × 103 M−1, respectively, were obtained. In another approach, negligible changes of absorption spectra were observed for soft divalent ions like Hg2+. Titration experiments were also performed for Na+ and the results indicated a Ka value of 4.7 × 103 M−1 for the 1/Na+ complex formation. The resulting metal ion dependent association strengths can be qualitatively rationalized by the amount of charge density, ρ, specified as ρ = g/(4/3πr3) where g and r are the corresponding formal charge (+1 or +2) and radius, respectively. Evidently, the value of ρ(Mg2+ ∼ 0.75, Ca2+ ∼ 0.24, Na+ ∼ 0.10 and Ba2+ ∼ 0.13)20 correlates well with the trend of Ka values deduced experimentally (see Table 1). Furthermore, negligible spectral changes were observed for K+ throughout [K+] of 10−2 M. This result can simply be rationalized by the mismatched sizes between K+ and the 1-aza-15-crown-5 ether.
In solid form 1 exhibits a 565 nm phosphorescent emission with a quantum yield as high as 0.25 (τp ∼ 12 µs). Thus, from the viewpoint of application, we have also examined whether a similar recognition capability can be applied in the heterogeneous solid film in aqueous solution. Note that 1 is insoluble in water. To simplify the process, a silica-based TLC plate was used as a solid support to soak 1 in CH3CN so that a complex 1 coated TLC plate was prepared with an optical density of ∼1.0 at 350 nm. This plate was then dipped into an aqueous solution (pH ∼ 7.0) containing ∼10−3 M CaClO4 for ∼30 s, and then vacuum-dried to remove water. With the use of a commercially available UV-lamp (366 nm) as an excitation source, a photograph in TOC demonstrates a salient change of emission from yellow-orange for the Ca2+-free, complex 1 coated TLC plate to a bright green emission upon complexation with the Ca2+ ion.
Conclusions
In conclusion, we demonstrate a novel metal ion sensor, complex 1, based on the rare metal-ion sensitive phosphorescence. In this case, the chromophores designed act as both recognition and signal-transducing units, while the center heavy metal, i.e. Ir(III), serves as a perturbing base to enhance the phosphorescence. Thus, the current system is versatile in that functional derivatization can be achieved with methods similar to those strategically designed for the singlet ππ* (i.e. fluorescence) ligand chromophores. In view of drastic oxygen quenching, in a steady state approach, one can then saturate the solution with N2 so that the enhanced phosphorescence can also serve as an additional signalling to distinguish it from the fluorescence interference. Alternatively, in a time-resolved manner, due to its much longer lifetime (even in the aerated solution) than that of typical fluorescence, the associated phosphorescence can be obtained free from fluorescence interferences in the solution simply by acquiring the phosphorescence only after a certain time delay of the excitation pulse. The success in the recognition of Ca2+(aq) in the TLC plate demonstrates its suitability for the future development of a practical device, such as a metal ion sensor anchored on cellular membranes. We thus believe that results presented in this study may spark a broad range of interest in both fundamental approach and applications relevant to the third-row transition metal complexes.Acknowledgements
This work was funded by the National Science Council of Taiwan, R.O.C. under grants: NSC 93-2113-M-007-012 and NSC 93-2752-M-002-002-PAE. We are also grateful to the National Center for High-Performance Computing for generous amounts of computing time.References
- (a) H.-G. Löhr and F. Vögtle, Acc. Chem. Res., 1985, 18, 65 CrossRef; (b) W.-S. Xia, R. H. Schmehl and C.-J. Li, J. Am. Chem. Soc., 1999, 121, 5599 CrossRef CAS; (c) B. Valeur and I. Leray, Coord. Chem. Rev., 2000, 205, 3 CrossRef; (d) A. P. de Silva, D. B. Fox, A. J. M. Huxley and T. S. Moody, Coord. Chem. Rev., 2000, 205, 41 CrossRef CAS; (e) A. P. de Silva, D. B. Fox, T. S. Moody and S. M. Weir, Pure Appl. Chem., 2001, 73, 503 CrossRef.
- (a) K. S. Schanze, D. B. MacQueen, T. A. Perkins and L. A. Cabana, Coord. Chem. Rev., 1993, 122, 63 CrossRef CAS; (b) G. W. Gokel, W. M. Leevy and M. E. Weber, Chem. Rev., 2004, 104, 2723 CrossRef CAS; (c) S.-W. Lai and C.-M. Che, Top. Curr. Chem., 2004, 241, 27 CAS.
- (a) A. P. de Silva, H. Q. Nimal Gunaratne, T. Gunnlaugsson, A. J. M. Huxley, C. P. McCoy, J. T. Rademacher and T. E. Rice, Chem. Rev., 1997, 97, 1515 CrossRef; (b) P. D. Beer and E. J. Hayes, Coord. Chem. Rev., 2003, 240, 167 CAS.
- (a) R. Slone, D. I. Yoon, R. M. Calhoun and J. T. Hupp, J. Am. Chem. Soc., 1995, 117, 11813 CrossRef CAS; (b) Y. Shen and B. P. Sullivan, Inorg. Chem., 1995, 34, 6235 CrossRef CAS; (c) V. W.-W. Yam, C.-L. Chan, C.-K. Li and K. M.-C. Wong, Coord. Chem. Rev., 2001, 216–217, 173 CrossRef CAS; (d) V. W.-W. Yam, R. P.-L. Tang, K. M.-C. Wong, X.-X. Lu, K.-K. Cheung and N. Zhu, Chem. Eur. J., 2002, 8, 4066 CrossRef CAS; (e) L. J. Charbonniere, R. F. Ziessel, C. A. Sams and A. Harriman, Inorg. Chem., 2003, 42, 3466 CrossRef CAS; (f) P. K. M. Siu, S.-W. Lai, W. Lu, N. Zhu and C.-M. Che, Eur. J. Inorg. Chem., 2003, 2749 CrossRef CAS; (g) Q.-Z. Yang, L.-Z. Wu, H. Zhang, B. Chen, Z.-X. Wu, L.-P. Zhang and C.-H. Tung, Inorg. Chem., 2004, 43, 5195 CrossRef CAS.
- (a) D. B. MacQueen and K. S. Schanze, J. Am. Chem. Soc., 1991, 113, 6108 CrossRef; (b) C. E. Whittle, J. A. Weinstein, M. W. George and K. S. Schanze, Inorg. Chem., 2001, 40, 4053 CrossRef CAS.
- (a) J. F. Michalec, S. A. Bejune, D. G. Cuttell, G. C. Summerton, J. A. Gertenbach, J. S. Field, R. J. Haines and D. R. McMillin, Inorg. Chem., 2001, 40, 2193 CrossRef CAS; (b) J. D. Lewis, L. Bussotti, P. Foggi, R. N. Perutz and J. N. Moore, J. Phys. Chem. A, 2002, 106, 12202 CrossRef CAS; (c) J. D. Lewis and J. N. Moore, Dalton Trans., 2004, 1376 RSC; (d) W.-S. Tang, X.-X. Lu, K. M.-C. Wong and V. W.-W. Yam, J. Mater. Chem., 2005, 15, 2714 RSC.
- A. B. Tamayo, B. D. Alleyne, P. I. Djurovich, S. Lamansky, I. Tsyba, N. N. Ho, R. Bau and M. E. Thompson, J. Am. Chem. Soc., 2003, 125, 7377 CrossRef CAS.
- (a) Y. Shen and B. P. Sullivan, J. Chem. Educ., 1997, 74, 685 CrossRef CAS; (b) P. D. Beer, S. W. Dent, N. C. Fletcher and T. J. Wear, Polyhedron, 1996, 15, 2983 CrossRef CAS; (c) M. E. Padilla-Tosta, J. M. Lloris, R. Martinez-Manez, M. D. Marcos, M. A. Miranda, T. Pardo, F. Sancenon and J. Soto, Eur. J. Inorg. Chem., 2001, 1475 CrossRef CAS.
- T. Lazarides, T. A. Miller, J. C. Jeffery, T. K. Ronson, H. Adams and M. D. Ward, Dalton Trans., 2005, 528 RSC.
- H. Maeda, S. Furuyoshi, Y. Nakatsuji and M. Okahara, Bull. Chem. Soc. Jpn., 1983, 56, 212 CAS.
- Y. Chen and G. L. Baker, J. Org. Chem., 1999, 64, 6870 CrossRef CAS.
- K. Y. Lee, J. M. Kim and J. N. Kim, Tetrahedron Lett., 2003, 44, 6737 CrossRef CAS.
- (a) M. Nonoyama, J. Organomet. Chem., 1975, 86, 263 CrossRef CAS; (b) C.-H. Yang, S.-W. Li, Y. Chi, Y.-M. Cheng, Y.-S. Yeh, P.-T. Chou, G.-H. Lee, C.-H. Wang and C.-F. Shu, Inorg. Chem., 2005, 44, 7770 CrossRef CAS.
- (a) P.-T. Chou, G.-R. Wu, C.-Y. Wei, C.-C. Cheng, C.-P. Chang and F.-T. Hung, J. Phys. Chem. B, 1999, 103, 10042 CrossRef CAS; (b) P.-T. Chou, G.-R. Wu, C.-Y. Wei, C.-C. Cheng, C.-P. Chang and F.-T. Hung, J. Phys. Chem. B, 2000, 104, 7818 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Montgomery, Jr., T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. G. Johnson, W. Chen, M. W. Wong, C. Gonzalez and J. A. Pople, GAUSSIAN 03 (Revision C.02), Gaussian, Inc., Wallingford, CT, 2004 Search PubMed.
- (a) A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS; (b) C. Lee, W. Yang and R. G. Parr, Phys. Rev. B, 1988, 37, 785 CrossRef CAS.
- (a) T. H. Dunning and P. J. Hay, In Modern Theoretical Chemistry, ed. H. F. Schaefer, III, Plenum Press, New York, 1976, vol. 3, pp. 11 Search PubMed; (b) P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 270 CrossRef CAS; (c) W. R. Wadt and P. J. Hay, J. Chem. Phys., 1985, 82, 284 CrossRef CAS; (d) P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 299 CrossRef CAS.
- P. C. Hariharan and J. A. Pople, Mol. Phys., 1974, 27, 209 CAS.
- (a) Y.-H. Song, S.-J. Yeh, C.-T. Chen, Y. Chi, C.-S. Liu, J.-K. Yu, Y.-H. Hu, P.-T. Chou, S.-M. Peng and G.-H. Lee, Adv. Funct. Mater., 2004, 14, 1221 CrossRef CAS; (b) F.-M. Hwang, H.-Y. Chen, P.-S. Chen, C.-S. Liu, Y. Chi, C.-F. Shu, F.-I. Wu, P.-T. Chou, S.-M. Peng and G.-H. Lee, Inorg. Chem., 2005, 44, 1344 CrossRef CAS.
- Q.-Z. Yang, Q.-X. Tong, L.-Z. Wu, Z. X. Wu, L. P. Zhang and C. H. Tung, Eur. J. Inorg. Chem., 2004, 1948 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2006 |