Synthesis of enantiopure C3- and C4-hydroxyretinals and their enzymatic reduction by ADH8 from Xenopus laevis
Marta
Domínguez
a,
Rosana
Álvarez
a,
Emma
Borràs
b,
Jaume
Farrés
b,
Xavier
Parés
b and
Angel R.
de Lera
*a
aDepartamento de Química Orgánica, Universidade de Vigo, 36310, Vigo, Spain. E-mail: qolera@uvigo.es; Fax: +34-986-811940; Tel: +34-986-812316
bDepartment of Biochemistry and Molecular Biology, Universitat Autònoma de Barcelona, E-08193 Bellaterra, Barcelona, Spain
First published on 30th November 2005
Abstract
(R)-all-trans-3-hydroxyretinal 1, (S)-all-trans-4-hydroxyretinal 3 and (R)-all-trans-4-hydroxyretinal 5 have been synthesized stereoselectively by Horner–Wadsworth–Emmons and Stille cross-coupling as bond-forming reactions. The CBS method of ketone reduction was used in the enantioface-differentiation step to provide the precursors for the synthesis of the 4-hydroxyretinal enantiomers. The kinetic constants of Xenopus laevis ADH8 with these retinoids have been determined.
Introduction
Vitamin A (retinol) and its metabolites (retinoids) are essential to the proper function of a number of biological processes. The visual cycle1 is perhaps the best characterized2 with 11-cis-retinal acting as the inverse agonist of the apoprotein opsin in vertebrates. Reproduction,3 cell growth and differentiation, embryonic development (e.g. limbs, nervous system, heart, kidney), immune response4 and intermediary metabolism are regulated by all-trans-retinoic acid and 9-cis-retinoic acid. These gene transcription regulators are the ligands for two classes of nuclear receptors, acting as ligand-dependent transcription factors: the retinoic acid receptors (RARα, β and γ) and the retinoid X receptors (RXRα, β and γ).5An increasing number of studies attribute a physiological role as signalling molecules to ring-oxidized derivatives at position C4, which are major metabolites of retinoids.6 In fact, C4-oxidized retinoids are natural ligands of several RAR receptors.6a, 6b, 7 Studies in Xenopus embryos revealed that 4-hydroxyretinol and 4-hydroxyretinoic acid can transactivate RAR receptors.7 Retinol metabolism to 4-hydroxy and 4-oxoretinol is detected in murine stem cells8 and a wide variety of cancer cell lines.7a, 9 In addition, 4-hydroxyretinol endogenously occurs in serum and liver of normal neonatal rats.10 A specific role in the onset of neuronal differentiation is postulated for 4-hydroxyretinoic acid,11 and both 4-hydroxyretinoic and 4-oxoretinoic acid exhibit biological activity in skin retinoid responsive systems in vivo.12 In Xenopus eggs and early embryos, the major bioactive retinoids are 4-oxoretinol and, particularly, 4-oxoretinal, which are critical for proper cell differentiation,6b whereas 4-oxoretinoic acid is a potent modulator of positional specification.6a
Retinoic acid is synthesized from retinol by a metabolic sequence involving two consecutive oxidation reactions, in which the reversible oxidation of retinol to retinal is considered to be the rate-limiting step. Further oxidation of retinal to retinoic acid appears to be irreversible.13 Three different enzyme types, grouped as superfamilies, have been suggested to be responsible for the reversible conversion of retinol to retinal: alcohol dehydrogenases (ADH) of the medium-chain dehydrogenase/reductase (MDR), retinol dehydrogenases of the short-chain dehydrogenase/reductase (SDR)13 and several members of the aldo-keto reductases (AKR).14
In vertebrates, the ADH family comprises eight different classes, involved in ethanol metabolism and also in the transformation of a variety of alcohols and aldehydes of physiological relevance, such as retinoids, steroids and cytotoxic aldehydes.15 ADH1 and ADH4 have been the best studied in mammals because of their wide expression in tissues and their activity towards ethanol and retinol. While ADH1 is present in all vertebrate groups, ADH4 has only been reported in mammals. In amphibians, an enzyme showing kinetic properties similar to those of ADH4 has been described, but it exhibits specificity towards NADP(H) instead of NAD(H), the common cofactor for these enzymes. ADH8, isolated from the stomach of Rana perezi, displayed high activity towards retinal.16 This property and the coenzyme requirement suggested that this enzyme might have a significant role in retinal metabolism, probably as a retinal reductase.
We are interested in deciphering the substrate specificity of ADHs towards retinoids in order to understand more deeply the biochemical pathways for the formation of vitamin A metabolites through oxidation/reduction reactions. Previous reports have dealt with the kinetic constants of ADH1 and ADH4 from humans and mice towards some ring-oxidized derivatives (rac-4-hydroxyretinol, 4-oxoretinal, 3,4-didehydroretinol and 3,4-didehydroretinal). All these compounds are good substrates for ADHs, judging from the low and similar Km values measured in all cases. The 4-hydroxyretinol derivative appeared to be the most active, showing a catalytic efficiency ca. 30 times higher than parent compound retinol. The data supported the involvement of the enzymes in the red-ox metabolism of the C4-oxidized retinoids.17
The availability of recombinant ADH8 from Xenopus laevis, the interest of this species to developmental studies, the high activity of ADH8 with retinal, and the physiological presence of several ring-oxidized retinoids in Xenopus prompted us to study the specificity and catalytic efficiency of this enzyme with chemically-modified retinals of biological significance.
Whereas in previous reports we determined the kinetic characteristics of racemic 4-hydroxyretinoids17, we consider the preparation of the corresponding enantiopure substrates to be of interest in order to determine whether enantiomeric discrimination by the enzyme can occur. In addition to the C4-hydroxyl derivatives, the study was extended to C3-hydroxyretinal, easily obtained from enantiopure starting material. The C4 and C3-ring-oxidized retinoids of the R configuration are components of the visual cycle in the animal kingdom, namely in the bioluminescent squid Watasenia scintillans18 and in most insects,19 respectively.
We report herein the stereoselective synthesis of (R)-all-trans-3-hydroxyretinal 1, (S)-all-trans-4-hydroxyretinal 3, (R)-all-trans-4-hydroxyretinal 5, and the kinetic constants measured upon incubation of these retinoids with Xenopus ADH8. Due to the pharmacological interest of 13-cis-retinoids, we also isolated the 13-cis isomers (2, 4 and 6, respectively) of the above compounds, which allowed a comparison of the catalytic efficiencies of C13–C14-double-bond stereoisomers (Fig. 1).
For the construction of the polyene side chain of these analogs, we took advantage of palladium-catalyzed cross-coupling reactions, a group of processes that have been found in general to be chemo- and stereoselective and to take place with retention of configuration of the coupling partners. We selected the Stille cross-coupling reaction for the construction of the C6–C7 bond [(R)-3-hydroxyretinal] and C8–C9 bond [(S)- and (R)-4-hydroxyretinal] guided by our comprehensive study on the scope and limitations of this process as applied to the synthesis of retinoids.20
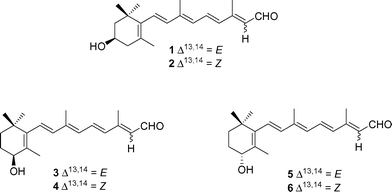 | ||
| Fig. 1 All-trans and 13-cis-isomers of enantiopure C3- and C4-hydroxyretinals. | ||
Results and discussion
Synthesis of (R)-3-hydroxyretinal
The Stille cross-coupling reaction of the already described triflate 721 and tributylstannyldienol 8,22 using Pd2dba3/AsPh3 as a catalyst and a LiCl additive, provided, after stirring for 14 h at 80 °C, alcohol 10 in 68% yield. Oxidation of 10 with catalytic quantities of tetra-n-propylammonium perruthenate (TPAP) and N-methylmorpholine N-oxide (NMO) as co-oxidant,23 afforded aldehyde 11 in good yield (Scheme 1).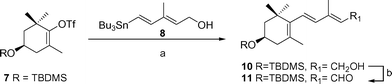 | ||
| Scheme 1 (a) Pd2dba3, AsPh3, NMP, tributylstannyldiene 8, LiCl, 80 °C, 14 h (68%); (b) TPAP, NMO, CH2Cl2, 25 °C (83%). | ||
The pentaene ester 13 was obtained by the Horner–Wadsworth–Emmons reaction between aldehyde 11 and phosphonate 12, using n-BuLi as a base in THF and DMPU as the cosolvent, in excellent yield after purification (99%).24 Deprotection of 13 with TBAF in THF at 0 °C afforded 14 in 58% yield (Scheme 2), which was uneventfully reduced to the corresponding alcohol with Dibal-H in THF at −78 °C and the latter oxidized using MnO2 (52% combined yield) to afford a 3 : 1 mixture of 125 and 2, which was separated by HPLC. No improvement could be achieved when the order of reactions (reduction–deprotection and oxidation) in the last functional group interconversion steps was reversed. Reduction of ester 13 with Dibal-H in THF at −78 °C afforded alcohol 15 in 89% yield, deprotection of which (68%), followed by oxidation with MnO2 under basic conditions at 25 °C afforded a mixture of 1 and 2 in lower yield (42%) and stereoselectivity (2 : 1 ratio) (Scheme 2).
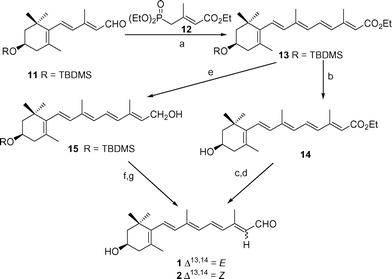 | ||
| Scheme 2 (a) THF, n-BuLi, DMPU, phosphonate 12, −78 − > 25 °C, 14 h (99%); (b) n-Bu4NF, THF, 25 °C (58%); (c) Dibal-H, THF, −78 °C (66%); (d) MnO2, Na2CO3, CH2Cl2, 25 °C (52%); (e) Dibal-H, THF, −78 °C (89%); (f) n-Bu4NF, THF, 25 °C (68%); (g) MnO2, Na2CO3, CH2Cl2, 25 °C (42%). | ||
Synthesis of (S) and (R)-4-hydroxyretinal
The C8–C9 Stille disconnection for the preparation of (S)-4-hydroxyretinal 3 and (R)-4-hydroxyretinal 5 required the condensation of known stannylated trienol 1620 (Scheme 4) and dienyliodides (S)-17 or (R)-17, both having trans geometries. The ketone 1817b already described was envisaged as the precursor of the electrophilic component of the Stille cross-coupling. For the enantioselective reduction of enynone 18, we selected the CBS chiral oxazaborolidine catalysts.26 Treatment of enynone 18 with (R)- or (S)-2-methyl-CBS-oxazaborolidine and BH3·SMe2 in THF at −30 °C afforded alcohols (S)-19 or (R)-19 in excellent yields (97% and 94%, respectively). Enantiomeric ratios [95% ee for (S)-19 and 91% ee for (R)-19] were determined by chiral HPLC (Chiralcel OD-H; 0,46 cm ϕ × 15 cm, cellulose on 5 µm silica gel and 95 : 5 hexane/MeOH as eluent) (Scheme 3). The CBS model for enantioface differentiation27 was adopted to assign the absolute configuration of the major enantiomer in each case.![(a) (R) or (S)-2-methyl-CBS-oxazaborolidine, BH3·SMe2, THF, 21 h, −30 °C [97% for (S)-19 and 94% for (R)-19]; (b) K2CO3, MeOH, 25 °C, 3.5 h [91% for (S)-20 and 98% for (R)-20]; (c) Ac2O, DMAP, Et3N, CH2Cl2 [96% for (S)-21 and 94% for (R)-21].](/image/article/2006/OB/b514273c/b514273c-s3.gif) | ||
| Scheme 3 (a) (R) or (S)-2-methyl-CBS-oxazaborolidine, BH3·SMe2, THF, 21 h, −30 °C [97% for (S)-19 and 94% for (R)-19]; (b) K2CO3, MeOH, 25 °C, 3.5 h [91% for (S)-20 and 98% for (R)-20]; (c) Ac2O, DMAP, Et3N, CH2Cl2 [96% for (S)-21 and 94% for (R)-21]. | ||
![(a) i. pinacol, BH3·SMe2, CH2Cl2, 0–>25 °C, ii. Enyne (S)-20 or (R)-20, CH2Cl2, 0–>50 °C [54% for (S)-22 and 57% for (R)-22]; (b) i. MeONa, MeOH, THF, −78 °C, ii. ICl, CH2Cl2, −78 °C [81% for (S)-17 and 86% for (R)-17]; (c) trienylstannane 16, Pd2(dba)3, AsPh3, NMP, 40 °C, 7 h [70% for (S)-23 and 65% for (R)-23]; (d) Dess–Martin Periodinane, CH2Cl2, pyridine, 25 °C, 6 h, [71% for (S) and 63% for (R)]; e) K2CO3, MeOH, 25 °C, 5 h [73% for 3 and 67% for 5].](/image/article/2006/OB/b514273c/b514273c-s4.gif) | ||
| Scheme 4 (a) i. pinacol, BH3·SMe2, CH2Cl2, 0–>25 °C, ii. Enyne (S)-20 or (R)-20, CH2Cl2, 0–>50 °C [54% for (S)-22 and 57% for (R)-22]; (b) i. MeONa, MeOH, THF, −78 °C, ii. ICl, CH2Cl2, −78 °C [81% for (S)-17 and 86% for (R)-17]; (c) trienylstannane 16, Pd2(dba)3, AsPh3, NMP, 40 °C, 7 h [70% for (S)-23 and 65% for (R)-23]; (d) Dess–Martin Periodinane, CH2Cl2, pyridine, 25 °C, 6 h, [71% for (S) and 63% for (R)]; e) K2CO3, MeOH, 25 °C, 5 h [73% for 3 and 67% for 5]. | ||
Deprotection of (S)-19 and (R)-19 with K2CO3 in MeOH provided efficiently unstable alkynes (91% and 98%, respectively), which were protected as acetates using Ac2O, DMAP and Et3N in CH2Cl2 in excellent yield (Scheme 3).
We attempted first to prepare the alkenyliodides by tin-iodide exchange of the corresponding alkenylstannanes, themselves obtained from the terminal alkynes by radical addition of tributyltin hydride,28 a methodology already used in the synthesis of the racemic material,17b but we encountered a number of difficulties, obtaining mixtures of alkenylstannanes with E and Z geometry as a result of the quality of the tributyltinhydride used being the most significant. In order to circumvent this problem, the alkenyl boronate was chosen instead of the alkenylstannane. Hydroboration of alkynes (S)-21 and (R)-21 using pinacol borane regio- and stereoselective provided the boronic esters (S)-22 and (R)-22 (54% and 57%, respectively, based on recovered starting material). In large-scale reactions, variable amounts of the minor regioisomer are obtained. Boron–halogen exchange with retention of configuration was effected by treatment of 22 with MeONa at −78 °C followed by addition of ICl. Alkenyl iodides (S)-17 and (R)-17 were isolated in very high yields, and their E geometry was confirmed by the value of the coupling constant (J = 14.8 Hz) for the alkenyl protons in their 1H-NMR spectrum (Scheme 4).
Coupling of iodides 17 with stannanyltrienol 16 under Farina's conditions [Pd2(dba)3, AsPh3, NMP] required 7 h at 40 °C and afforded (S)-23 and (R)-23 in 70 and 65% yield, respectively. Oxidation of diols (S)-23 and (R)-23 was carried out with the Dess–Martin reagent in CH2Cl2-pyridine to minimize isomerization at the terminal double bond. Despite these precautions, 8.5 : 1 and 6.5 : 1 mixtures of the trans:13-cis-isomers for the (S) and (R) enantiomers were obtained in 71 and 63% yield, respectively. Although they could be separated at this stage by column chromatography if desired, additional isomerization of the C13–C14 bond took place upon deprotection of the acetates with K2CO3 in MeOH, affording 3 : 1 mixtures of all-trans:13-cis-isomers of both (S)-4-hydroxyretinal and (R)-4-hydroxyretinal. These isomers were separated by HPLC (Preparative Nova Pak® HR silica, 60 Å, 19 × 300 mm and 95:5 hexane/ethyl acetate as eluent) (Scheme 4). The ee measured for the final compounds (89% ee for 3 and 93% ee for 4)29 indicates that enantiopurity has been mostly preserved along the sequence.
Kinetics of ADH8 with retinoids
The kinetics of Xenopus ADH8 with these C3- and C4-oxidized derivatives of all-trans-retinal and 13-cis-retinal are shown in Table 1. All the hydroxyretinals assayed were very active, suggesting that they can be physiological substrates for ADH8. When compared with the corresponding parent compounds, the kcat values for the ring-oxidized retinals showed a 10-fold increase, while Km values remained essentially constant. This effect was independent of the position of the hydroxyl group since no substantial differences were found between the C3- and the C4-hydroxyl derivatives of either all-trans-retinal or 13-cis-retinal. Furthermore, no differences were observed between R and S enantiomers of the C4-hydroxyl derivatives of both all-trans-retinal and 13-cis-retinal, thus concluding that ADH8 does not exhibit chiral discrimination, at least at position C4. On the other hand, the trans-ring-oxidized retinal stereoisomers showed 2.4- to 3.2-fold higher kcat values with respect to the derivatives of 13-cis-retinal, known to be a poor substrate for several ADH enzymes studied.17a, 30| Substrate | K m/µM | k cat/min−1 | k cat/Km/mM−1·min−1 |
|---|---|---|---|
| a Activities were determined in 0.1 M sodium phosphate, pH 7.5, 0.02% Tween 80, using 0.6 mM NADPH, at 25 °C. b Constants measured for the ADH8 enzyme of Rana perezi.16 | |||
| All-trans-retinal | 21.5 ± 3.5 | 270 ± 13 | 12560 ± 2100 |
| (R)-all-trans-4-hydroxy-retinal | 13.2 ± 1.8 | 2080 ± 90 | 157600 ± 23500 |
| (S)-all-trans-4-hydroxyretinal | 12.6 ± 1.3 | 2360 ± 70 | 187300 ± 20000 |
| (R)-all-trans-3-hydroxyretinal | 10.1 ± 0.9 | 2460 ± 70 | 243600 ± 23800 |
| 13-cis-retinalb | 15 | 63 | 4200 |
| (R)-13-cis-4-hydroxyretinal | 23.7 ± 2.3 | 860 ± 30 | 36300 ± 3700 |
| (S)-13-cis-4-hydroxyretinal | 20.3 ± 2.3 | 860 ± 30 | 42400 ± 5000 |
| (R)-13-cis-3-hydroxyretinal | 21.9 ± 2.5 | 760 ± 30 | 34700 ± 4200 |
| Hexanal | 79.4 ± 11.8 | 18600 ± 900 | 234000 ± 37000 |
The kcat values of ADH8 toward C3- and C4-hydroxyretinals are the highest among all retinoid substrates assayed for this enzyme. Similarly, C4-oxo- and C4-hydroxyretinoids were the most active retinoids for ADH1 and ADH4.17b,31 In general, ADH shows low turnover numbers (kcat) with retinol isomers, in comparison to those for aliphatic substrates.17a,32 For retinoids the release of product may be a significant rate-limiting step, in contrast to the kinetics with aliphatic substrates, where the limiting step is, in general, the cofactor release.33 For ADH8 the kcat for ring-oxidized retinals (about 2000 min−1, Table 1) is still lower than that for the best aliphatic aldehydes (kcat for hexanal = 18600 min−1 and 14000 min−1 for Xenopus laevis (Table 1) and Rana perezi enzymes, respectively). Thus, it can be suggested that the kcat for the ring-oxidized-retinals is higher than that for the parent compounds because the extra polarity facilitates the delivery to the aqueous environment of the reduced product, but still this product release is rate limiting in the reduction of hydroxy-retinals by ADH8.
The fact that ADH8 does not discriminate between the hydroxyl groups at the C3 or C4 positions of the cyclohexene ring, or between the C4-hydroxyretinal enantiomers, is consistent with docking studies using several retinoids as ligands and the crystallographic ADH8 structure from Rana perezi34 and other ADH structures.35 While the functional group of the retinoid interacts with the active site Zn, buried in the inner part of the molecule, the cyclohexene ring localizes in the wide entrance of the substrate-binding pocket, accessible to the solvent, and presumably with little structural constraints.
Finally, this is the first time that ring-oxidized derivatives of the 13-cis-retinal isomer have been assayed for ADH activity, and they also show a great increase in their kcat values when compared with 13-cis-retinal. The rate increase is about 10-fold, similar to that observed for the hydroxyl derivatives of all-trans-retinal. The 13-cis-isomers are among the worst retinoid substrates for ADHs because of their low kcat values, being virtually inactive with ADH1.17a In fact, ADH8 is the most active known ADH with 13-cis-retinoids.16
Conclusions
The catalytic constant (kcat) values of Xenopus laevis ADH8 with enantiopure C3- and C4-hydroxyretinals were about 10-fold those for the parent all-trans-retinal. The kinetic studies revealed neither enantiomeric discrimination (4R vs 4S) nor positional discrimination (3R vs 4R) by the enzyme, consistent with an external position of the cyclohexene ring in the ADH-retinoid complex structure. The corresponding 13-cis isomers of the ring oxidized derivatives, obtained as isomerization by-products in the synthetic sequence, showed from 2.4- to 3.2-fold lower kcat than the trans-isomers.The OH substitution at the cyclohexene ring would facilitate the study of the kinetic effect of the double bond isomerization, even using compounds, such as 13-cis-retinoids, with poor activity with ADHs.
Experimental
General
Solvents were dried according to published methods and distilled before use. HPLC grade solvents were used for the HPLC purification. All other reagents were commercial compounds of the highest purity available. All reactions were carried out under an argon atmosphere, and those not involving aqueous reagents were carried out in oven-dried glassware. Analytical thin layer chromatography (TLC) was performed on aluminium plates with Merck Kieselgel 60F254 and visualised by UV irradiation (254 nm) or by staining with solution of phosphomolibdic acid. Flash column chromatography was carried out using Merck Kieselgel 60 (230–400 mesh) under pressure. High performance liquid chromatography was performed using a Waters instrument using a dualwave detector (254 and 300 nm) with a Preparative Nova Pak® HR silica, 60 Å, 19 × 300 mm and 95 : 5 hexane/ethyl acetate as eluent. Enantiomeric excess was calculated by chiral HPLC with a Waters™ 996 (Photodiode Array) detector with a Chiralcel OD–H Column 0,46 cm × 15 cm cellulose on 5 µm silica gel and 95 : 5 hexane/MeOH as eluent. UV–Vis spectra were recorded on a Cary 100 Bio spectrophotometer using MeOH as solvent. Infrared spectra were obtained on JASCO FT-IR 4200 spectrophotometer, from a thin film deposited onto a NaCl glass. Specific rotation was obtained on JASCO P-1020. Mass spectra were obtained on a Hewlett-Packard HP59970 instrument operating at 70 eV by electron ionisation. High resolution mass spectra were taken on a VG Autospec instrument. 1H NMR spectra were recorded in CDCl3, C6D6 and (CD3)2CO at ambient temperature on a Bruker AMX-400 spectrometer at 400 MHz with residual protic solvent as the internal reference (CDCl3, δH = 7.26 ppm; C6D6, δH = 7.16 ppm; (CD3)2CO, δH = 2.05 ppm); chemical shifts (δ) are given in parts per million (ppm), and coupling constants (J) are given in Hertz (Hz). The proton spectra are reported as follows: δ (multiplicity, coupling constant J, number of protons, assignment). 13C NMR spectra were recorded in CDCl3, C6D6 and (CD3)2CO at ambient temperature on the same spectrometer at 100 MHz, with the central peak of CDCl3 (δC = 77.0 ppm), C6D6 (δC = 128.0 ppm) or (CD3)2CO (δC = 30.8 ppm) as the internal reference. DEPT135 are used to aid in the assignment of signals in the 13C NMR spectra.![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 1083 cm−1. UV (MeOH): λmax 280 nm.
O), 1149 cm−1. UV (MeOH): λmax 348 nm (ε = 26800).
O), 1151 cm−1. UV (MeOH): λmax 353 nm (ε = 47000).
O), 1083 cm−1. UV (MeOH): λmax 280 nm.
O), 1149 cm−1. UV (MeOH): λmax 348 nm (ε = 26800).
O), 1151 cm−1. UV (MeOH): λmax 353 nm (ε = 47000).
To a solution of this compound (0.04 g, 0.14 mmol) in CH2Cl2 (2.6 mL) was added MnO2 (0.22 g, 2.54 mmol) and Na2CO3 (0.27 g, 2.54 mmol), and the suspension was stirred for 5 h. The mixture was filtered throught Celite and the solvents were removed. The residue was purified by column chromatography (silica gel, 97 : 3 hexane/ethyl acetate) to afford 0.022 g (52%) as a yellow oil identified as a mixture of 1 and 2 in a 3 : 1 ratio, which were separated by HPLC.
Data for (−)-(R)-all-trans-3-hydroxyretinal 1: [α]D20
− 68.6 (c 0.022, MeOH). 1H-NMR (400.16 MHz, (CD3)2CO): δ 9.81 (d, J = 8.0 Hz, 1H, H15), 6.98 (dd, J = 15.1, 11.4 Hz, 1H, H11), 6.17 (d, J = 15.1 Hz, 1H, H12), 6.03 (d, J = 16.1 Hz, 1H, H7), 5.98 (d, J = 11.5 Hz, 1H, H10), 5.90 (d, J = 16.1 Hz, 1H, H8), 5.59 (d, J = 8.0 Hz, 1H, H14), 3.7–3.5 (m, 1H, H3), 2.49 (s, 3H, C13–CH3), 2.1–2.0 (m, 1H, H4), 1.74 (s, 3H, C9–CH3), 1.7–1.6 (m, 1H, H4), 1.4–1.3 (m, 1H, H2), 1.41 (s, 3H, C5–CH3), 1.09 (t, J = 11.9 Hz, 1H, H2), 0.75 (s, 3H, C1–CH3), 0.74 (s, 3H, C1–CH3). 13C-NMR (100.62 MHz, (CD3)2CO): δ 190.3 (d), 154.2 (s), 140.3 (s), 137.7 (d), 137.1 (s), 135.0 (d), 132.2 (d), 129.9 (d), 128.9 (d), 128.3 (d), 127.4 (s), 63.3 (d), 48.5 (t), 42.5 (t), 36.5 (s), 29.7 (q), 27.9 (q), 20.8 (q), 12.0 (q), 11.9 (q). MS (EI+): m/z (%) 300 (M+, 72), 171 (63), 159 (25), 157 (26), 147 (25), 145 (27), 133 (25), 131 (26), 119 (43), 105 (39), 95 (36), 91 (40), 69 (100). HRMS (EI+): Calcd. for C20H28O2, 300.2089; found, 300.2086. IR (NaCl): ν 3550–3150 (br, OH), 2957 (s, C–H), 2918 (s, C–H), 2850 (s, C–H), 1658 (s, CO), 1574 cm−1. UV (MeOH): λmax 378 nm (ε = 28000).
Data for (−)-(R)-13-cis-3-hydroxyretinal 2: [α]D20
− 64.8 (c 0.012, MeOH). 1H-NMR (400.16 MHz, (CD3)2CO): δ 10.25 (d, J = 7.9 Hz, 1H, H15), 7.50 (d, J = 15.0 Hz, 1H, H12), 7.20 (dd, J = 15.0, 11.5 Hz, 1H, H11), 6.37 (d, J = 15.9 Hz, 1H, H7), 6.35 (d, J = 11.1 Hz, 1H, H10), 6.23 (d, J = 15.9 Hz, 1H, H8), 5.80 (d, J = 7.9 Hz, 1H, H14), 3.9–3.8 (m, 1H, H3), 2.4–2.3 (m, 1H, H2), 2.17 (s, 3H, C13–CH3), 2.04 (s, 3H, C9–CH3), 2.0–1.9 (m, 1H, H2), 1.8–1.7 (m, 1H, H4), 1.74 (s, 3H, C5–CH3), 1.42 (t, J = 12.0 Hz, 1H, H4), 1.08 (s, 3H, C1–CH3), 1.07 (s, 3H, C1–CH3). MS (EI+): m/z (%) 300 (M+, 100), 147 (22), 121 (24), 119 (40), 105 (25), 95 (24), 91 (29), 77 (19). HRMS (EI+): Calcd. for C20H28O2, 300.2089; found, 300.2086. IR (NaCl): ν 3580–3150 (br, OH), 2956 (s, C–H), 2927 (s, C–H), 2855 (s, C–H), 1658 (s, CO), 1575 cm−1. UV (MeOH): λmax 374 nm (ε = 22000).
![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C), 1249 cm−1. Elemental analysis: calcd. for C14H25OSi: C, 71.12; H, 10.23; found: C, 71.12; H, 10.27.
C) cm−1.
C), 1735 (s, CO), 1236 cm−1. MS (EI+): m/z (%) 206 (M+, 8), 147 (31), 146 (56), 132 (43), 131 (100), 129 (59), 128 (53), 116 (75), 115 (75), 108 (44), 91 (76), 77 (41), 65 (12), 63 (12). HRMS (EI+): Calcd. for C13H18O2, 206.1307; found, 206.1310.
O), 1619, 1349, 1242, 1142 cm−1.
O), 1241 cm−1.
O), 1241 cm−1. UV (MeOH): λ 323 nm (ε = 37000).
C), 1249 cm−1. Elemental analysis: calcd. for C14H25OSi: C, 71.12; H, 10.23; found: C, 71.12; H, 10.27.
C) cm−1.
C), 1735 (s, CO), 1236 cm−1. MS (EI+): m/z (%) 206 (M+, 8), 147 (31), 146 (56), 132 (43), 131 (100), 129 (59), 128 (53), 116 (75), 115 (75), 108 (44), 91 (76), 77 (41), 65 (12), 63 (12). HRMS (EI+): Calcd. for C13H18O2, 206.1307; found, 206.1310.
O), 1619, 1349, 1242, 1142 cm−1.
O), 1241 cm−1.
O), 1241 cm−1. UV (MeOH): λ 323 nm (ε = 37000).
Data for (−)-(S)-all-trans-4-acetoxyretinal (S)-24. [α]D24 − 19.4 (c 0.036, MeOH).1H-NMR (400.16 MHz, C6D6): δ 10.01 (d, J = 7.8 Hz, 1H, H15), 6.80 (dd, J = 15.0, 11.5 Hz, 1H, H11), 6.20 (s, 2H, H7 + H8), 6.04 (d, J = 15.1 Hz, 1H, H12), 6.1–5.9 (m, 2H, H10 + H14), 5.50 (t, J = 4.6 Hz, 1H, H4), 1.83 (s, 3H, COOCH3), 1.78 (s, 3H, C–CH3), 1.73 (s, 3H, C–CH3), 1.72 (s, 3H, C–CH3), 1.7–1.6 (m, 2H, 2H3), 1.4–1.2 (m, 2H, 2H2), 1.05 (s, 3H, C1–CH3), 0.96 (s, 3H, C1–CH3) ppm. 13C-NMR (100.62 MHz, (CD3)2CO): δ 190.3 (d), 170.1 (s), 155.1 (s), 143.5 (s), 139.9 (s), 138.5 (d), 135.5 (d), 132.0 (d), 130.7 (d), 129.0 (d), 127.6 (d), 127.2 (s), 71.5 (d), 34.4 (t), 32.5 (s), 28.6 (q), 26.6 (q), 24.9 (t), 20.1 (q), 17.6 (q), 12.0 (q), 11.9 (q). MS (EI+): m/z (%) 342 (M+, 62), 300 (50), 283 (37), 282 (M+–OAc, 100), 187 (34), 119 (51), 105 (44), 95 (45), 91 (45), 77 (24). HRMS (EI+): Calcd. for C22H30O3, 342.2195; found, 342.2193. IR (NaCl): δ 2953 (s, C–H), 2924 (s, C–H), 2854 (m, C–H), 1735 (m, C
O), 1661, 1457, 1240 cm−1. UV (MeOH): λ 368 nm (ε = 23300).
Data for (S)-13-cis-4-acetoxyretinal (S, 13Z)-24. 1H-NMR (400.13 MHz, C6D6): δ 10.13 (d, J = 7.3 Hz, 1H, H15), 7.10 (d, J = 15.2 Hz, 1H, H12), 6.71 (dd, J = 15.2, 11.5 Hz, 1H, H11), 6.21 (s, 2H, H7 + H8), 6.00 (d, J = 11.5 Hz, 1H, H10), 5.75 (d, J = 7.3 Hz, 1H, H14), 5.49 (t, J = 4.5 Hz, 1H, H4), 1.84 (s, 3H, COOCH3), 1.77 (s, 3H, C–CH3), 1.71 (s, 3H, C–CH3), 1.58 (s, 3H, C–CH3), 1.6–1.2 (m, 4H, 2H3 + 2H2), 1.05 (s, 3H, C1–CH3), 0.96 (s, 3H, C1–CH3) ppm.
Data for (R)-24. [α]24 + 17.6 (c 0.034, MeOH)
Data for (−)-(S)-all-trans-4-hydroxyretinal 3. [α]D24 − 89.0 (c 0.016, MeOH).1H-NMR (400.13 MHz, (CD3)2CO): δ 10.13 (d, J = 8.0 Hz, 1H, H15), 7.31 (dd, J = 15.1, 11.5 Hz, 1H, H11), 6.51 (d, J = 15.1 Hz, 1H, H12), 6.37 (d, J = 16.1 Hz, 1H, H7), 6.32 (d, J = 11.5 Hz, 1H, H10), 6.24 (d, J = 16.1 Hz, 1H, H8), 5.92 (d, J = 8.0 Hz, 1H, H14), 3.94 (t, J = 4.5 Hz, 1H, H4), 2.38 (s, 3H, C13–CH3), 2.07 (s, 3H, C9–CH3), 1.9–1.8 (m, 1H, H3), 1.82 (s, 3H, C5–CH3), 1.7–1.6 (m, 2H, H3 + H2), 1.4–1.3 (m, 1H, H2), 1.04 (s, 3H, C1–CH3), 1.03 (s, 3H, C1–CH3) ppm. 13C-NMR (100.62 MHz, (CD3)2CO): δ 190.3 (d), 154.2 (s), 140.2 (s), 139.7 (s), 137.9 (d), 135.1 (d), 132.1 (d), 131.9 (s), 130.1 (d), 128.9 (d), 128.5 (d), 68.9 (d), 34.7 (t), 34.2 (s), 28.5 (t), 28.2 (q), 27.1 (q), 17.9 (q), 12.0 (q), 11.9 (q). MS (EI+): m/z (%) 300 (M+, 72), 203 (15), 190 (14), 175 (15), 161 (26), 119 (43), 105 (37), 91 (37), 86 (69). HRMS (EI+): Calcd. for C20H28O2, 300.2089; found, 300.2086. IR (NaCl): δ 3500–3100 (br, OH), 2918 (s, C–H), 2850 (m, C–H), 1656 (m, C
O), 1575, 1164, 996 cm−1. UV (MeOH): λmax 375 nm (ε = 31900).
Data for (−)-(S)-13-cis-4-hydroxyretinal 4. [α]D24 − 75.8 (c 0.018, MeOH).1H-NMR (400.13 MHz, (CD3)2CO): δ 10.25 (d, J = 7.8 Hz, 1H, H15), 7.52 (d, J = 15.0 Hz, 1H, H12), 7.20 (dd, J = 15.0, 11.3 Hz, 1H, H11), 6.38 (d, J = 16.2 Hz, 1H, H7), 6.36 (d, J = 11.3 Hz, 1H, H10), 6.25 (d, J = 16.2 Hz, 1H, H8), 5.80 (d, J = 8.0 Hz, 1H, H14), 3.94 (t, J = 4.5 Hz, 1H, H4), 2.18 (d, J = 0.9 Hz, 3H, C13–CH3), 2.07 (s, 3H, C9–CH3), 1.9–1.8 (m, 1H, H3), 1.82 (s, 3H, C5–CH3), 1.7–1.6 (m, 2H, H3 + H2), 1.4–1.3 (m, 1H, H2), 1.05 (s, 3H, C1–CH3), 1.03 (s, 3H, C1–CH3) ppm. MS (EI+): m/z (%) 300 (M+, 100), 161 (25), 135 (25), 119 (38), 107 (25), 105 (33), 95 (25), 91 (33), 69 (31). HRMS (EI+): Calcd. for C20H28O2, 300.2089; found, 300.2087. IR (NaCl): ν 3580–3150 (br, OH), 2956 (s, C–H), 2927 (s, C–H), 2855 (s, C–H), 1658 (s, C
O), 1575 cm−1. UV (MeOH): λmax 368 nm (ε = 28500).
Data for (+)-(R)-all-trans-4-hydroxyretinal 5: [α]D24 + 87.0 (c 0.018, MeOH).
Data for (+)-(R)-13-cis-4-hydroxyretinal 6: [α]D24 + 73.2 (c 0.012, MeOH).
Cloning, expression and purification of Xenopus laevis ADH8
ADH8 cDNA sequence from Xenopus laevis was obtained by nested-PCR, using two sets of degenerated primers based on the sequence of the orthologous enzyme from Rana perezi,16 followed by 3′-end RACE-PCR amplification. The sequence was deposited in the GenBank data base under the accession no. AJ566764. The full-length cDNA was then generated by PCR amplification, cloned in the expression vector pGEX-4T-2 (Amersham Biosciences) and used to transform E. coli BL21 cells, as described previously.17a Expression, cell lysis and purification of Xenopus ADH8 as a fusion protein with glutathione-S-transferase (GST) were conducted as described for the Rana perezi enzyme.36 Finally, the homogeneity of the purified protein was assessed by SDS-PAGE followed by Coomassie Brilliant Blue (Sigma) staining.Enzyme kinetics
Enzymatic activities of purified Xenopus ADH8 were determined in a Varian Cary 400 spectrophotometer, at 25 °C. Standard activity was measured with 1.92 mM octanol (Merck) and 2.4 mM NADP+ (Roche) in 0.1 M glycine, pH 10.0, at 340 nm, in 1-cm pathlength cuvettes. A specific activity of 34.02 U mg−1 for octanol was considered. One unit (U) of ADH activity is defined as the amount of enzyme required to transform 1 µmol of substrate or cofactor per min at 25 °C. Activities for retinal reduction kinetics were determined at 400 nm, with 0.6 mM NADPH (Roche), and 0.1 M sodium phosphate, pH 7.5, 0.02% Tween 80 (assay buffer), in 0.2-cm pathlength cuvettes. Retinoid concentration ranged from 0.1 × Km to 10 × Km and each individual rate measurement was run in duplicate. Kinetic constants were calculated using the GraFit 5.0 program (Erithacus Software Limited), and the results were expressed as the mean value ± SEM. A molecular weight of 80,000 for ADH dimer was used to calculate kcat values.Molar absorption coefficients in the assay buffer, used to calculate retinoid concentration, were ε400 = 29500 M−1 cm−1 for all-trans-retinal (Sigma) and ε370 = 27000 M−1 cm−1 for 13-cis-retinal (Sigma). Since the absorption coefficients for the ring-oxidized retinals were not known, these values were first estimated in methanol, and then calculated in the assay buffer: ε375 = 30300 M−1 cm−1 for 4-hydroxyretinal, ε378 = 26600 M−1 cm−1 for 3-hydroxyretinal, ε368 = 28500 M−1 cm−1 for 4-hydroxy-13-cis-retinal, and ε374 = 25400 M−1 cm−1 for 13-cis-3-hydroxyretinal. Substrate solutions were prepared by diluting 1 mg retinal, dissolved in 150–500 µL methanol, with the appropriate volume of the assay buffer to have a final concentration of approximately 200 µM, at 4 °C and under dim red light. The stability of the retinoid was checked spectrophotometrically.
Acknowledgements
Supported by grants from Ministerio de Educación y Ciencia (FPU Fellowship to M.D., Ramón y Cajal Contract to R.A.), the Spanish Dirección General de Investigación (BMC2002-02659, BMC2003-09606 and SAF-2004-07131) and the Generalitat de Catalunya (2001SGR 00198). We thank Dr. Gregg Duester for his help in the preparation of ADH8 cDNA. Graphical Abstract graphics were kindly provided by Oriol Gallego.References
- (a) R. R. Rando, Chem. Rev., 2001, 101, 1881 CrossRef CAS; (b) J. K. McBee, K. Palczewski, W. Baehr and D. R. Pepperberg, Prog. Retinal Eye Res., 2001, 20, 469 Search PubMed.
- J. C. Saari, in The Retinoids: Biology, Chemistry and Medicine, 2nd Edition, ed. M. B. Sporn, A. B. Roberts and D. S. Goodman, Raven, New York, 1993, pp. 351–385 Search PubMed.
- J. T. Davis and D. E. Ong, Biol. Reprod., 1995, 52, 356 Search PubMed.
- J. L. Napoli, Biochim. Biophys. Acta, 1999, 1440, 139 CrossRef CAS.
- (a) R. V. Weatherman, R. J. Fletterick and T. S. Scanlan, Annu. Rev. Biochem., 1999, 68, 559 CrossRef CAS; (b) W. Bourquet, P. Germain and H. Gronemeyer, Trends Pharmacol. Sci., 2000, 21, 381 CrossRef CAS.
- (a) W. W. M. Pijnappel, H. F. J. Hendriks, G. E. Folkers, C. E. van der Brink, E. J. Dekker, C. Edelenbosch, P. T. van der Saag and A. J. Durston, Nature, 1993, 366, 340 CrossRef CAS; (b) B. Blumberg, J. Bolado, F. Derguini, A. Grey Craig, T. A. Moreno, D. Chakravarti, R. A. Heyman, J. Buck and R. M. Evans, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 4873 CrossRef CAS; (c) C. K. Schmidt, J. Volland, G. Hamscher and H. Nau, Biochim. Biophys. Acta, 2002, 1583, 237 CAS.
- (a) C. C. Achkar, F. Derguini, B. Blumberg, A. Langston, A. A. Levin, J. Speck, R. M. Evans, J. Bolado, K. Nakanishi, J. Buck and L. J. Gudas, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 4879 CrossRef CAS; (b) N. Idres, J. Marill, M. A. Flexor and G. G. Chabot, J. Biol. Chem., 2002, 277, 31491 CrossRef CAS.
- M. A. Lane, A. C. Chen, S. D. Roman, F. Derguini and L. J. Gudas, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 13524 CrossRef CAS.
- (a) A. C. Chen, X. Guo, F. Derguini and L. J. Gudas, Cancer Res., 1997, 57, 4642 CAS; (b) T. N. Faria, R. Rivi, F. Derguini, P. P. Pandolfi and L. J. Gudas, Cancer Res., 1998, 58, 2007 CAS.
- R. Ruhl, G. Hamscher, A. L. Garcia, H. Nau and F. J. Schweigert, Life Sci., 2005, 76, 1613 CrossRef.
- E. Sonneveld, C. E. van den Brink, L. G. Tertoolen, B. van der Burg and P. T. van der Saag, Dev. Biol., 1999, 213, 390 CrossRef CAS.
- N. J. Reynolds, G. J. Fisher, C. E. Griffiths, A. Tavakkol, H. S. Talwar, P. E. Rowse, T. A. Hamilton and J. J. Voorhees, J. Pharmacol. Exp. Ther., 1993, 266, 1636 CAS.
- G. Duester, Biochemistry, 1996, 35, 12221 CrossRef CAS.
- B. Crosas, D. J. Hyndman, O. Gallego, S. Martras, X. Parés, T. G. Flynn and J. Farrés, Biochem. J., 2003, 373, 973 CrossRef CAS.
- G. Duester, J. Farrés, M. R. Felder, R. S. Holmes, J. O. Höög, X. Parés, B. V. Plapp, S. J. Yin and H. Jörnvall, Biochem. Pharmacol., 1999, 58, 389 CrossRef CAS.
- J. M. Peralba, E. Cederlund, B. Crosas, A. Moreno, P. Julià, S. E. Martínez, B. Persson, J. Farrés, X. Parés and H. Jörnvall, J. Biol. Chem., 1999, 274, 26021 CrossRef CAS.
- (a) S. Martras, R. Álvarez, S. E. Martínez, D. Torres, O. Gallego, G. Duester, J. Farrés, A. R. de Lera and X. Parés, Eur. J. Biochem., 2004, 271, 1660 CrossRef CAS; (b) M. Domínguez, R. Álvarez, S. Martras, J. Farrés, X. Parés and A. R. de Lera, Org. Biomol. Chem., 2004, 2, 3368 RSC.
- Y. Katsuta, K. Yoshihara, K. Nakanishi and M. Ito, Tetrahedron Lett., 1994, 35, 905 CrossRef CAS.
- T. Seki, K. Isono, M. Ito and Y. Katsuta, Eur. J. Biochem., 1994, 226, 691 CrossRef CAS.
- B. Domínguez, B. Iglesias and A. R. de Lera, Tetrahedron, 1999, 55, 15071 CrossRef CAS.
- (a) M. Ito, Y. Hirata, Y. Shibata and K. Tsukida, J. Chem. Soc., Perkin Trans. 1, 1990, 197 RSC; (b) Y. Yamano and M. Ito, J. Chem. Soc., Perkin Trans. 1, 1993, 1599 RSC; (c) M. Ito, Y. Yamano, S. Sumiya and A. Wada, Pure Appl. Chem., 1994, 66, 939 CrossRef CAS; (d) Y. Yamano, C. Tode and M. Ito, J. Chem. Soc., Perkin Trans. 1, 1995, 1895 RSC.
- J. F. Betzer, F. Delalage, B. Muller, A. Pancrazi and J. Prunet, J. Org. Chem., 1997, 62, 7768 CrossRef CAS.
- W. P. Griffith, S. V. Ley, G. P. Withcombe and A. D. White, J. Chem. Soc., Chem. Commun., 1987, 1625 RSC.
- (a) Y. L. Bennani and M. F. Boehm, J. Org. Chem., 1995, 60, 1195 CrossRef; (b) Y. L. Bennani, J. Org. Chem., 1996, 61, 3542 CrossRef CAS; (c) B. Domínguez, B. Iglesias and A. R. de Lera, J. Org. Chem., 1998, 63, 4135 CrossRef CAS.
- (a) Compound 1 has been synthesised by conventional double-bond forming reactions: V. H. Mayer and J. M. Santer, Helv. Chim. Acta, 1980, 63, 1467 Search PubMed; (b) H. Ok, C. Caldwell, D. R. Schroeder, A. K. Singh and K. Nakanishi, Tetrahedron Lett., 1988, 29, 2275 CrossRef CAS.
- (a) E. J. Corey, R. K. Bakshi and S. Shibata, J. Am. Chem. Soc., 1987, 109, 5551 CrossRef CAS; (b) E. J. Corey, R. K. Bakshi, S. Shibata, C. P. Chen and V. K. Singh, J. Am. Chem. Soc., 1987, 109, 7925 CrossRef CAS; (c) E. J. Corey, S. Shibata and R. K. Bakshi, J. Org. Chem., 1988, 53, 2861 CrossRef CAS; (d) E. J. Corey, Pure Appl. Chem., 1990, 62, 1209 CrossRef CAS.
- E. J. Corey and C. Helal, Angew. Chem., Int. Ed., 1998, 37, 1986 CrossRef CAS.
- S. M. L. Chen, R. E. Schaub and C. V. Grudzinskas, J. Org. Chem., 1978, 43, 3450 CrossRef CAS.
- Resolution of racemic 4-hydroxyretinal has been achieved through formation of the diastereomeric camphanic esters: Y. Katsuta, K. Yoshihara, K. Nakanishi and M. Ito, Tetrahedron Lett., 1994, 35, 905 Search PubMed.
- Z. N. Yang, G. F. Davis, T. D. Hurley, C. L. Stone, T. K. Li and W. F. Bosron, Alcohol. Clin. Exp. Res., 1994, 18, 587 Search PubMed.
- S. Martras, R. Álvarez, O. Gallego, M. Domínguez, A. R. de Lera, J. Farrés and X. Parés, Arch. Biochem. Biophys., 2004, 430, 210 CrossRef CAS.
- B. Crosas, A. Allali-Hassani, S. E. Martínez, S. Martras, B. Persson, H. Jörnvall, X. Parés and J. Farrés, J. Biol. Chem., 2000, 275, 25180 CrossRef CAS.
- B. V. Plapp, J. L. Mitchell and K. B. Berst, Chem. Biol. Int., 2001, 130, 445 Search PubMed.
- A. Rosell, E. Valencia, X. Parés, I. Fita, J. Farrés and W. F. Ochoa, J. Mol. Biol., 2003, 330, 75 CrossRef CAS.
- (a) M. Foglio and G. Duester, Biochim. Biophys. Acta, 1999, 1432, 239 CAS; (b) N. Y. Kedishvili, W. F. Bosron, C. L. Stone, T. D. Hurley, C. F. Peggs, H. R. Thomasson, K. M. Popov, L. G. Carr, H. J. Edenberg and T. K. Li, J. Biol. Chem., 1995, 270, 3625 CrossRef CAS.
- E. Valencia, A. Rosell, C. Larroy, J. Farrés, J. A. Biosca, I. Fita, X. Parés and W. F. Ochoa, Acta Crystallogr., 2003, D59, 334 CAS.
| This journal is © The Royal Society of Chemistry 2006 |