Chemoenzymatic synthesis of GM3 and GM2 gangliosides containing a truncated ceramide functionalized for glycoconjugate synthesis and solid phase applications†
Sandra Jacques, Jamie R. Rich, Chang-Chun Ling and David R. Bundle*
Alberta Ingenuity Centre for Carbohydrate Science, Department of Chemistry, University of Alberta, Edmonton, Alberta, Canada T6G 2G2. E-mail: dave.bundle@ualberta.ca
First published on 30th November 2005
Abstract
Analogues of GM3 and GM2 gangliosides were chemoenzymatically synthesized on a multifunctional ceramide-type tether designed to facilitate diverse strategies for glycoconjugate synthesis. The truncated ceramide aglycon maintains the stereogenic centres of natural ceramide while avoiding extensive hydrophobicity that can hamper synthesis and purification of the glycolipids. Tetanus toxoid and BSA glycoconjugates of these two gangliosides were prepared for immunization of mice, and for solid phase assays to screen for ganglioside-specific antibodies. Inhibition experiments showed that antibodies generated by tetanus toxoid conjugates of GM3 and GM2 exhibited specificity for the carbohydrate epitope and the stereogenic centres of the ceramide.
Introduction
Oligosaccharides at the surface of mammalian cells act as key elements in recognition processes occurring during embryogenesis, metastasis, inflammation and pathogen recognition. Among other glycoconjugates at the cell surface, gangliosides, a group of complex sialylated glycosphingolipids, play a significant role in a variety of cell functions. They have been proposed to control growth and differentiation of cells, mediate cell–cell interactions, regulate cell signalling by acting as receptors for growth factors, and act as specific receptors for various microbial toxins.1 Furthermore, these glycolipids have found wide applications in oligosaccharide microarrays,2 investigations of sialic acid binding proteins,3 immunotherapy,4 and in the study of cell membrane events. Their importance in biological recognition requires efficient methods for their preparation.The synthesis of gangliosides is particularly challenging due to their amphiphilic nature and the difficulties associated with stereoselective coupling of N-acetyl neuraminic acid.5 Enzymatic methods have proved to be very useful in the synthesis of sialylated structures,6 avoiding many of the problems associated with chemical approaches. Due to their hydrophobic lipid chain and their hydrophilic head group, gangliosides exhibit surfactant-like properties such as low solubility in organic solvents and aqueous solutions, as well as aggregation, resulting in low reaction yields and purification problems.7 Several lines of evidence in the literature relating to ganglioside recognition by proteins, implicate the ceramide portion of glycolipids either directly or via its influence on the orientation of the polar head group when the ganglioside is located in the mammalian cell membrane.8 In order to build a capability to include tandem recognition of the ceramide and the carbohydrate, we developed a truncated version of sphingosine.9 This moiety confers increased aqueous solubility and allows for the elaboration with different lipid chains at a late stage of the synthesis, thereby facilitating manipulation of the intermediates involved.
Gangliosides, in particular GM3, GM2, GD3 and GD2 (Fig. 1) are highly overexpressed in melanoma and other tumors of neuroectodermal origin, making these structures interesting targets for active cancer immunotherapy.4a,c,10 Unfortunately, gangliosides are poorly immunogenic, making their use as immunogens impractical and the preparation of anti-ganglioside antibodies for passive immunization difficult. Conjugation of an oligosaccharide epitope to an immunogenic carrier protein has been widely used as a strategy to enhance the immunogenicity of otherwise T-cell independent carbohydrate antigens, and has been applied to augment the immunogenicity of gangliosides.11 Thus far, vaccines based on naturally occurring gangliosides or semi-synthetic gangliosides conjugated to keyhole limpet hemocyanin (KLH) have displayed only limited success in clinical trials.4b,c,12 These results have motivated new strategies for the design of ganglioside-based vaccines in order to improve their immunogenicity.4b,c,12,13 In this context, ganglioside analogues containing functional groups that facilitate flexible conjugation strategies, constitute a valuable tool for the design and development of cancer vaccines as well as for the study and modulation of ganglioside functions at membrane interfaces.
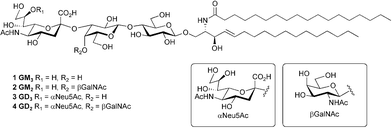 | ||
| Fig. 1 Structure of gangliosides GM3 (1), GM2 (2), GD3 (3), and GD2 (4). | ||
Here we address some of these issues and report the chemoenzymatic synthesis of versatile GM3 and GM2 ganglioside analogues designed for conjugation via their ceramide moiety. These compounds contain a truncated ceramide aglycon9 which enhances water solubility of the molecule while conserving the important stereochemical and structural features of naturally occurring ceramides. The inherent flexibility to functionalize the ganglioside analogues with different linkers for covalent attachment to proteins, surfaces, or fluorescent probes, is demonstrated by the synthesis of tetanus toxoid (TT) and bovine serum albumin (BSA) glycoconjugates of GM3 and GM2. The immune response to both conjugates shows a good IgG response in mice, directed against the sialylated portion of the molecule and the ceramide moiety.
Results and discussion
Our chemoenzymatic approach was centred on a versatile lactosyl ceramide analogue 6, which could be elaborated to gangliosides of higher complexity or functionalized with different acyl chains at the sphingosine amine. Intermediate 6 was synthesized as outlined in Scheme 1. The azide 5 was reduced in the presence of hydrogen sulfide in a mixture of pyridine, water and triethylamine.9,14 The use of the free amine in the next enzymatic step has proven not to be very practical in our hands. Therefore, we envisioned that protection of the amine group would ease purification of the compound in the subsequent enzymatic reactions. Due to the acid lability of the N-acetyl neuraminic acid residue,15 an amine protecting group such as trifluoroacetamide, which could be removed under mild basic conditions, was preferred. The free amine was acylated by reaction with methyl trifluoroacetate to yield 6.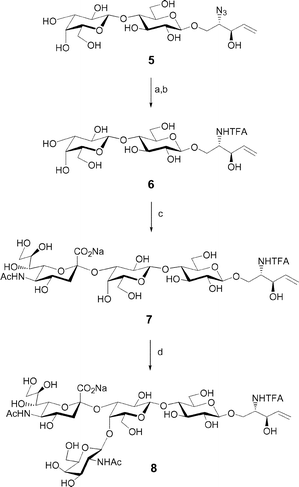 | ||
| Scheme 1 Reagents and conditions: (a) H2S, pyridine–H2O–Et3N (10 : 1 : 0.3), 87%; (b) methyl trifluoroacetate, MeOH, 75%; (c) α-(2,3)-Neu5Ac transferase, CMP-Neu5Ac, 88%; (d) β-(1,4)-GalNAc transferase, UDP-GlcNAc 4-epimerase, UDP-GlcNAc, 95%. | ||
Enzymatic synthesis of oligosaccharides obviates many of the problems associated with chemical synthesis, including laborious protecting group manipulations and separation of numerous diastereomeric glycosylation products. We envisioned elaborating our ganglioside analogues from a lactose core using sialyltransferase- and N-acetyl-galactosaminyl transferase-catalyzed reactions. A major drawback of glycosylation reactions catalyzed by glycosyltransferases is their requirement for expensive nucleotide sugar donors. However, we have employed recombinant CMP-Neu5Ac synthetase and UDP-GlcNAc 4-epimerase in the production of glycosyl donors. The crude product obtained was subsequently used to effect sialylation of the acceptor 6 catalyzed by a recombinant α-(2,3)-sialyltransferase from Campylobacter jejuni to obtain 7 in good yield.16 The carbohydrate portion of intermediate 7 was further elaborated via an enzymatic glycosylation yielding the GM2 analogue 8 (Scheme 1). Using a recombinant β-(1,4)-N-acetylgalactosaminyl transferase6 and UDP-GlcNAc 4-epimerase17 from Campylobacter jejuni, GM2 analogue 8 was produced from acceptor 7 in excellent yield.
The immobilization of oligosaccharides on solid surfaces and particles as well as their conjugation to proteins, are important tools in glycobiology for the study and modulation of carbohydrate functions. Oligosaccharide intermediates bearing a functional group designed for conjugation are valuable compounds for such undertakings. To this end, GM3 and GM2 analogues containing a functionalized N-acyl chain were synthesized from intermediates 7 and 8 (Scheme 2). Deprotection of the sphingosine amine followed by reaction with a succinimide-activated octadecanoate18 yielded compound 9. It can be envisioned that the long acyl chain could be used to non-covalently coat microtiter plates via hydrophobic interactions.19 Furthermore, following protection, the truncated ceramide could be extended to a natural ceramide via olefin metathesis.20 Alternatively, the alkene portion of the truncated ceramide could serve as a handle for protein conjugation via radical addition of cysteamine to the terminal olefin.9,21 In an analogous fashion, compounds 7 and 8 were converted to GM3 and GM2 mimics 10 and 11, which contain an azide group at the N-acyl chain terminus. These ganglioside analogues could be covalently attached to alkyne-functionalized surfaces via 1,4-dipolar cycloaddition or coupled to proteins or functionalized surfaces upon reduction of the azide to an amine. Similarly, compounds 7 and 8 were converted to 12 and 13, which bear a terminal thioester. Upon deprotection of the thiol, these molecules would be suitable for attachment to gold surfaces or nanoparticles, and for conjugation to proteins.
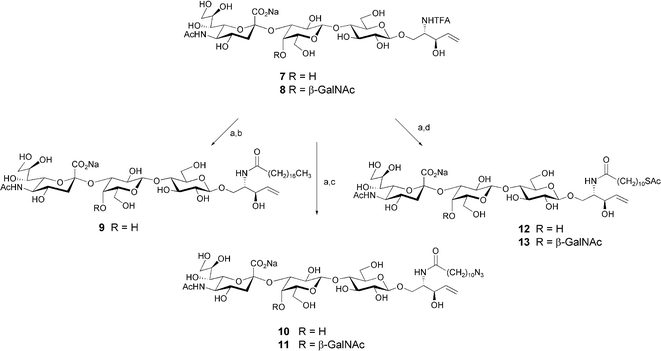 | ||
| Scheme 2 Reagents and conditions: (a) NaOMe, MeOH, 40 °C; (b) N-(octadecanoyloxy)succinimide, 4-N,N-dimethylaminopyridine, pyridine–H2O (9 : 1), 40 °C, 55%; (c) N-(11-azidoundecanoyloxy)succinimide, 4-N,N-dimethylaminopyridine, pyridine–H2O (9 : 1), 40 °C, 78% for 10, 76% for 11; (d) N-(11-thioacetylundecanoyloxy)succinimide, 4-N,N-dimethylaminopyridine, pyridine–H2O (9 : 1), 40 °C, 80% for 12, 95% for 13. | ||
The method described above for accessing GM3 and GM2 analogues from a lactose core, was particularly useful for the synthesis of compounds incorporating a long hydrophobic chain at the sphingosine nitrogen. However, compounds functionalized with a shorter acyl chain could be synthesized via an alternate pathway as shown in Scheme 3. The azide group of 14 was reduced under Staudinger conditions.9 Acylation of the amine with p-nitrophenyl 6-trifluoroacetamidohexanoate followed by deprotection, afforded compound 15. Alternatively, the benzoylated lactosyl azidosphingosine 14 could be deprotected and reduced in the presence of H2S. Reaction of the amine with butyric anhydride afforded 16. The lactosyl ceramides functionalized with shorter acyl chains were soluble in aqueous solutions and thus could be used as acceptors in enzymatic glycosylation reactions. Reaction in the presence of α-(2,3)-sialyltransferase yielded GM3 analogues 17 and 18, which could be further elaborated to GM2 analogues 19 and 20 using β-(1,4)-N-acetylgalactosaminyl transferase.
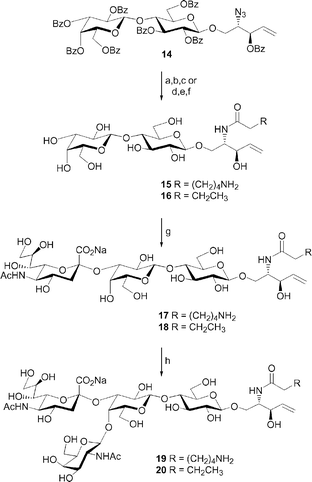 | ||
| Scheme 3 Reagents and conditions: (a) PPh3, pyridine–H2O (9 : 1), 50 °C; (b) p-nitrophenyl 6-trifluoroacetamidohexanoate, pyridine–H2O (9 : 1), 50 °C; (c) NaOMe, MeOH, 58%, three steps; (d) NaOMe, MeOH; (e) H2S, pyridine–H2O–Et3N (10 : 1 : 0.3); (f) butyric anhydride, MeOH, 40 °C, 71%, three steps; (g) α-(2,3)-Neu5Ac transferase, CMP-Neu5Ac, 80% for 17, 79% for 18; (h) β-(1,4)-GalNAc transferase, UDP-GlcNAc 4-epimerase, UDP-GlcNAc, 79% for 19, 83% for 20. | ||
Recent work from our group has focused on the incorporation of thiooligosaccharides in anti-cancer vaccines as a means to increase their immunogenicity.22 We hypothesized that the metabolic stability of the S-linked glycans23 could increase the immunogenicity of the conjugates, while producing antibodies cross-reactive with the corresponding O-linked antigen. In order to compare the immunogenicity, cross-reactivity, and metabolic stability of the S-linked GM3 and GM2 antigens, their O-linked counterparts were required. Evaluation of the response against the immunogen by ELISA necessitates the synthesis of a set of glycoconjugates, incorporating heterologous proteins and linkers as well as water-soluble oligosaccharides. The methods described herein were used to rapidly access GM3 (25 and 26), and GM2 (27) conjugates for immunization and solid phase assays (ELISA).
The use of tetanus toxoid in commercial carbohydrate conjugate vaccines and our recent successes in inducing high antibody titers against tetanus toxin glycoconjugates, motivated the use of this carrier protein for our studies.24 Typical strategies for the generation of a conjugate vaccine involve the coupling of a carbohydrate antigen via a linker to reactive groups at the surface of a carrier protein.25 Ideally, the coupling chemistry should allow for appropriate levels of substitution, while using a minimal amount of precious synthetic hapten. To this end, an adipate-based linker26 was chosen to effect conjugation of haptens 17 and 19, without introducing a potentially immunogenic tether. The generation of the GM3 and GM2 conjugates (Scheme 4) first involved the introduction of an amine-terminated spacer unit. Treatment of intermediates 7 and 8 with sodium methoxide in methanol yielded the corresponding amines. N-Acylation with 6-trifluoroacetamidohexanoic anhydride afforded intermediates 21 and 22. Following deprotection of the primary amine, compounds 17 and 19 were functionalized for conjugation to proteins. Reaction of 17 and 19 with p-nitrophenyl adipate diester in a mixture of N,N-dimethylformamide and triethylamine followed by reverse phase HPLC purification in the presence of 1% acetic acid afforded activated ganglioside analogues 23 and 24 for protein coupling. Incubation of tetanus toxoid with approximately 30 equivalents of the activated sugars yielded glycoconjugates 25 and 27 with hapten incorporation levels of 6 and 7 respectively.
 | ||
| Scheme 4 Reagents and conditions: (a) NaOMe, MeOH, 40 °C; (b) 6-trifluoroacetamidohexanoic anhydride, MeOH, 78% for 21, 78% for 22, two steps; (c) NaOMe, MeOH, 40 °C, 72% for 17, 73% for 19; (d) p-nitrophenyl adipate diester, DMF–Et3N; (e) tetanus toxoid, PBS, pH 7.2 (f) bovine serum albumin, water. | ||
The tetanus toxoid glycoconjugates 25 and 27 were used to immunize mice and their ability to raise an immune response was evaluated by ELISA. Synthetic glycoconjugates possessing a heterologous protein were required to detect carbohydrate-specific antibodies by solid phase assays (ELISA). For this purpose, ganglioside analogue 17 was coupled to bovine serum albumin using a similar approach to the one used to generate tetanus toxoid conjugates (Scheme 4). A hapten incorporation level of 5 was obtained for the GM3-bovine serum albumin conjugate. In order to perform a more stringent assay for carbohydrate-specific antibodies and to exclude the possibility of tether-specific antibodies, we sought a glycoconjugate consisting of the same carbohydrate hapten conjugated to a heterologous protein via a tether that was distinct from that of the immunizing agent. Bovine serum albumin conjugates containing a squarate linker were prepared from intermediates 17 and 19 to address this requirement.22b
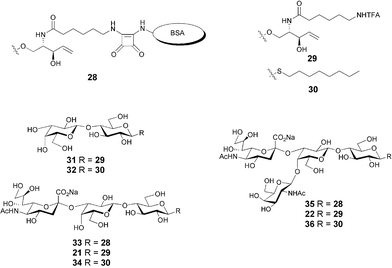
Preliminary experiments established that mice immunized with either GM3-TT conjugate 25 or GM2-TT conjugate 27 had a high titer of glycolipid-specific antibodies.24c Mice immunized with GM3-TT conjugate 25 responded with an IgG titer of 104.24c In order to establish that the antibodies recognize sialoside-containing epitopes and not just the internal segment of the oligosaccharide hapten, inhibition of antibody binding to glycoconjugate-coated ELISA plates was performed with a panel of oligosaccharides (Fig. 2). These results showed that antibody binding had a strict requirement for the terminal sialic acid residue. GM3 derivative 21 was at least 1000-fold more effective inhibitor than the lactosyl derivative 31 (IC50 of 0.39 µM vs. >1000 µM, data not shown). The ceramide portion of the molecule also has an effect on antibody binding, since substitution of the truncated sphingosine aglycon by an S-octyl analogue resulted in a 100-fold decrease in the IC50 (Fig. 3).
 | ||
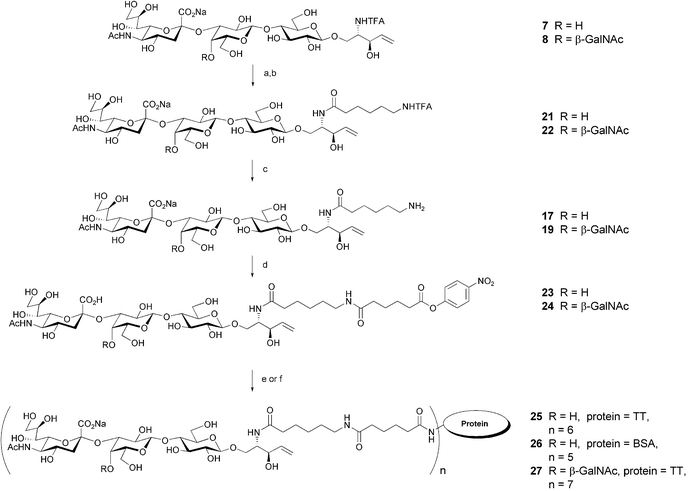
| Fig. 2 Structure of immobilized glycoconjugates (33 and 35) and soluble inhibitors (21, 22, 31, 32, 34 and 36) used for inhibition ELISA experiments. | ||
 | ||
| Fig. 3 ELISA inhibition experiments with serum obtained from mice immunized with GM3-TT conjugate 25. Synthetic oligosaccharides 21 and 34 were used to inhibit antibody binding to immobilized GM3 conjugate 33. | ||
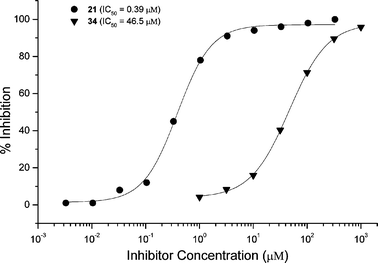
Similar results were observed for mice immunized with GM2-TT conjugate 27. Sera from immunized mice were titered against an immobilized GM2-BSA conjugate (35). The mice responded to immunization with an IgG titer of ∼105.24c Further study of the binding epitope by ELISA inhibition experiments indicated that both the terminal sialic acid residue and the N-acetylgalactosamine residue were required for antibody binding (Fig. 4). Comparison of IC50 values for compounds containing the truncated ceramide and for compounds containing the S-octyl aglycon again indicated that the ceramide portion of the molecule is also recognized by antibodies. Substitutions at the ceramide end of the molecule affect antibody binding to a smaller extent than either deletion of the sialic acid or the N-acetylgalactosamine residues.
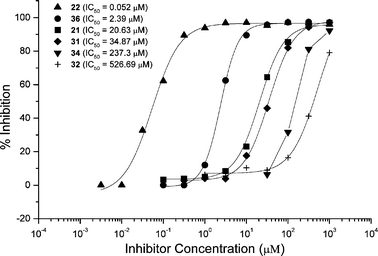 | ||
| Fig. 4 ELISA inhibition experiments with serum obtained from mice immunized with GM2-TT conjugate 27. Synthetic oligosaccharides 21, 22, 31, 32, 34, and 36 were used to inhibit antibody binding to immobilized GM2 conjugate 35. | ||
Conclusions
We have demonstrated a versatile approach for the chemoenzymatic synthesis of ganglioside analogues in a form suitable for conjugation. The truncated ceramide aglycon allows coupling of the molecule via a wide variety of conjugation strategies, while maintaining the relevant stereochemical and structural features of natural ceramide. Mice immunized with GM3-TT and GM2-TT conjugates gave a high IgG hapten-specific response. The preponderance of the IgG subtype is indicative of an antibody class switch and activation of TH cells. Immunochemical data obtained with the synthetic antigens created by this approach indicate that ceramide elements are recognized by antibodies induced by glycoconjugates 25 and 27. This suggests that it can be important to include at least the stereogenic centres of the ceramide portion of gangliosides in conjugate vaccine design. Although the main thrust of the current work was directed toward ganglioside analogues in the context of tumor immunotherapy, the design of the tether is not limited to vaccine applications. Through the introduction of different terminal functionalities, these ganglioside mimics may be tailored for applications that present glycolipids in ordered arrays and model membranes.Experimental
General methods
All chemical reagents were of reagent grade and used as supplied from Sigma-Aldrich, without further purification unless otherwise indicated. Organic solvents used in reactions were distilled under an inert atmosphere, except N,N-dimethylformamide, which was stored over 3 Å molecular sieves and subjected to reduced pressure for several minutes (<0.5mm Hg) prior to use. Pyridine was dried over solid KOH and used without further purification. Analytical thin layer chromatography (TLC) was conducted on silica gel 60-F254 (Merck). Plates were visualized under UV light, and/or by treatment with either acidic cerium ammonium molybdate or 5% ethanolic sulfuric acid, followed by heating. HPLC purification was conducted using a UV absorbance detector. Separations were performed on Beckman C18-silica, Kromasil C18-silica or Hamilton PRP-1 reverse-phase semi-preparative columns with combinations of water and methanol as eluent (flow rate 1.5–2.0 mL min−1). Size exclusion chromatography purification was performed using Biorad P-4 Biogel resin and 5% methanol–water as eluent (flow rate 0.4 mL min−1).1H NMR spectra were recorded at 500 or 600 MHz and 13C NMR spectra were recorded at 125 MHz. Chemical shifts are reported in δ (ppm) units using 13C and residual 1H signals from deuterated solvents as references. 1H and 13C NMR spectra were assigned with the assistance of COSY, HMQC, HMBC, and TOCSY spectra. NMR data for compounds containing sphingosine/ceramide and derivatives such as the aglycon were labelled as follows. Hydrogen atoms of sphingosine are denoted H–Xsphing, where numbering begins with H-1 at the glycosyl linkage. The cis and trans olefinic hydrogen atoms of the truncated lipid are denoted H–Xcsphing and H–Xtsphing respectively. Similarly, the axial and equatorial protons at the 3-position of the neuraminic acid unit are denoted H-3a and H-3e respectively. For some compounds, the coupling pattern of the H-2 of the glucopyranosyl unit is reported as a multiplet, due to high order coupling.
Electro-spray ionization (ESI) and MALDI mass spectra were recorded by the mass spectrometry laboratory service at the University of Alberta. For high resolution mass determination, spectra were obtained by voltage scan over a narrow range at a resolution of approximately 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000. Optical rotations were determined in a 10 cm cell at 22 ± 2 °C. [α]D values are given in units of 10−1 deg cm2 g−1.
000. Optical rotations were determined in a 10 cm cell at 22 ± 2 °C. [α]D values are given in units of 10−1 deg cm2 g−1.
Enzymatic synthesis
000 rpm, 4 °C for 60 min. The supernatant, containing CMP-Neu5Ac synthetase, was used to produce the CMP-Neu5Ac needed for enzymatic sialylation reactions.000 rpm at 4 °C for 30 min followed by ultracentrifugation of the supernatant at 40000 rpm at 4 °C for 60 min. The enzyme was recovered in the pellet, which was resuspended in 50 mM HEPES, pH 7.5 containing 10% (v/v) glycerol. The suspension was used without any further purification in subsequent sialylation reactions.000 rpm, 4 °C for 60 min. The supernatant, containing the enzyme, was used in subsequent reactions without any further purification.000 rpm, 4 °C for 20 min. The supernatant, containing the enzyme, was collected and used in subsequent reactions without any further purification.The activity of the bifunctional UDP-GlcNAc/Glc 4-epimerase was determined by a radiochemical assay in which the formation of labeled UDP-GalNAc is monitored through a glycosyltransferase reaction using octyl O-(α-D-fucopyranosyl)-(1 → 2)-O-β-D-galactopyranoside as an acceptor.
One unit of enzyme activity was defined as the amount that catalyzes the formation of 1 µmol of product per minute at 37 °C using the corresponding acceptor.
000 rpm for 20 min, the supernatant was then collected and lyophilized. The crude solid obtained was used in sialylation reactions without any further purification assuming 90% conversion of starting material.Immunization protocol
BALB/c mice were immunized by interperitoneal injection of tetanus toxoid conjugates 25 and 27 (50 µg in 100 µL PBS and 100 µL Freund's complete adjuvant, homogenized). Subsequent injections (50 µg in 100 µL PBS and 100 µL Freund's incomplete adjuvant, homogenized) were made subcutaneously on days 21 and 28. Mice were bled on day 38 and the sera were isolated by centrifugation and stored at −20 °C.Enzyme-linked immunosorbent assay (ELISA)
BSA-carbohydrate-protein conjugates (10 µg mL−1 in PBS) were used to coat 96-well microtiter plates (MaxiSorp, Nunc) overnight at 4 °C. The plate was washed 5 times with PBST (PBS containing 0.05% (v/v) Tween 20). Sera were diluted with PBST containing 0.1% BSA. The solutions were distributed on the coated microtiter plate and incubated at room temperature for 2 hours. The plate was washed with PBST (5 times) and goat anti-mouse IgG antibody conjugated to horseradish peroxidase (Kirkegaard & Perry Laboratories; 1 : 2000 dilution in 0.1% BSA–PBST; 100 µL per well) was added. The mixture was then incubated for 1 hour. The plate was washed 5 times with PBST before addition of a 1 : 1 mixture of 3,3′,5,5′-tetramethylbenzidine (0.4 g L−1) and 0.02% H2O2 solution (Kirkegaard & Perry Laboratories; 100 µL per well). After 2 minutes, the reaction was stopped by addition of 1 M phosphoric acid (100 µL per well). Absorbance was read at 450 nm.Oligosaccharide inhibition of enzyme-linked immunosorbent assay (ELISA)
Carbohydrate-protein conjugates (10 µg mL−1 in PBS) were used to coat 96-well microtiter plates (MaxiSorp, Nunc) overnight at 4 °C. The plate was washed 5 times with PBST (PBS containing 0.05% (v/v) Tween 20). The serum solution (1 : 2000 dilution in 0.1% BSA–PBST) was mixed with inhibitor dissolved in 0.1% BSA–PBST at concentrations ranging from 0.001 µM to 1 mM. The resulting solutions were added to the coated microtiter plate in triplicates, and incubated at room temperature for 2 hours. The plate was washed with PBST (5 times) and goat anti-mouse IgG antibody conjugated to horseradish peroxidase (Kirkegaard & Perry Laboratories; 1 : 2000 dilution in 0.1% BSA–PBST; 100 µL per well) was added. The mixture was then incubated for 1 hour. The plate was washed 5 times with PBST before addition of a 1 : 1 mixture of 3,3′,5,5′-tetramethylbenzidine (0.4 g L−1) and 0.02% H2O2 solution (Kirkegaard & Perry Laboratories; 100 µL per well). After 2 minutes, the reaction was stopped by addition of 1 M phosphoric acid (100 µL per well). Absorbance was read at 450 nm and percent inhibition was calculated relative to wells containing sera without inhibitor.000 rpm for 30 min and the supernatant was applied to a Sep-Pak (C18; 10 g). Gradient elution (MeOH–H2O) afforded 7. Further purification by size exclusion chromatography (Biogel P-4 resin; 50 cm × 2 cm) using 5% MeOH–H2O as an eluent yielded 7 (18.9 mg; 22.2 µmol; 88%): [α]D +5.1 (c 1.08, H2O). 1H NMR (500 MHz, D2O): δH 5.83 (ddd, 1H, J4,5t = 17.2 Hz, J4,5c = 10.4 Hz, J4,3 = 7.2 Hz, H-4sphing), 5.33 (m, 1H, H-5tsphing), 5.28 (m, 1H, H-5csphing), 4.51 (d, 1H, J1,2 = 7.8 Hz, H-1Gal), 4.47 (d, 1H, J1,2 = 8.0 Hz, H-1Glc), 4.30 (dd, 1H, J3,2 = 7.2 Hz, J3,4 = 7.2 Hz, H-3sphing), 4.22 (ddd, 1H, J2,3 = 7.1 Hz, J2,1a = 7.1 Hz, J2,1b = 3.6 Hz, H-2sphing), 4.11–4.07 (m, 2H, H-1asphing, H-3Gal), 3.98 (dd, 1H, J6a,6b = 12.2 Hz, J6a,5 = 2.1 Hz, H-6aGlc), 3.94 (d, 1H, J4,3 = 3.05 Hz, H-4Gal), 3.91–3.79 (m, 9H, H-1bsphing, H-8Neu, H-9aNeu, H-5Gal, H-9bNeu, H-6Neu, H-7Neu, H-2Gal, H-6bGlc), 3.75–3.56 (m, 6H, H-4Neu, H-6aGal, H-6bGal, H-5Neu, H-3Glc, H-4Glc, H-5Glc), 3.32 (m, 1H, H-2Glc), 2.75 (dd, 1H, J3e,3a = 12.2 Hz, J3e,4 = 4.7 Hz, H-3eNeu), 2.02 (s, 3H, C(O)CH3), 1.79 (dd, 1H, J3a,3e = 12.2 Hz, J3a,4 = 12.2 Hz, H-3aNeu). 13C NMR (125 MHz, D2O): δC 175.9, 174.7, 160.0, 136.4, 119.4, 116.7 (q, JC,F = 285 Hz), 103.6, 100.7, 79.2, 76.4, 76.1, 75.7, 75.1, 73.8, 73.6, 72.7, 72.4, 70.2, 69.2, 69.0, 68.6, 68.4, 63.5, 61.9, 60.9, 54.8, 52.6, 49.8, 40.5, 22.9. ESI HRMS calcd for C30H46N2O21F3: 827.25448. Found: 827.25507.000 rpm, the supernatant was collected and applied to a Sep-Pak (C18; 5g). Elution with a gradient of water–methanol afforded 8. A portion (18.5 mg; 17.9 µmol) was further purified by size exclusion chromatography (Biogel P-4 resin; 50 cm × 2 cm) and eluted with 5% MeOH–H2O to yield 8 (17.7 mg; 17.2 µmol; 95%): [α]D +14.2 (c 0.32, H2O). 1H NMR (600 MHz, D2O): δH 5.85 (ddd, 1H, J4,5t = 17.2 Hz, J4,5c = 10.4 Hz, J4,3 = 7.0 Hz, H-4sphing), 5.33 (m, 1H, H-5tsphing), 5.30 (d, 1H, H-5csphing), 4.74 (d, overlapped with HOD, 1H, H-1GalNAc), 4.52 (d, 1H, J1,2 = 8.0 Hz, H-1Gal), 4.49 (d, 1H, J1,2 = 8.0 Hz, H-1Glc), 4.32 (dd, 1H, J3,2 = 7.0 Hz, J3,4 = 7.0 Hz, H-3sphing), 4.23 (ddd, 1H, J2,3 = 7.0 Hz, J2,1a = 7.0 Hz, J2,1b = 3.9 Hz, H-2sphing), 4.14 (dd, 1H, J3,2 = 9.8 Hz, J3,4 = 2.6 Hz, H-3Gal), 4.12–4.10 (m, 2H, H-1asphing, H-4Gal), 3.99 (d, 1H, J6a,6b = 12.1 Hz, H-6aGlc), 3.93–3.86 (m, 4H, H-4GalNAc, H-2GalNAc, H-1bsphing, H-9aNeu), 3.83–3.59 (m, 16H, H-5Neu, H-6bGlc, H-4Neu, H-5Gal, H-6aGal, H-6bGal, H-5GalNAc, H-6aGalNAc, H-6bGalNAc, H-8Neu, H-3GalNAc, H-3Glc, H-9bNeu, H-4Glc, H-7Neu, H-5Glc), 3.49 (d, 1H, J6,5 = 10.1 Hz, H-6Neu), 3.37–3.31 (m, 2H, H-2Gal, H-2Glc), 2.66 (dd, 1H, J3e,3a = 12.3 Hz, J3e,4 = 4.5 Hz, H-3eNeu), 2.03, 2.02 (2 × s, 2 × 3H, 2 × C(O)CH3), 1.92 (dd, 1H, J3a,3e = 12.3 Hz, J3a,4 = 11.8 Hz, H-3aNeu). 13C NMR (125 MHz, D2O): δC 175.9, 175.7, 174.9, 159.8, 136.4, 119.4, 116.7 (q, JC,F = 285 Hz), 103.7, 103.5, 103.0, 102.5, 79.5, 78.0, 75.7, 75.6, 75.2, 75.1, 74.9, 74.0, 73.5, 73.1, 72.5, 72.2, 70.9, 69.6, 68.9, 65.1, 63.7, 62.1, 61.5, 61.0, 54.8, 53.2, 52.5, 49.8, 37.9, 23.6, 22.9. ESI HRMS calcd for C38H59N3O26F3: 1030.33444. Found: 1030.33555.Procedure (A) for deprotection and acylation of sphingosine amine using succinimide esters
Oligosaccharide 7 or 8 (5–40 µmol) was dissolved in dry methanol (2.0 mL). Sodium methoxide (∼3.0 eq.) was added and the mixture, stirred for 3 days at room temperature until completion of the reaction. The pH of the reaction mixture was adjusted to 9.0 using 0.5 M HCl and the methanol was evaporated. The compound was purified by HPLC (Hamilton semi-preparative PRP-1 column) using a MeOH–H2O gradient. A portion of the amine (5–15 µmol), the acylating agent (2.0–5.0 eq.) and 4-N,N-dimethylaminopyridine (1.0–3.0 eq.) were dissolved in a 9 : 1 pyridine–water mixture (1.0 mL). The reaction mixture was stirred overnight at 40 °C and concentrated. The residue was redissolved in a solution of 5% NaHCO3–H2O and applied to a C18 Sep-Pak cartridge. Gradient elution (MeOH–H2O) afforded the target compound which was further purified by size exclusion chromatography (Biogel P-4 resin; 50 cm × 2 cm) using 5% MeOH–H2O as an eluent.000 rpm for 30 min to remove cellular debris. The supernatant was applied directly to a size exclusion column (Biogel P-4; 50 × 2 cm). Elution with 5% aqueous methanol afforded 17 (31 mg; 36 µmol; 80%): [α]D +2.7 (c 0.24, H2O). 1H NMR (500 MHz, D2O): δH 5.84 (ddd, 1H, J4,5t = 17.2 Hz, J4,5c = 10.4 Hz, J4,3 = 7.0 Hz, H-4sphing), 5.31 (ddd, 1H, J5t,4 = 17.2 Hz, J5t,5c = 1.2 Hz, J5t,3 = 1.2 Hz, H-5tsphing), 5.25 (ddd, 1H, J5c,4 = 10.4 Hz, J5c,5t = 1.2 Hz, J5c,3 = 1.2 Hz, H-5csphing), 4.51 (d, 1H, J1,2 = 7.9 Hz, H-1Gal), 4.46 (d, 1H, J1,2 = 7.9 Hz, H-1Glc), 4.21 (dd, 1H, J3,4 = 7.0 Hz, J3,2 = 6.6 Hz, H-3sphing), 4.12–4.08 (m, 2H, H-2sphing, H-3Gal), 4.04 (dd, 1H, J1a,1b = 10.5 Hz, J1a,2 = 6.4 Hz, H-1asphing), 3.98 (dd, 1H, J6a,6b = 12.3 Hz, J6a,5 = 2.2 Hz, H-6aGlc), 3.94 (d, 1H, J4,3 = 3.1 Hz, H-4Gal), 3.90–3.73 (m, 5H, H-8Neu, H-5Neu, H-9aNeu, H-6bGlc, H-1bsphing), 3.71–3.67 (m, 3H, H-4Neu, H-6aGal, H-6bGal), 3.66–3.61 (m, 5H, H-5Gal, H-3Glc, H-4Glc, H-9bNeu, H-6Neu), 3.59–3.55 (m, 3H, H-5Glc, H-7Neu, H-2Gal), 3.32 (m, 1H, H-2Glc), 2.97 (t, 2H, J = 7.6 Hz, CH2NH2), 2.75 (dd, 1H, J3e,3a = 12.2 Hz, J3e,4 = 4.6 Hz, H-3eNeu), 2.28 (t, 2H, J = 7.4 Hz, CH2C(O)), 2.03 (s, 3H, C(O)CH3), 1.79 (dd, 1H, J3a,3e = 12.2 Hz, J3a,4 = 12.1 Hz, H-3aNeu), 1.69–1.59 (m, 4H, CH2CH2NH2, CH2CH2C(O)), 1.41–1.34 (m, 2H, CH2). 13C NMR (125 MHz, D2O): δC 177.6, 175.9, 174.7, 137.2, 118.9, 103.6, 103.2, 100.7, 79.2, 76.4, 76.1, 75.6, 75.1, 73.8, 73.7, 72.8, 72.7, 70.2, 69.5, 69.2, 69.0, 68.4, 63.5, 61.9, 60.9, 53.8, 52.6, 40.6, 40.2, 36.4, 27.4, 26.0, 25.6, 22.9. ESI HRMS calcd for C34H60N3O21: 846.37193. Found: 846.37177.000 rpm for 30 min and the supernatant was applied to a Sep-Pak (C18; 5 g) which was eluted with water (30 mL), followed by a gradient of water–methanol. The product obtained was further purified by HPLC (Kromasil C18-silica semi-preparative column) using a gradient of water and methanol as eluents to afford 18 (24.9 mg; 31.0 µmol; 79%): [α]D +2.4 (c 0.30, H2O). 1H NMR (600 MHz, D2O): δH 5.85 (dddd, 1H, J4,5t = 17.3 Hz, J4,5c = 10.5 Hz, J4,3 = 7.1 Hz, J4,2 = 1.1 Hz, H-4sphing), 5.32 (m, 1H, H-5tsphing), 5.27 (m, 1H, H-5csphing), 4.52 (dd, 1H, J1,2 = 7.8 Hz, J1.3 = 0.9 Hz, H-1Gal), 4.47 (dd, 1H, J1,2 = 7.9 Hz, J1,3 = 0.9 Hz, H-1Glc), 4.22 (ddd, 1H, J3,4 = 7.1 Hz, J3,2 = 7.1 Hz, J3,1 = 0.9 Hz, H-3sphing), 4.12–4.05 (m, 3H, H-3Gal, H-2sphing, H-1asphing), 3.99 (d, 1H, J6a,6b = 11.4 Hz, J6a,5 = 1.8 Hz, H-6aGlc), 3.95 (d, 1H, J4,3 = 2.7 Hz, H-4Gal), 3.90–3.73 (m, 5H, H-8Neu, H-5Neu, H-9aNeu, H-6bGlc, H-1bsphing), 3.71–3.62 (m, 8H, H-4Neu, H-6aGal, H-6bGal, H-5Gal, H-3Glc, H-4Glc, H-9bNeu, H-6Neu), 3.60–3.55 (m, 3H, H-5Glc, H-7Neu, H-2Gal), 3.33 (m, 1H, H-2Glc), 2.75 (dd, 1H, J3e,3a = 12.3 Hz, J3e,4 = 4.6 Hz, H-3eNeu), 2.22 (t, 2H, J = 7.1 Hz, CH2C(O)), 2.03 (s, 3H, C(O)CH3), 1.79 (dd, 1H, J3a,3e = 12.3 Hz, J3a,4 = 12.1 Hz, H-3aNeu), 1.62–1.56 (m, 4H, CH2CH2C(O), CH2CH2CH2C(O)), 0.89 (t, 3H, J = 9.3 Hz, CH3). 13C NMR (125 MHz, D2O): δC 178.1, 175.9, 174.8, 137.1, 119.0, 103.6, 103.3, 100.7, 79.2, 76.4, 76.1, 75.6, 75.1, 73.8, 73.7, 72.74, 72.66, 70.3, 69.5, 69.2, 69.0, 68.4, 63.5, 61.9, 61.0, 53.9, 52.6, 40.7, 38.7, 22.9, 19.9, 13.7. ESI HRMS calcd for C32H53N2O21: 801.31336. Found: 801.31463.000 rpm. The supernatant was applied to a Sep-Pak (C18; 5 g), washed with water and eluted with a water–methanol gradient. The residue thus obtained was applied to a Biogel P-4 column (50 cm × 2 cm) and eluted with 5% MeOH–H2O to yield 19 (23 mg; 22 µmol; 79%): [α]D +27.1 (c 0.24, H2O). 1H NMR (600 MHz, D2O): δH 5.85 (ddd, 1H, J4,5t = 17.2 Hz, J4,5c = 10.4 Hz, J4,3 = 7.0 Hz, H-4sphing), 5.32 (m, 1H, H-5tsphing), 5.27 (m, 1H, H-5csphing), 4.73 (d, 1H, J1,2 = 7.3 Hz, H-1GalNAc), 4.52 (d, 1H, J1,2 = 7.9 Hz, H-1Gal), 4.48 (d, 1H, J1,2 = 8.0 Hz, H-1Glc), 4.23 (dd, 1H, J3,4 = 7.0 Hz, J3,2 = 7.0 Hz, H-3sphing), 4.14 (dd, 1H, J3,2 = 9.8 Hz, J3,4 = 3.0 Hz, H-3Gal), 4.12–4.09 (m, 2H, H-4Gal, H-2sphing), 4.06 (dd, 1H, J1a,1b = 10.4 Hz, J1a,2 = 6.2 Hz, H-1asphing), 3.99 (dd, 1H, J6a,6b = 12.2 Hz, J6a,5 = 1.9 Hz, H-6aGlc), 3.93–3.90 (m, 2H, H-4GalNAc, H-2GalNAc), 3.87 (dd, 1H, J9a,9b = 12.0 Hz, J9a,8 = 2.2 Hz, H-9aNeu), 3.83–3.58 (m, 17H, H-5Neu, H-6bGlc, H-1bsphing, H-4Neu, H-5Gal, H-6aGal, H-6bGal, H-5GalNAc, H-6aGalNAc, H-6bGalNAc, H-8Neu, H-3GalNAc, H-3Glc, H-9bNeu, H-4Glc, H-7Neu, H-5Glc), 3.49 (dd, 1H, J6,5 = 10.1 Hz, J6,7 = 2.1 Hz, H-6Neu), 3.36 (dd, 1H, J2,3 = 9.5 Hz, J2,1 = 7.8 Hz, H-2Gal), 3.32 (dd, 1H, J2,3 = 9.2 Hz, J2,1 = 8.1 Hz, H-2Glc), 2.98 (t, 2H, J = 7.6 Hz, CH2NH2), 2.66 (dd, 1H, J3e,3a = 12.3 Hz, J3e,4 = 4.3 Hz, H-3eNeu), 2.28 (t, 2H, J = 7.3 Hz, CH2C(O)), 2.03, 2.01 (2 × s, 2 × 3H, 2 × C(O)CH3), 1.92 (dd, 1H, J3a,3e = 12.3 Hz, J3a,4 = 12.3 Hz, H-3aNeu), 1.69–1.60 (m, 4H, CH2CH2NH2, CH2CH2C(O)), 1.40–1.36 (m, 2H, CH2). 13C NMR (125 MHz, D2O): δC 177.6, 175.9, 175.7, 174.9, 137.2, 118.9, 103.6, 103.5, 103.2, 102.5, 79.5, 78.0, 75.6 (2C), 75.2, 75.1, 74.9, 74.0, 73.6, 73.1, 72.7, 72.1, 70.9, 69.6, 69.5, 68.9, 68.7, 63.7, 62.0, 61.5, 61.0, 53.8, 53.2, 52.5, 40.2, 37.9, 36.4, 27.4, 26.0, 25.6, 23.5, 22.9. ESI HRMS calcd for C42H73N4O26: 1049.45131. Found: 1049.45134.000 rpm. The supernatant was applied to a C18 Sep-Pak cartridge (C18; 5 g), washed with water and eluted with a water–methanol gradient. The residue thus obtained was applied to a Biogel P-4 column in 5% aqueous methanol (50 cm × 2 cm) to yield 20 (13 mg; 13 µmol; 83%): [α]D +14.8 (c 0.73, H2O). 1H NMR (600 MHz, D2O): δH 5.85 (ddd, 1H, J4,5t = 17.3 Hz, J4,5c = 10.5 Hz, J4,3 = 7.0 Hz, H-4sphing), 5.32 (m, 1H, H-5tsphing), 5.26 (m, 1H, H-5csphing), 4.73 (d, overlapped with HOD, 1H, H-1GalNAc), 4.51 (d, 1H, J1,2 = 8.0 Hz, H-1Gal), 4.47 (d, 1H, J1,2 = 8.0 Hz, H-1Glc), 4.22 (dd, 1H, J3,4 = 7.0 Hz, J3,2 = 7.0 Hz, H-3sphing), 4.14 (dd, 1H, J3,2 = 9.8 Hz, J3,4 = 3.0 Hz, H-3Gal), 4.11–4.05 (m, 3H, H-4Gal, H-2sphing, H-1asphing), 3.99 (dd, 1H, J6a,6b = 12.2 Hz, J6a,5 = 1.9 Hz, H-6aGlc), 3.92–3.89 (m, 2H, H-4GalNAc, H-2GalNAc), 3.87–3.58 (m, 18H, H-9aNeu, H-5Neu, H-6bGlc, H-1bsphing, H-4Neu, H-5Gal, H-6aGal, H-6bGal, H-5GalNAc, H-6aGalNAc, H-6bGalNAc, H-8Neu, H-3GalNAc, H-3Glc, H-9bNeu, H-4Glc, H-7Neu, H-5Glc), 3.48 (dd, 1H, J6,5 = 10.1 Hz, J6,7 = 1.8 Hz, H-6Neu), 3.37–3.31 (m, 2H, H-2Gal, H-2Glc), 2.65 (dd, 1H, J3e,3a = 12.6 Hz, J3e,4 = 4.5 Hz, H-3eNeu), 2.22 (t, 2H, J = 7.3 Hz, CH2C(O)), 2.02, 2.01 (2 × s, 2 × 3H, 2 × C(O)CH3), 1.91 (dd, 1H, J3a,3e = 12.2 Hz, J3a,4 = 12.2 Hz, H-3aNeu), 1.61–1.55 (m, 4H, CH2CH2CH2C(O), CH2CH2C(O)), 0.89 (t, 3H, J = 7.3 Hz, CH3). 13C NMR (125 MHz, D2O): δC 178.0 (2C), 175.9, 174.9, 137.1, 119.0, 103.6, 103.5, 103.2, 102.5, 79.5, 78.0, 75.60, 75.59, 75.2, 75.1, 74.9, 74.0, 73.6, 73.1, 72.7, 72.2, 70.9, 69.6, 69.5, 68.9, 68.7, 63.7, 62.0, 61.4, 61.0, 53.9, 53.2, 52.5, 38.7, 37.9, 23.5, 22.9, 19.9, 13.7. ESI HRMS calcd for C40H66N3O26: 1004.39404 Found: 1004.39400.Procedure (B) for deprotection and acylation of sphingosine amine using anhydride
Oligosaccharide 7 or 8 (17–40 µmol) was dissolved in dry methanol (2.0 mL). Sodium methoxide (∼2 eq.) was added and the mixture was stirred for 3 days at room temperature until completion of the reaction. The pH of the reaction mixture was adjusted to 9.0 using 0.5 M HCl and the methanol was evaporated. The compound was purified by HPLC (Hamilton semi-preparative PRP-1 column) using a MeOH–H2O gradient. A portion of the amine (15–18 µmol), 6-trifluoroacetamidohexanoic anhydride (∼5.0 eq.), and triethylamine (2–4 eq.) were dissolved in dry degassed methanol (3.0 mL). The solution was stirred overnight at room temperature. The mixture was concentrated, redissolved in water, and applied to a C18 Sep-Pak cartridge. Gradient elution (MeOH–H2O) afforded the target compound, which was further purified by HPLC (Hamilton semi-preparative PRP-1 column) using a MeOH–H2O gradient.Preparation of tetanus toxoid conjugates 25 and 27
Purified monomeric tetanus toxoid (∼6.6 mg mL−1) in PBS, pH 7.2 (840 µL for 23, 1.07 mL for 24) was added to the activated sugars 23 or 24 (∼30 eq.). The mixture was gently tumbled overnight before being dialyzed against PBS (5 × 2 L) at 4 °C. The incorporation level was determined by MALDI mass spectrometry. Results are presented in Scheme 4.Preparation of bovine serum albumin conjugate 26
The activated sugar 23 (∼30 eq.) was dissolved in water and bovine serum albumin (4.3 mg) was added. The mixture was gently tumbled overnight before being dialyzed against water (5 × 2 L) at 4 °C. The incorporation level was determined by MALDI mass spectrometry. Results are presented in Scheme 4.Acknowledgements
The authors would like to thank Dr Frédéric Carrel for helpful discussions, Dr Warren Wakarchuk and Dr Michel Gilbert, Institute for Biological Sciences, National Research Council of Canada for providing clones of the enzymes used in this work, Angie Morales-Izquierdo for acquiring mass spectra and Blake Zheng for help with enzyme expression and assay of glycosyltransferase activity. S. Jacques thanks the Fond de la recherche en santé du Québec for a Master's Training Scholarship, and J. R. Rich thanks the Natural Sciences and Engineering Research Council of Canada (NSERC) and the Alberta Heritage Foundation for Medical Research for graduate studentships. This work was funded by NSERC and the Alberta Ingenuity Centre for Carbohydrate Science.References
- (a) S. I. Hakomori and Y. Igarashi, Adv. Lipid Res., 1993, 25, 147 Search PubMed; (b) S. I. Hakomori, Biochem. Soc. Trans., 1993, 21, 583 CAS; (c) A. Varki, Glycobiology, 1993, 3, 97 CrossRef CAS; (d) C. Traving and R. Schauer, Cell. Mol. Life Sci., 1998, 54, 1330 CrossRef CAS; (e) E. V. Dyatlovitskaya and A. G. Kandyba, Russ. J. Bioorg. Chem., 2004, 30, 201 Search PubMed; (f) S. Hakomori and Y. M. Zhang, Chem. Biol., 1997, 4, 97.
- (a) O. Blixt, S. Head, T. Mondala, C. Scanlan, M. E. Huflejt, R. Alvarez, M. C. Bryan, F. Fazio, D. Calarese, J. Stevens, N. Razi, D. J. Stevens, J. J. Skehel, I. van Die, D. R. Burton, I. A. Wilson, R. Cummings, N. Bovin, C. H. Wong and J. C. Paulson, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 17033 CrossRef CAS; (b) Y. Fang, A. G. Frutos and J. Lahiri, Langmuir, 2003, 19, 1500 CrossRef CAS.
- (a) S. A. Kalovidouris, O. Blixt, A. Nelson, S. Vidal, W. B. Turnbull, J. C. Paulson and J. F. Stoddart, J. Org. Chem., 2003, 68, 8485 CrossRef CAS; (b) B. S. Bochner, R. A. Alvarez, P. Mehta, N. V. Bovin, O. Blixt, J. R. White and R. L. Schnaar, J. Biol. Chem., 2005, 280, 4307 CAS.
- (a) S. J. Danishefsky and J. R. Allen, Angew. Chem., Int. Ed., 2000, 39, 836 CrossRef CAS; (b) P. Fredman, K. Hedberg and T. Brezicka, BioDrugs, 2003, 17, 155 Search PubMed; (c) R. J. Bitton, M. D. Guthmann, M. R. Gabri, A. J. L. Carnero, D. F. Alonso, L. Fainboim and D. E. Gomez, Oncol. Rep., 2002, 9, 267 CAS; (d) S. M. Andersen, C.-C. Ling, P. Zhang, K. Townson, H. J. Willison and D. R. Bundle, Org. Biomol. Chem., 2004, 2, 1199 RSC; (e) H. J. Willison, K. Townson, J. Veitch, J. Boffey, N. Isaacs, S. M. Andersen, P. Zhang, C.-C. Ling and D. R. Bundle, Brain, 2004, 127, 680.
- G. J. Boons and A. V. Demchenko, Chem. Rev., 2000, 100, 4539 CrossRef CAS.
- O. Blixt, D. Vasiliu, K. Allin, N. Jacobsen, D. Warnock, N. Razi, J. C. Paulson, S. Bernatchez, M. Gilbert and W. Wakarchuk, Carbohydr. Res., 2005, 340, 1963 CrossRef CAS.
- A. S. Timen and P. Somfai, J. Org. Chem., 2003, 68, 9958 CrossRef.
- (a) T. Kawano, J. Q. Cui, Y. Koezuka, I. Toura, Y. Kaneko, K. Motoki, H. Ueno, R. Nakagawa, H. Sato, E. Kondo, H. Koseki and M. Taniguchi, Science, 1997, 278, 1626 CrossRef CAS; (b) T. I. Prigozy, O. Naidenko, P. Qasba, D. Elewaut, L. Brossay, A. Khurana, T. Natori, Y. Koezuka, A. Kulkarni and M. Kronenberg, Science, 2001, 291, 664 CrossRef CAS; (c) D. B. Moody, B. B. Reinhold, M. R. Guy, E. M. Beckman, D. E. Frederique, S. T. Furlong, S. Ye, V. N. Reinhold, P. A. Sieling, R. L. Modlin, G. S. Besra and S. A. Porcelli, Science, 1997, 278, 283 CrossRef CAS; (d) K. Miyamoto, S. Miyake and T. Yamamura, Nature, 2001, 413, 531 CrossRef CAS; (e) C. A. Lingwood, Glycoconjugate J., 1996, 13, 495 CrossRef CAS.
- D. R. Bundle, C.-C. Ling and P. Zhang, Chem. Abstr., 2003, 140, 16928.
- S. I. Hakomori, Cancer Res., 1985, 45, 2405 CAS.
- G. Ada and D. Isaacs, Clin. Microbiol. Infect., 2003, 9, 79 Search PubMed.
- (a) P. Fredman, K. Hedberg and T. Brezicka, BioDrugs, 2003, 17, 155 Search PubMed; (b) P. O. Livingston, S. L. Zhang and K. O. Lloyd, Cancer Immunol. Immunother., 1997, 45, 1 Search PubMed; (c) P. O. Livingston and G. Ragupathi, Cancer Immunol. Immunother., 1997, 45, 10 Search PubMed.
- (a) P. O. Livingston, Semin. Cancer Biol., 1995, 6, 357 CrossRef CAS; (b) I. C. le Poole, M. A. T. Gerberi and W. M. Kast, Curr. Opin. Oncol., 2002, 14, 641 CrossRef CAS; (c) G. Ragupathi, F. Koide, N. Sathyan, E. Kagan, M. Spassova, W. Bornmann, P. Gregor, C. Reis, H. Clausen, S. J. Danishefsky and P. Livingston, Cancer Immunol. Immunother., 2003, 52, 608 Search PubMed.
- J. Ohlsson and G. Magnusson, Tetrahedron, 2000, 56, 9975 CrossRef CAS.
- J. L. Sonnenburg, H. van Halbeek and A. Varki, J. Biol. Chem., 2002, 277, 17502 CrossRef CAS.
- M. Gilbert, R. Bayer, A. M. Cunningham, S. Defrees, Y. H. Gao, D. C. Watson, N. M. Young and W. W. Wakarchuk, Nat. Biotechnol., 1998, 16, 769 CAS.
- M. Gilbert, J. R. Brisson, M. F. Karwaski, J. Michniewicz, A. M. Cunningham, Y. Y. Wu, N. M. Young and W. W. Wakarchuk, J. Biol. Chem., 2000, 275, 3896 CrossRef CAS.
- Y. Lapidot, S. Rappoport and Y. Wolman, J. Lipid Res., 1967, 8, 142 CAS.
- (a) M. C. Bryan, O. Plettenburg, P. Sears, D. Rabuka, S. Wacowich-Sgarbi and C.-H. Wong, Chem. Biol., 2002, 9, 713 CrossRef CAS; (b) F. Fazio, M. C. Bryan, O. Blixt, J. C. Paulson and C. H. Wong, J. Am. Chem. Soc., 2002, 124, 14397 CrossRef CAS; (c) F. Fazio, M. C. Bryan, H.-K. Lee, A. Chang and C.-H. Wong, Tetrahedron Lett., 2004, 45, 2689 CrossRef CAS.
- (a) A. N. Rai and A. Basu, Org. Lett., 2004, 6, 2861 CrossRef CAS; (b) S. Torssell and P. Somfai, Org. Biomol. Chem., 2004, 2, 1643 RSC; (c) C.-C. Ling, Y. Cai and D. R. Bundle, unpublished work.
- R. T. Lee and Y. C. Lee, Carbohydr. Res., 1978, 37, 193.
- (a) J. R. Rich and D. R. Bundle, Org. Lett., 2004, 6, 897 CrossRef CAS; (b) J. R. Rich, W. W. Wakarchuk and D. R. Bundle, Chem.–Eur. J. DOI:10.1002/chem.200500518.
- (a) J. C. Wilson, M. J. Kiefel, D. I. Angus and M. von Itzstein, Org. Lett., 1999, 1, 443 CrossRef CAS; (b) L. M. Mikkelsen, M. J. Hernaiz, M. Martin-Pastor, T. Skrydstrup and J. Jimenez-Barbero, J. Am. Chem. Soc., 2002, 124, 14940 CrossRef; (c) M. Qian, S. Spinelli, H. Driguez and F. Payan, Protein Sci., 1997, 6, 2285 CrossRef CAS.
- (a) M. Nitz, PhD Thesis, University of Alberta, 2001; (b) X. Wu and D. R. Bundle, J. Org. Chem., 2005, 70, 7381 CrossRef CAS; (c) D. R. Bundle, J. R. Rich, S. Jacques, H. N. Yu, M. Nitz and C.-C. Ling, Angew. Chem., Int. Ed., 2005, 44, 7725 CrossRef.
- G. T. Hermanson, Bioconjugate Techniques, Academic Press, San Diego, 1996 Search PubMed.
- X. Y. Wu, C.-C. Ling and D. R. Bundle, Org. Lett., 2004, 6, 4407 CrossRef CAS.
- M. F. Karwaski, W. W. Wakarchuk and M. Gilbert, Protein Expression Purif., 2002, 25, 237 CrossRef CAS.
- S. Bernatchez, C. M. Szymanski, N. Ishiyama, J. J. Li, H. C. Jarrell, P. C. Lau, A. M. Berghuis, N. M. Young and W. W. Wakarchuk, J. Biol. Chem., 2005, 280, 4792 CAS.
- M. M. Palcic, L. D. Heerze, M. Pierce and O. Hindsgaul, Glycoconjugate J., 1988, 5, 49 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures for all novel compounds. See DOI: 10.1039/b513595h |
| This journal is © The Royal Society of Chemistry 2006 |