Nucleophilic identity substitution reactions. The reaction between hydrogen fluoride and protonated alkyl fluorides†
Received
21st September 2005
, Accepted 8th November 2005
First published on 28th November 2005
Abstract
The gas phase reactions between HF and the protonated alkyl fluorides MeFH+, EtFH+, PriFH+, and ButFH+ have been studied using ab initio methods. The potential energy profiles for both nucleophilic substitution (SN2) and elimination (E2) pathways have been investigated. Both backside Walden inversion and frontside nucleophilic substitution reaction profiles have been generated. Backside substitution is very favourable, but shows relatively little variation with the alkyl group. Frontside substitution reaction barriers are only slightly higher than the barrier for backside substitution for HF + MeFH+, and the difference in barrier heights for frontside and backside displacement seems negligible for the larger alkyl groups. Reaction barrier trends have been analysed and compared with the results of similar studies of the H2O/ROH2+ and NH3/RNH3+ systems (R = Me, Et, Pri, and But). Compared to the two other classes, protonated fluorides have extreme structures which, with the exception of the Me substrate, are weakly bound complexes between an alkyl cation and HF. The results nourish the idea that nucleophilic substitution reactions are better understood in view of competition between frontside and backside substitution than from the traditional SN1/SN2 perspective.
Introduction
Many chemical reactions are catalyzed by acids,1,2 and very often the key step is protonation of the substrate molecule at a hetero atom. Typically, this leads to bond activation of polar bonds like C–O, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, C–N and C–X (X = F, Cl, Br, I). As a direct result of protonation, the heterolytic bond dissociation energies are lowered. A most remarkable feature of protonation is encountered for aryl and alkyl fluorides, both in the gas phase and in super acidic solutions. While bond distances between carbon and electronegative atoms normally increase somewhat upon protonation, C–F bonds are extreme in becoming very long, in some cases close to being broken.3–11 This stands in stark contrast to the native C–F bond which is very robust, as in Teflon.
O, C–N and C–X (X = F, Cl, Br, I). As a direct result of protonation, the heterolytic bond dissociation energies are lowered. A most remarkable feature of protonation is encountered for aryl and alkyl fluorides, both in the gas phase and in super acidic solutions. While bond distances between carbon and electronegative atoms normally increase somewhat upon protonation, C–F bonds are extreme in becoming very long, in some cases close to being broken.3–11 This stands in stark contrast to the native C–F bond which is very robust, as in Teflon.
Recently, we studied the reactions between water and protonated alcohols in the gas phase, both experimentally and theoretically.12,13 The alcohols become much more susceptible to nucleophilic substitution and elimination upon protonation. Our study gave one very surprising result, namely that the SN2 reactivity trend with regard to alkyl substitution is opposite to what is normally found in solution. This challenges the idea of steric hindrance as the reason behind the normal reactivity trend (CH3 > CH3CH2 > (CH3)2CH > (CH3)3C). Obviously, factors other than the size of the alkyl group are playing a role. We have also studied the analogous reaction between ammonia and protonated amines.14 In this reaction the normal reactivity trend is encountered. We note that the polarity of the C–N bond, both in the neutral and protonated amines, is lower than in the corresponding C–O bonds, so the electronegativity of the heteroatom seems to play a key role in alkyl group reactivity trends.
Another interesting finding of the calculations is that for reactions of water with protonated isopropanol or tertiary butanol, the potential energy barriers for frontside substitutions are only slightly higher than for the normal Walden backside substitution.12,13 Again, this appears atypical, at least for reactions of negatively charged nucleophiles on methyl substrates, which are known to have high barriers for frontside substitution.15,16 The feasibility of frontside substitution in sufficiently activated substrates is very interesting, and calls for mechanistic alternatives to classical SN1 and SN2. Clearly, more investigations into this phenomenon are required.
On the basis of these observations, it is logical to consider a more electronegative element than oxygen, and fluorine is the obvious choice. The identity nucleophilic substitution reaction
| | | HF + RFH+
→ HFR+
+ FH, | (1) |
with R = CH
3, CH
3CH
2, (CH
3)
2CH, and (CH
3)
3C was studied at an appropriate level of theory. This study parallels our previous theoretical investigations of the corresponding reactions with protonated
alcohols and
amines.
12–14 The main advantage of choosing an identity reaction is that the absence of thermodynamic driving forces provides insight into the intrinsic reactivity.
17,18 In addition to backside and frontside nucleophilic substitution we wanted to study the likelihood for elimination reactions when
hydrogen fluoride reacts with a protonated alkyl halide. While the chemical reactions under study are only achievable in the gas phase or in superacid solution, the results will be generally valid in defining a borderline for chemical reactivity within or beyond the S
N2/S
N1 paradigm.
Computational details
Quantum chemical calculations were carried out using the program system GAUSSIAN 03.19 All relevant critical points (reactants, transition structures, intermediates and products) of the potential energy surface were characterized by complete optimization of the molecular geometries at the MP2/6-31++G(d,p) level of theory. Geometry optimizations were performed with the full analytical Hessian calculated at every geometry step and without any geometrical or symmetry constraints. Harmonic frequencies were also calculated using this wave function by diagonalizing the mass-weighed Cartesian force constant matrix, calculated from the analytical second derivatives of the total energy (the Hessian). The energies presented here are 0 K energies including the zero point vibrational energies (ZPVEs). Proton affinities at room temperature (PAs) were estimated by adding 5/2 RT (with T = 298 K) to the 0 K enthalpy differences between the base and the corresponding acid, thereby ignoring heat capacity differences.| | | A + H+
→ AH+, PA = −
ΔHo | (2) |
Results and discussion
Accuracy considerations and choice of method
The compound G2 and G3 methods provide estimates of calculations at the QCISD(T)/6-311 + G(3df,2p) and QCISD/G3Large levels, respectively, through a series of lower level calculations.20,21 It has previously been established that both methods are excellent in reproducing the appropriate thermochemical quantities as well as barrier heights with an accuracy of the order of 5 kJ mol−1.13,14,22 This applies for both SN2 and E2 gas phase reactions. One limitation of G2 and G3 is that geometry optimization is carried out at the relatively modest MP2/6-31G(d) level. Usually this provides a very good fundament for accurate energetics for a majority of simple organic molecules since molecular geometries of first, second and third row atoms normally give very reasonable bond lengths and angles with MP2/6-31G(d). It is convenient that errors in geometries scale to the square in energy, but the major advantage in calculations of relative energies is that most of the errors due to displaced geometries cancel out because geometrical errors tend to be systematic. The present case, however, poses a challenge in this respect, since it is well known that small basis sets, lacking diffuse functions, often give quite poor molecular geometries of fluorine containing molecules. The polar C–F and H–F bonds, and in particular the low polarizability of the fluorine atom, may create difficulties in describing molecules with weak bonding such as complexes and transition structures.
The problems with the 6-31G(d) basis set became evident after some time. A first indication was encountered in the case of the HF dimer–one product of a potential elimination reaction. Geometry optimization with MP2/6-31G(d) gives rise to a rhombic Cs arrangement, in which the two H–F dipoles are oriented parallel, but pointing oppositely. Klopper et al. demonstrated that inclusion of diffuse functions in the basis set is prerequisite for reproducing the spectroscopically known non-linear, head-to-tail arrangement.23 We found that by using MP2/6-31++G(d,p) this characteristic is fully accounted for. We therefore expect that the geometrical arrangements in the many weakly bound species involved in this study are more correctly described with this wave function than MP2/6-31G(d). In addition to this, MP2/6-31++G(d,p) should give accurate relative energies, although not as accurate as G2/G3-like calculations based on MP2/6-31++G(d,p) geometries.
To validate our calculated energetics, we compare calculated PAs with experimental. As evident from Table 1 the comparison is very favourable.
Table 1 Proton affinities (kJ mol−1)
| Molecule |
MP2/6-31++G(d,p) |
Experimenta |
|
Reference 26.
|
| HF |
480 |
484 |
| (HF)2 |
600 |
|
| Ethene |
683 |
683 |
| Propene |
745 |
752 |
| Isobutene |
802 |
802 |
The geometry of methyl fluoride is correctly reproduced with MP2/6-31G(d). The C–F bond length is calculated to be 1.392 Å, compared to the experimental re = 1.380 Å. Upon protonation the MP2/6-31G(d) bond length becomes 1.606 Å, which should be compared with the MP2/6-31++G(d,p) value of 1.631 Å. The MP2/6-31G(d) geometry used in the G2 method should therefore provide a solid basis for the higher level calculations, and we would expect the G2 barrier height for the reaction
| | | HF + CH3FH+
→ CH3FH+
+ HF, | (3) |
of −34 kJ mol
−1 as a highly reliable estimate of the true value.
24 The MP2/6-31++G(d,p) value of −37 kJ mol
−1 is therefore very satisfying. On the basis of these
indicators we would expect that barriers and relative energies are somewhat less accurately described with MP2/6-31++G(d,p) than with a G2- or G3-like scheme based on the more accurate MP2/6-31++G(d,p) geometries. We estimate the average error to be less than 10 kJ mol
−1.
After having established the reliability of our quantum chemical scheme, the rest of the manuscript will be devoted to presentation and discussion of the MP2/6-31++G(d,p) data.
General results
Optimized geometries for the stationary points on the MP2/6-31++G(d,p) potential energy surface (PES) for reactions eqn (1) and the competing E2 reactions are presented in Figs. 1–4. Cartesian co-ordinates for all species and imaginary frequencies of vibrations for the transition structures are given in the electronic supplementary information (ESI)†. The corresponding potential energy profiles are given in Figs. 5–8.
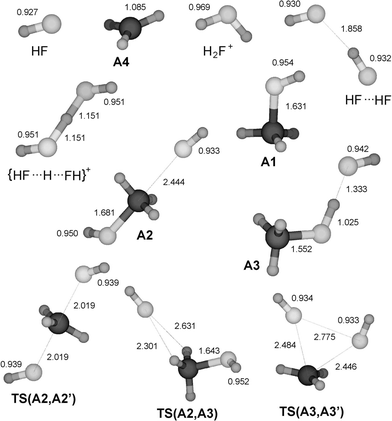 |
| | Fig. 1 Structures of stationary points on the PES for the substitution reaction between HF and MeFH+ as well as HF, H2F+, and their adduct (the protonated dimer of HF) calculated at the MP2/6-31++G(d,p) level. All bond lengths are given in Å. Cartesian co-ordinates for these structures have been included as ESI†. | |
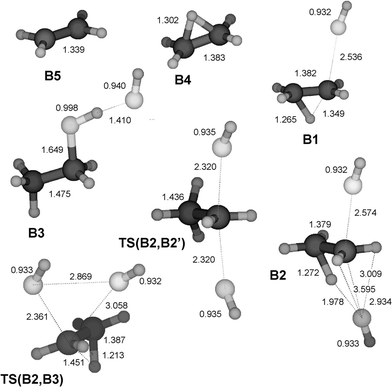 |
| | Fig. 2 Structures of stationary points on the PES for the substitution and elimination reaction between HF and EtFH+ calculated at the MP2/6-31++G(d,p) level. All bond lengths are given in Å. Cartesian co-ordinates for these structures have been included as ESI†. | |
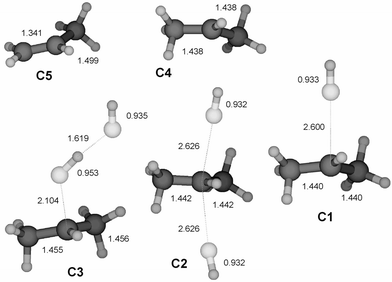 |
| | Fig. 3 Structures of stationary points on the PES for the substitution and elimination reaction between HF and PriFH+ calculated at the MP2/6-31++G(d,p) level. All bond lengths are given in Å. Cartesian co-ordinates for these structures have been included as ESI†. | |
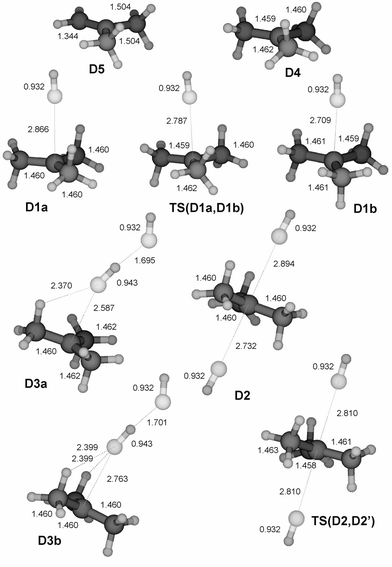 |
| | Fig. 4 Structures of stationary points on the PES for the substitution and elimination reaction between HF and ButFH+ calculated at the MP2/6-31++G(d,p) level. All bond lengths are given in Å. Cartesian co-ordinates for these structures have been included as ESI†. | |
Dissociation RFH+
→ R+
+ HF
Protonated ethyl, isopropyl and tertiary butyl fluoride (structures B1, C1 and D1) display similar behaviour towards dissociation, while protonated methyl fluoride, CH3FH+ (A1), is an exception. Despite a significant increase in the C–F bond length upon protonation, the latter does not dissociate and keeps its integrity as a covalently bonded molecule, although the bond dissociation energy for the processis rather low (Table 2). On the basis of the data of Table 2 the rest of the molecules must be considered to be weakly (largely electrostatically) bound complexes R+⋯FH, having bond dissociation energies below 50 kJ mol−1. This is illustrated by the fact that the interaction energy between an ion and a permanent dipole of 2.1 D (corresponding to the hydrogen fluoride molecule) is calculated to be 20 kJ mol−1 at a distance of 3 Å. As a consequence of this, we would expect the chemical properties of these R+⋯FH species to be closer to that of the separate constituents than of a covalently bonded RFH+.
Table 2 Binding in protonated alkyl fluorides at the MP2/6-31++G(d,p) level compared with available experimental data
| Molecule |
C–F bond distance in RFH+/Å |
Calc. heterolytic BDE vs. exp./kJ mol−1 |
|
References 26,33
|
| MeF |
1.631 |
124 (125a) |
| EtF |
2.536 |
43 (55a) |
| PriF |
2.600 |
46 (43a) |
| ButF |
2.866 |
42 |
Reactions HF + MeFH+
The gross features of the PES of HF/CH3FH+ are the same as for the previously studied systems NH3/RNH3+ and H2O/ROH2+ (Fig. 5). As will be evident below, the situation is not the same for the higher alkylated systems. Encounters between HF and CH3FH+ (A1) may give rise to the backside complex HF⋯CH3FH+(A2) or the frontside complex CH3FH+⋯FH(A3), the latter being the more stable. A barrier due to the symmetric transition structure TS(A2,A3) separates the two complexes. All these species have potential energies well below the reactants. From the backside complex there is a route viaTS(A2,A2′) accomplishing nucleophilic substitution in the traditional backside fashion. The reaction is energetically very favourable, with the transition structure at −37.3 kJ mol−1, only 5.2 kJ mol−1 above the complex A2. It appears there are no reports on experiments of reactions between HF and protonated alkyl fluorides. However, it is known that HF in CH3FH+ is readily displaced by xenon.25
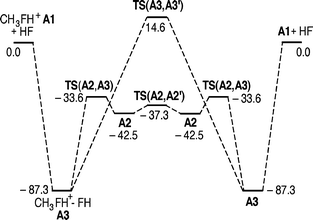 |
| | Fig. 5 Potential energy diagram for the substitution reaction between HF and MeFH+ calculated at the MP2/6-31++G(d,p) level. All relative energies are given in kJ mol−1 at 0 K. | |
It is remarkable that there is also a possible route for frontside SN2 substitution at methyl viaTS(A3,A3′) with a barrier as low as +14.6 kJ mol−1. The signature of the reaction co-ordinate is clearly that of one HF molecule substituting the other. This was confirmed by finding that the two intrinsic reaction co-ordinate (IRC) paths starting at the TS end at A3 and A3′. Although frontside substitution is quite possible for the identity reactions between water and protonated alcohols having large alkyl groups, R = (CH3)2CH, and (CH3)3C,12,13 the transition structures for the homologous frontside substitutions
| | | H2O + CH3OH2+
→ CH3OH2+
+ H2O, | (5) |
| | | NH3
+ CH3NH3+
→ CH3NH3+
+ NH3, | (6) |
are unreachable under thermal conditions, lying at 120 and 226 kJ mol
−1, respectively.
12–14 The corresponding values for the backside TSs are at 3 and 56 kJ mol
−1.
Reactions HF + EtFH+
The structure resulting from geometry optimization of protonated ethyl fluoride depends strongly on the quantum chemical method. MP2/6-31G(d) gives the classical structure CH3CH2FH+, while MP2/6-31++G(d,p) which includes the important low exponent functions in the basis set, gives the non-classical structure B1 illustrated in Fig. 2. Protonated ethyl fluoride forms a backside complex (B2) with HF (having a non-classical ethyl moiety), as well as a frontside complex (B3) (having a classical ethyl moiety). One should notice that HF association reinforces the C–F bond considerably by shortening it by 0.9 Å and by strengthening it (Fig. 6). Also for this reaction system, we find a very attractive TS for SN2 backside displacement at TS(B2,B2′), lying at −27.7 kJ mol−1. Upon searching for a transition structure for a frontside substitution we found a transition structure at −26.6 kJ mol−1 which from its geometry appeared to be a candidate. However, running the IRC revealed this not to be the case. As a matter of fact, the IRC from this TS ends up at B2 and B3, respectively, showing that the proper description is TS(B2,B3).
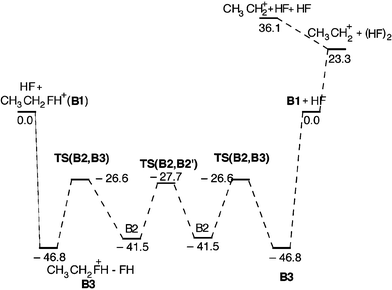 |
| | Fig. 6 Potential energy diagram for the substitution and elimination reaction between HF and EtFH+ calculated at the MP2/6-31++G(d,p) level. All relative energies are given in kJ mol−1 at 0 K. | |
It was not possible to locate a proper transition structure for frontside displacement, and the looseness of a transition structure resembling B3 could hamper its location if it exists. Most likely, however, it is a multi-step reaction. In the extreme case, frontside substitution can be envisaged to occur in three formal steps, involving full bond dissociation:
| CH3CH2FH+⋯FH (B3) → CH3CH2+
+ (FH,FH) → |
| | | → CH3CH2+
+ (FH,FH) → CH3CH2FH+⋯ FH(B3′) | (7) |
The theoretical upper limit for frontside displacement is therefore 23.3 kJ mol
−1 (corresponding to the energy of CH
3CH
2+
+ (HF)
2,
Fig. 6). Most likely the necessary reorientation of the two HF molecules may occur within a bonded complex at a potential energy below zero, including both the species
B2 and
B3. This option will be discussed below in conjunction with the two largest alkyl fluorides.
Elimination products were observed in the experiments with water and protonated alcohols. In reactions between hydrogen fluoride and ethyl fluoride, we therefore investigated the possibility for an E2 reaction. From Fig. 6 it is evident that forming the ethyl cation plus the dimer (HF)2 requires 23.3 kJ mol−1. Elimination would require subsequent or concerted transfer of a proton from ethyl to this dimer. Since the proton affinity of the HF dimer is calculated to be 601 kJ mol−1 (Table 1) the total elimination reaction
| | | HF + CH3CH2FH+
→ CH2CH2
+ (HF⋯H⋯FH)+ | (8) |
would be around 100 kJ mol
−1 endothermic, since
ethylene has a
proton affinity of 680.5 kJ mol
−1.
26 For the higher alkyl fluoride the reaction becomes even less realistic, since the
proton affinities of the corresponding
alkenes are higher.
Reactions HF +PriFH+
For protonated isopropyl fluoride (C1) the reactivity trends reported above are enforced (Fig. 7). Interestingly, the backside complex (C2) is now slightly lower in potential energy than the frontside complex (C3). Moreover, a genuine transition structure for a backside Walden inversion no longer exists. The backside complex and the TS have collapsed into one stable symmetric minimum structure (C2). This is exactly the same situation that Ruggiero and Williams have described for He/CH3He+ and Ne/CH3Ne+.27 It should also be mentioned in this context that for third and fourth row nucleophiles, symmetric minimum energy structures of the type [Nu⋯R⋯Nu] appear to be the rule, rather than the exception.28–30 In a new paper we have discussed this in more general terms, especially in relationship to the periodic table.24
Also for this system it is not possible to localize a proper TS for frontside substitution. However, the reaction is very likely to take place, probably with nearly the same probability as backside substitution. This can be inferred from a limited number of HF/3-21G Born Oppenheimer dynamics calculations of the reaction.31 From these simulations it appears that frontside nucleophilic substitution reactions between hydrogen fluoride and protonated isopropyl fluoride occurs via a mechanism which has close resemblance to a billiard ball game, since both the incoming and the departing HF interact with the central C3H7+ moiety with weak electrostatic forces. Momentum transfer seems to be an essential part of the mechanism. The simulations show that also backside substitution happens via a sequence dominated by momentum transfer events.
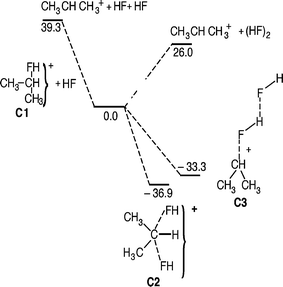 |
| | Fig. 7 Potential energy diagram for the substitution and elimination reaction between HF and PriFH+ calculated at the MP2/6-31++G(d,p) level. All relative energies are given in kJ mol−1 at 0 K. | |
Reactions HF +ButFH+
The interactions within protonated butyl fluoride (CH3)3C⋯FH+(D1) and the collision complexes HF/D1 turned out to be so weak that the binding within these structures and their reactions are subject to the conformation of the central tertiary butyl cation. Protonated t-butyl fluoride has two conformers, D1a and D1b (Fig. 4). An almost degenerate transition structure TS(D1a,D1b) separates the two, with all three stationary points effectively at the same potential energy. The conformers differ by having two or one C–H bonds pointing outwards to the fluorine. Exactly the same phenomenon is observed for the frontside complexes, D3a and D3b. These two species are effectively of the same potential energy. Slight shortening of the C⋯F contact can be observed upon formation of the frontside complexes. The backside complex (D2) is of the same energy as the frontside complexes (Fig. 8). The C⋯F distances are slightly different, one (the shortest) approximately as in D1a, while the other corresponds to D1b. The bond lengths are in accordance with the positions of the C–H bonds of the methyl groups relative to each of the fluorine atoms, as in D1a and D1b. The transition structure TS(D2,D2′) which separates D2 from its mirror image D2′, is formally that of an SN2 reaction, with a symmetric arrangement of the to HF groups relative to the alkyl. In reality, however, the arrangement in TS(D2,D2′) is the result of rotation of one of the methyl groups, as for TS(D1a,D1b). The potential energy does not change significantly as the results of these movements, and the potential energy surface is essentially flat. Here, as demonstrated by dynamics calculations, nucleophilic substitution has more or less degenerated into billiard ball ligand exchange.31
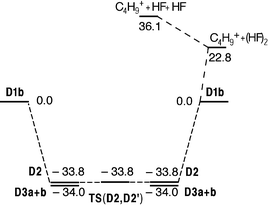 |
| | Fig. 8 Potential energy diagram for the substitution reaction between HF and ButFH+ calculated at the MP2/6-31++G(d,p) level. All relative energies are given in kJ mol−1 at 0 K. | |
Properties of protonated alkyl fluorides
In many respects, protonated alkyl fluorides lie in between protonated alkanes and protonated alkyl chlorides;4 the proton affinity of methyl fluoride is the arithmetic mean of that of methane and that of methyl chloride. The relative increase in the C–F bond upon protonation appears to be amid C–Cl and C–H. For example, the C–Cl bond in methyl chloride increases quite modestly (0.07 Å)24 while methane gives rise to a non-classical side-on adduct between H2 and a methyl cation.32 From Fig. 1, we see that the C–F binding in A1 is in between. However, in the second row of the periodic table (alcohols, amines, alkyl fluorides) fluorine is at the extreme, being the most electronegative element. With the exception of the cationic noble gas species RE+ (E = He, Ne) and protonated alkanes, protonated fluorides give rise to structures which are closest to naked alkyl cations. In this respect, HF exchange appears to be close to the archetype SN1 reaction.
Nucleophilic substitution mechanisms
The weakness of the SN2/SN1 paradigm of nucleophilic substitution is not that it requires the free alkyl cation as the limiting situation, but that mechanism and stereochemistry are linked together in an artificial way. Upon going from SN2 to SN1, somewhere along the line there is an abrupt change from a bimolecular reaction to a unimolecular reaction, or a gradual shift from SN2 to SN1. In the latter, the mixed mechanism has usually been used to account for the normal result of enantiomeric mixtures different from 50 : 50 and 100 : 0, and different mechanistic modifications are necessary to explain salt effects and solvent participation.1 As our examples show, a much simpler and more realistic picture is obtained by invoking the topographically distinct backside and frontside substitution situations, and realizing that frontside substitution becomes gradually more competitive when the alkyl group becomes bigger and the leaving group/nucleophile becomes better.
Conclusion
Comparing the present results on HF/RFH+ with the earlier ones on alcohols (H2O/ROH2+)12,13 and amines (NH3/RNH3+)14 the following trends are discovered.
For a given alkyl group, the barrier for identity SN2 reaction (backside displacement) is highest for the protonated amines, decreasing via the protonated alcohols to the protonated alkyl fluorides. This has recently been explained as resulting from differences in the electronegativity of the hetero atom.24 For the amines, the normal textbook “steric effect” is observed with barriers increasing with size (Me < Et < Pri < But). For the alcohols a practically inverse behaviour is observed, while the fluorides show little variation with alkyl substitution.
Within each class (protonated amines, alcohols and fluorides) the barriers for frontside displacement decrease with increasing size of the alkyl group, following the general pattern (Me > Et > Pri > But). By making the alkyl group larger and the hetero atom more electronegative, the barrier for frontside substitution becomes gradually closer to that for backside substitution. While the difference for MeNH3+ is 172 kJ mol−1, it has disappeared for ButFH+.
Elimination (to give the corresponding alkene; CH2CH2, CH3CHCH2 > (CH3)2CCH2) becomes less favourable the larger the alkyl group is, since the thermodynamical factor, i.e.PA of alkene dominates, except for CH3CH2OH2+ which has a small barrier for the proton transfer.12,13 The combination of a favourable enthalpy for heterolytic C–XH+ bond dissociation enthalpy and PA (for XH as well at its dimer) makes elimination easier for the alcohols compared to the corresponding amines and fluorides.
Acknowledgements
We are grateful for a generous grant of computing time from NOTUR (The Norwegian High Performance Computing Consortium).
References
-
T. H. Lowry and K. S. Richardson, Mechanism and Theory in Organic Chemistry, Harper & Row, 1981 Search PubMed
 .
.
- E. Uggerud, Mass Spectrom. Rev., 1992, 11, 389 CAS .
- D. M. Brouwer, Rec. Trav. Chim. Pays-Bas, 1968, 87, 342 CAS .
- B. A. Hess, Jr. and R. Zahradnik, J. Am. Chem. Soc., 1990, 112, 5731 CrossRef .
- J. Hrusak, D. Schroeder, T. Weiske and H. Schwarz, J. Am. Chem. Soc., 1993, 115, 2015 CrossRef CAS .
- J. L. M. Abboud, R. Notario, E. Ballesteros, M. Herreros, O. Mo, M. Yanez, J. Elguero, G. Boyer and R. Claramunt, J. Am. Chem. Soc., 1994, 116, 2486 CrossRef CAS .
- H. Yamaoka, R. H. Fokkens, S. Tajima, H. Yamataka and N. M. M. Nibbering, Bull. Soc. Chim. Bel., 1997, 106, 399 Search PubMed .
- L. Gonzalez, O. Mo and M. Yanez, J. Phys. Chem. A, 1998, 102, 1356 CrossRef CAS .
- D. Schroeder, I. Oref, J. Hrusak, T. Weiske, E. E. Nikitin, W. Zummack and H. Schwarz, J. Phys. Chem. A, 1999, 103, 4609 CrossRef CAS .
- N. Solca and O. Dopfer, J. Am. Chem. Soc., 2003, 125, 1421 CrossRef CAS .
- O. Sekiguchi, D. Watanabe, S. Nakajima, S. Tajima and E. Uggerud, Int. J. Mass Spectrom., 2003, 222, 1 CrossRef CAS .
- L. Bache-Andreassen and E. Uggerud, Chem.–Eur. J., 1999, 5, 1917 CrossRef CAS .
- J. K. Laerdahl and E. Uggerud, Org. Biomol. Chem., 2003, 1, 2935 RSC .
- J. K. Laerdahl, L. Bache-Andreassen and E. Uggerud, Org. Biomol. Chem., 2003, 1, 2943 RSC .
- M. N. Glukhovtsev, R. D. Bach, A. Pross and L. Radom, Chem. Phys. Lett., 1996, 260, 558 CrossRef CAS .
- S. Harder, A. Streitwieser, J. T. Petty and P. v. R. Schleyer, J. Am. Chem. Soc., 1995, 117, 3253 CrossRef CAS .
- S. Wolfe, D. J. Mitchell and H. B. Schlegel, J. Am. Chem. Soc., 1981, 103, 7692 CrossRef CAS .
- S. Wolfe, D. J. Mitchell and H. B. Schlegel, J. Am. Chem. Soc., 1981, 103, 7694 CrossRef CAS .
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Montgomery, Jr., T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. Ochterski, P. Y. Ayala, K. Morokuma, G.
A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. G. Johnson, W. Chen, M. W. Wong, C. Gonzalez and J. A. Pople, GAUSSIAN 03 (Revision C.02), Gaussian, Inc., Wallingford, CT, 2004 Search PubMed .
- L. A. Curtiss, K. Raghavachari, G. W. Trucks and J. A. Pople, J. Chem. Phys., 1991, 94, 7221 CrossRef CAS .
- L. A. Curtiss, K. Raghavachari, P. C. Redfern, V. Rassolov and J. A. Pople, J. Chem. Phys., 1998, 109, 7764 CrossRef CAS .
- J. K. Laerdahl and E. Uggerud, Int. J. Mass Spectrom., 2002, 214, 277 CrossRef CAS .
- W. Klopper, M. Quack and M. A. Suhm, J. Chem. Phys., 1998, 108, 10096 CrossRef CAS .
-
E. Uggerud, Chem.–Eur. J., in press, 2005. DOI10.1002/chem.200500639 Search PubMed .
- D. Holtz and J. L. Beauchamp, Science, 1971, 173, 1237 CrossRef CAS .
-
S. G. Lias, H. M. Rosenstock, K. Deard, B. W. Steiner, J. T. Herron, J. H. Holmes, R. D. Levin, J. F. Liebman, S. A. Kafafi, J. E. Bartmess and E. F. Hunter, in ‘NIST-webbook’, (http://webbook.nist.gov/chemistry/), 1997 Search PubMed.
- G. D. Ruggiero and I. H. Williams, J. Chem. Soc., Perkin Trans. 2, 2002, 591 RSC .
- T. I. Solling, S. B. Wild and L. Radom, Inorg. Chem., 1999, 38, 6049 CrossRef CAS .
- T. I. Solling and L. Radom, Eur. J. Mass Spectrom., 2000, 6, 153 CrossRef CAS .
- T. I. Solling and L. Radom, Chem.–Eur. J., 2001, 7, 1516 CrossRef CAS .
-
K. A. Jakobsen, ‘Dynamiske simuleringer av SN2-og E2-reaksjoner i gassfase’, Cand. scient., University of Oslo, Oslo, 2004 Search PubMed .
- E. T. White, J. Tang and T. Oka, Science, 1999, 284, 135–137 CrossRef CAS .
- S. G. Lias, J. E. Bartmess, J. F. Liebman, J. H. Holmes, R. D. Levin and W. G. Mallard, J. Phys. Chem. Ref. Data, 1988, 17, 1 .
Footnote |
| † Electronic supplementary information (ESI) available: Cartesian co-ordinates for all species and imaginary frequencies of vibrations for the transition structures. See DOI: 10.1039/b513315g |
|
| This journal is © The Royal Society of Chemistry 2006 |
Click here to see how this site uses Cookies. View our privacy policy here.