Electrophilic fluorination of organosilanes
Matthew
Tredwell
and
Véronique
Gouverneur
*
University of Oxford, Chemistry Research laboratory, 12, Mansfield Road, Oxford, UK OX1 3TA. E-mail: veronique.gouverneur@chem.ox.ac.uk; Fax: +44 (0)1865 275 644
First published on 23rd November 2005
Abstract
The fluorination of organosilanes with the silyl groups directly attached or adjacent to an aryl or alkenyl group has been only very recently examined despite the fact that the corresponding fluorinated products are synthetically useful building blocks. In these reactions, the silyl group enhances the reactivity of the π-nucleophile and controls the sense of regiochemistry upon addition of the electrophilic source of fluorine. These reactions take advantage of the β effect of the silicon–carbon bond and recent results from the literature revealed that this chemistry allows for the preparation of a variety of novel fluorinated building blocks including enantioenriched derivatives.
 Matthew Tredwell | Matthew Tredwell studied for his MChem at St Catherine's College, Oxford. He completed his Part II research project under the supervision of Dr Gouverneur, where he worked on biocatalysis applied to the preparation of versatile enantiopure building blocks. At present he is carrying out his DPhil, funded by the EPSRC (GR/S43283/01), in the Gouverneur group, where his research focuses on the electrophilic fluorodesilylation of chiral allylsilanes and the synthesis of fluorinated biologically active targets. |
 Véronique Gouverneur Véronique Gouverneur | Véronique Gouverneur has been a University Lecturer in Organic Chemistry at the University of Oxford since 1998. Since her appointment in Oxford, she also holds a Tutorial Fellowship at Merton College. After her PhD work with Professor Léon Ghosez at the Université Catholique de Louvain, she left Belgium and moved to a postdoctoral position with Professor Richard Lerner at the Scripps Research Institute (California, USA) where her work culminated with the generation of exo Diels–Alderase antibodies. She returned to Europe in 1994 where she accepted a position of Maître de Conférence at the Université Louis Pasteur in Strasbourg (France) with Dr Charles Mioskowski, then moved to her current position. Her research on fluorine chemistry and her specific interests in new reaction design, enantioselective catalysis and the synthesis of bio-relevant targets has been recently recognised with an AstraZeneca Research Award in Organic Chemistry (2005). |
Introduction
Ever since the first synthesis of hydrofluoric acid by Margraf in 1764 and of elemental fluorine by Moissan in 1886, fluorine chemistry has come a long way as reflected by the impressive range of applications of fluorinated compounds in our daily life. Very recently, several books, reviews and perspective papers have highlighted the importance of fluorine chemistry and speculated on its future. They discussed why fluorine-containing organic materials are commonly used in materials, medicinal, pharmaceutical and agrochemical science and how the incorporation of fluorine into an organic compound perturbs its chemical, physical and biological properties.1 Because naturally occurring organofluorine compounds are rare, the preparation of fluorine-substituted target molecules depends heavily on modern synthetic chemistry. Key strategies for their preparation include the “fluorination method”, the “building block method” and a combination of these two approaches. The former allows for the synthesis of an increasing repertoire of fluorinated target molecules along with intermediates that provide scope for the “building block” approach. For some applications, such as the preparation of fluorine-18 labelled tracers for positron emission tomography, the fluorination method is the most suitable strategy as the short half-life of fluorine-18 (t1/2 = 110 minutes) necessitates the introduction of the fluorine atom at as late a stage as possible in the synthetic sequence.2 The involvement of many research groups in synthetic fluorine chemistry has been driven by the unusual reactivity sometimes observed when using fluorinated building blocks or by the need to control product outcome and overcome selectivity issues when working with fluorinating reagents. As a result, ingenious methods for the preparation of fluorinated compounds have been disclosed to address some of these challenges.3 With the appearance of “easy to handle” electrophilic fluorinating reagents and the importance of organometallic chemistry, it is noteworthy that, putting aside the preparation of α-fluorinated carbonyl derivatives, relatively little progress has been made to access fluorinated targets or intermediates by substitution of metals especially for the preparation of compounds with the fluorine on an sp3 hybridised carbon. In this account, we briefly outline some of the problems associated with the fluorination of organometallic species and we describe how organosilanes other than silyl enol ethers are emerging as a new class of suitable precursors for the preparation of structurally diverse fluorinated targets or building blocks, including homochiral compounds with the fluorine atom on a stereogenic centre.Organosilanes versus other organometallic derivatives as precursors for the preparation of fluorinated compounds
The reactivity of organometallics such as lithium, tin, germanium and silicon in the presence of an electrophilic source of fluorine has been examined mainly for the formation of aryl fluorides probably due to the importance of this motif in the pharmaceutical industry (Scheme 1).4 | ||
| Scheme 1 Electrophilic fluorination of organometallics for the preparation of aryl fluorides. | ||
Vinyllithium, tin, and boron derivatives have also been converted into the corresponding fluoroalkenes upon treatment with an electrophilic source of fluorine. Molecular fluorine, perchloryl fluoride, caesium fluoroxysulfate, xenon difluoride or diverse N–F reagents were found to be suitable reagents for these transformations allowing for the preparation of fluoroalkenes with good yields (Scheme 2).5
 | ||
| Scheme 2 Electrophilic fluorination of trifluoroborates, vinylstannanes and vinyllithium. | ||
With the exception of α-fluorinated carbonyl derivatives, the use of organometallic precursors for the preparation of fluorinated compounds, for which the fluorine atom is attached to an sp3-hybridised carbon has not been examined to a great extent, despite the synthetic value of the resulting fluorinated products. Until recently, only organolithium, Grignard reagents, organothallium and organomercury derivatives were successfully reacted in the presence of electrophilic sources of fluorine for the preparation of simple fluorinated compounds with the fluorine atom attached to an sp3-hybridised carbon (Scheme 3).6
 | ||
| Scheme 3 Direct substitution of metals on an sp3-hybridised carbon with fluorine. | ||
Altogether, the following trends emerge from these results. In reaction with an electrophilic source of fluorine, organolithium reagents are advantageous in comparison with Grignard reagents due to the lack of a halogen partner present to neutralise the bivalent magnesium metal. The presence of a bromide or iodide ion complicates the electrophilic fluorination process as oxidation of the halogen ion, particularly iodide can occur as a competitive process. The group 1 organolithium reagents can be prepared halogen-free and present the added advantage of being generally more reactive than the group 2 organometallics. However, the basicity of these lithiated precursors prevents the use of some N–F reagents that decompose under strong basic conditions. The chemistry of the milder organolead, tin, mercury and thallium species has been little examined probably due the high toxicity of their derivatives. Arylgermanium compounds have been used as an alternative to aryltin but the yields obtained upon fluorination with elemental fluorine and acetyl hypofluorite are generally lower due to the stronger C–Ge bond. The lower reactivity coupled with the high cost of germanium is probably the reason why little research has been done in this area.7 Organosilanes present the advantage of being safe to handle but in comparison with the corresponding tin or germanium derivatives, the increase of carbon–metal bond energy and the decrease in carbon–metal bond lengths result in lower reactivity. This is likely the reason why this class of compounds was overlooked as an alternative to other organometallic species. However, recent results suggest that these compounds are now emerging as valuable precursors for the preparation of a large variety of structurally diverse fluorinated compounds. Organosilanes with the silyl groups directly attached or adjacent to an aryl or alkenyl group are very useful synthetic intermediates as they can react with electrophiles to give structurally diverse products. In these reactions, the silyl group enhances the reactivity of the π-nucleophile and controls the sense of regiochemistry upon addition of the electrophile as illustrated with a representative allylsilane in Scheme 4. These reactions take advantage of the β effect of a silicon center and when using electrophilic sources of fluorine, recent results from the literature revealed that structurally diverse fluorinated compounds are now accessible from organosilanes, including enantioenriched targets.8 In this account, we will not cover the long-established reactivity of silyl enol ethers.
 | ||
| Scheme 4 Regioselective fluorination via a silicon-stabilised carbocation. | ||
Arylsilanes
The ipso-substitution of arylsilanes for the formation of aryl fluorides has been achieved using relatively strong fluorinating agents such as F2, AcOF, caesium fluoroxysulfate (CFS) and XeF2.9 Early reports described the fluorination of arylsilanes for the purpose of introducing 18F into radiopharmaceuticals.10 In general, when using molecular fluorine for the fluorodesilylation of arylsilanes, a large excess of substrate to fluorine is used to minimise unwanted side-products resulting from subsequent fluorination on the product. This limitation can be overcome by carrying out the reaction in a polar solvent in the presence of boron trifluoride, an additive that promotes polarisation of the F–F bond. Under these optimised conditions, a stoichiometric amount of the substrate can be used. For these reactions, secondary products are sometimes observed arising from fluorodeprotonation and/or 1,2-migration of the trimethylsilyl group. These 1,2-shifts are driven by the fact that the carbocation produced after migration of the silyl group is stabilised both by the β-silicon effect and the mesomeric effect of the newly introduced α-fluorine. Xenon difluoride in C6F6 has also been used as the fluorinating agent for the fluorodesilylation of arylsilane.9c For these reactions, it has been suggested that the mechanism of reaction involves an aryl radical as the key intermediate (Scheme 5). | ||
| Scheme 5 ipso-Electrophilic fluorosubstitution of para-substituted arylsilanes. | ||
N–F Reagents including 1-chloromethyl-4-fluoro-1,4-diazoniabicyclo[2.2.2]octane bis(tetrafluoro-borate) (Selectfluor) are not really suitable for the fluorodesilylation of arylsilanes as very low yields of the desired products are obtained even after prolonged reaction times. In addition, side products are observed, resulting from a competitive flurorodeprotonation process.11 These data suggest that only strong electrophilic fluorinating agents are suitable for the ipso fluorodesilylation of arylsilanes. This is likely to be the result of the loss of aromaticity of the ring upon formation of the β-silyl cation. In addition, the Si–C σ bond and the π orbital to be stabilised are initially orthogonal and consequently, stabilisation from the β-effect occurs only late in the approach to the transition state.
Vinylsilanes
Vinylsilanes, which are slightly more reactive toward electrophiles than arylsilanes, still require a highly reactive fluorinating agent for the fluorodesilylation process to occur. Recent studies demonstrated that vinylsilanes can be converted to fluoroalkenes upon treatment with Selectfluor in acetonitrile at room temperature (Scheme 6).12 | ||
| Scheme 6 Mono- and difluorination of vinylsilanes. | ||
When the starting vinylsilane is used as a single geometrical Z or E isomer, the corresponding fluoroalkene is formed as a mixture of stereoisomers with retention of stereochemistry as the predominant pathway [Scheme 6, eqn (1)]. The loss of stereochemical integrity is more important for vinylsilanes bearing substituents capable of stabilising inductively or mesomerically the presumably formed positively charged reaction intermediates. The formation of a mixture of stereoisomers and the faster reaction with the more nucleophilic vinylsilanes are consistent with an addition–elimination mechanistic pathway via a carbocationic intermediate followed by loss of the silyl group to restore neutrality. However the results could not rule out the possibility of a single electron transfer mechanism. The isolated yields for these reactions are moderate. Prolonged reaction time did not improve the yields as secondary products identified as difluoroamides are formed, resulting from further fluorination of the fluoroalkenes followed by reaction with acetonitrile. When alkenylsilanes are treated with more than one equivalent of Selectfluor, the corresponding difluoroamides are formed in good yields according to a Ritter-type fluoro-functionalisation with acetonitrile [Scheme 6, eqn (2)]. This reaction is relatively limited in scope and can be applied only to activated vinylsilanes. Indeed, vinylsilanes substituted with a single alkyl group do not deliver the corresponding difluorinated amide in the presence of an excess of Selectfluor because the reaction halted at the first stage affording the fluoroalkene as the only product. When the electrophilic fluorodesilylation of activated vinylsilanes was carried out in aqueous acetonitrile or in a mixture of methanol and acetonitrile, difluorinated alcohols or ethers are obtained with high chemical yields [Scheme 6, eqn (3)]. For these reactions, traces of the difluoroamides are always detected but could be easily separated by chromatography. The electrophilic double fluorination of vinylsilane bearing a strategically positioned alcohol group allows for the preparation of tetrahydrofurans and tetrahydropyrans bearing a difluoromethyl group, resulting from the intramolecular trapping of the difluorinated carbocationic intermediate with the primary alcohol functionality [Scheme 6, eqn (4)]. This is the only route to these compounds featuring the introduction of the fluorine as the last step of the synthetic sequence.
Allylsilanes
Allylsilanes, which are typically more reactive than vinyl- or arylsilanes, are perhaps the most useful type of silyl nucleophiles as they react with a wide variety of electrophiles.13 With a σ–π hyperconjugative stabilisation of the β-carbonium ion of approximately 30 kcal mol−1 combined with steric factors, the site of attack of the electrophile is at C-γ (C-3) with the development of a carbocation at C-β (C-2). The resulting carbocationic intermediate can undergo rapid loss of the silyl group allowing for the formation of a product with a net transposition of the double bond. The overall process is taking place according to an SE2′ mechanism. The electrophilic fluorodesilylation of allylsilanes has been the subject of three recent reports. Research conducted in our laboratories has established the utility of this process for the preparation of structurally diverse allylic fluorides including enantioenriched derivatives. The initial report described how diverse fiunctionalised allylic fluorides are prepared using a simple two-step process involving a cross-metathesis reaction of allyltrimethylsilane with different olefinic partners followed by a fluorodesilylation of the corresponding functionalised allylsilanes with Selectfluor (Scheme 7).14 The fluorodesilylation process occurs with clean transposition of the double bond as expected for a reaction taking place according to an SE2′ mechanism. The methodology has been applied to the preparation of terminal secondary and tertiary allylic fluorides bearing numerous functionalities such as ether, ester, acetal, protected amine as well as imido groups. With no side products detected in the crude reaction mixture, the isolated yields of allylic fluorides are generally very good. The reaction does not allow for the preparation of α-fluorinated carbonyl derivatives as the corresponding allylsilanes featuring an electron-deficient enone functionality is not sufficiently reactive toward Selectfluor.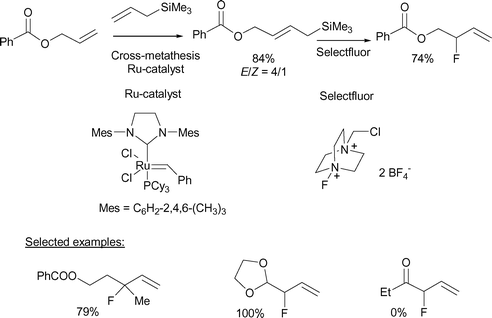 | ||
| Scheme 7 Sequential cross-metathesis/fluorodesilylation. | ||
The methodology has also been expanded to the preparation of enantioenriched allylic fluorides using two complementary strategies. The first approach relies on the use of non-racemic chiral organosilanes combined with achiral fluorinating reagent and the second involves the treatment of prochiral allylsilanes with chiral fluorinating reagents. The first approach has been applied to chiral allylsilanes possessing their stereogenic centre on the allylic carbon not substituted by the silyl group because these precursors could be readily prepared as single E-isomers by cross-metathesis of commercially available allyltrimethylsilane with olefinic partners which are the products resulting from an asymmetric deconjugative alkylation of known acetylated oxazolidinone (Scheme 8).15 Upon treatment with Selectfluor, the desired allylic fluorides were obtained as a mixture of diastereomers that could be separated cleanly by column chromatography. Hydrolytic cleavage of the chiral auxiliary is taking place in high yields affording the β-fluorinated carboxylic acids as single diastereo- and enantiomer. The corresponding alcohol was obtained by reduction of these fluorinated acids with lithium aluminium hydride. It is noteworthy that the direct reductive cleavage of the chiral auxiliary of the fluorinated oxazolidinones was not successful as a competitive elimination process occurred under these conditions.
 | ||
| Scheme 8 Asymmetric synthesis of allylic fluorides. | ||
A regio- and enantioselective synthesis of allylic fluorides has also been reported based on the use of enantiopure N–F reagents (Scheme 9).16 Allylsilanes derived from indanone and tetralone were reacted in the presence of chiral N-fluorocinchona alkaloids, which were prepared in situ by mixing the commercially available cinchona alkaloids with Selectfluor. Amongst the numerous alkaloids screened for this reaction, the reagent derived from (DHQ)2PYR was found to be most promising with enantiomeric excess up to 96% for the benzyl-substituted allylsilane derived from indanone 1b. In general, higher enantiomeric excesses were obtained for allylsilanes derived from indanones in comparison with the ones derived from tetralones. The substitution pattern of the substrate was also important with the best results obtained for substrates substituted by large groups. This study also showed that the replacement of the three methyl groups attached to the silicon by three phenyl groups was beneficial as reflected in increased enantiomeric excesses.
 | ||
| Scheme 9 Enantioselective fluorodesilylation of prochiral allylsilanes. | ||
Allenylmethylsilanes
The SE2′ reaction of allenylmethylsilanes in the presence of Selectfluor has also been recently investigated and allowed for the preparation of 2-fluoro-1,3-dienes (Scheme 10).17 | ||
| Scheme 10 Synthesis of fluorodienes from allenylmethylsilanes. | ||
For these reactions, acetone was found to be the solvent of choice in order to facilitate the work-up procedure and the purification of the product. This is the first route to these valuable compounds that is not based on the use of a fluorinated building block. To prevent the formation of the non-fluorinated diene, a side product resulting from a protodesilylation process, it is essential to carry out the reaction in the presence of 1.2 equivalent of NaHCO3. Using these reaction conditions, the best yields were obtained for substrates with a substituent that reinforces the β-effect of the trimethylsilyl group upon addition of Selectfluor. Racemic di- and trisubstituted allenylmethylsilanes led to the formation of the desired fluorodienes as mixtures of E/Z dienes (roughly 2 : 1) with the E isomer as the major compound. Assuming that an SE2′ mechanism is operating, the preferential formation of the E isomer indicates that Selectfluor approached the central carbon of the allenylmethylsilane from the opposite side to the sterically demanding R substituent, with the organosilane adopting a reactive conformation allowing early stabilisation of the developing positive charge. As the two E and Z isomers are difficult to separate, the methodology is best applied to fluorodienes that cannot be formed as mixtures of isomers unless the stereochemistry of the double bond is unimportant for subsequent transformations.
Outlook
The spectrum of organosilanes containing σ-bonded silyl groups attached to a participating π-bond and their use in organic synthesis has grown considerably over the past years. Until recently, little was done on the use of organosilanes for the preparation of fluorocompounds probably because preliminary work was centered on the less reactive aryl silanes and revealed that within this series, aryltin and arylgermanes were more suitable substrates. Vinylsilanes, allylsilanes and allenylmethyl silanes have now been identified as highly valuable vinylic, allylic and dienylic transfer reagents onto Selectfluor allowing for the formation of structurally diverse fluorinated building blocks. More recently, the fluorodesilylation of allenylsilanes has also been studied allowing for the preparation of propargylic fluorides.18 This chemistry takes advantage of the long-established β-effect of the silicon and opens unprecedented pathways for the stereoselective synthesis of cyclic and acyclic fluorinated targets. Exciting developments in this field in the near future are expected, including studies aimed at elucidating unambiguously the reaction mechanism of these reactions.References
- (a) P. Kirsch, Modern Fluoroorganic Chemistry, VCH, Wenheim, 2004 Search PubMed; (b) R. D. Chambers, Fluorine in Organic Chemistry, Blackwell Publishing CRC Press, 2004 Search PubMed.
- (a) M. R. Kilbourn, Fluorine-18 Labelling of Pharmaceuticals, Nuclear Science Series, NAS-NS-3203, National Academy Press, Washington, DC, 1990 Search PubMed; (b) M.-C. Lasne, C. Perrio, J. Rousden, L. Barré, D. Roeda, F. Dolle and C. Crouzel, Top. Curr. Chem., 2002, 222, 201–258 CAS; (c) W. C. Eckelman, Drug Discovery Today, 2003, 8, 404–410 CrossRef CAS.
- (a) J.-A. Ma and D. Cahard, Chem. Rev., 2004, 104, 6119–6146 CrossRef CAS; (b) R. P. Singh and J. M. Shreeve, Synthesis, 2002, 17, 2561–2578; (c) S. D. Taylor, C. C. Kotoris and G. Hum, Tetrahedron, 1999, 55, 12431–12477 CrossRef CAS; (d) S. T. Purrington, B. S. Kagen and T. B. Patrick, Chem. Rev., 1986, 86, 997–1018 CrossRef CAS.
- (a) M. J. Adam, T. J. Ruth, S. Jivan and B. D. Pate, Can. J. Chem., 1983, 61, 658–660 CAS; (b) M. Namavari, N. Satyamurthy and J. R. Barrio, J. Fluorine Chem., 1995, 74, 113–121 CrossRef CAS; (c) H. H. Coenen and S. M. Moerlein, J. Fluorine Chem., 1987, 36, 63–75 CrossRef CAS; (d) W. E. Barnette, J. Am. Chem. Soc., 1984, 106, 452–454 CrossRef CAS; (e) M. A. Tius and J. K. Kawakami, Tetrahedron, 1995, 51, 3997–4010 CrossRef CAS; (f) F. Füchtner and J. Steinbach, Appl. Radiat. Isot., 2003, 58, 575–578 CrossRef CAS; (g) V. Snieckus, F. Beaulieu, K. Mohri, W. Han, C. K. Murphy and F. A. Davis, Tetrahedron Lett., 1994, 35, 3465–3468 CrossRef CAS.
- (a) H. F. Hodson, D. J. Madge and D. A. Widdowson, Synlett, 1992, 831–832 CrossRef CAS; (b) M. A. Tius and J. K. Kawakami, Synlett, 1993, 207–208 CrossRef CAS; (c) E. Differding and H. Ofner, Synlett, 1991, 187–189 CrossRef CAS; (d) S. H. Lee and J. Schwartz, J. Am. Chem. Soc., 1986, 108, 2445–2447 CrossRef CAS; (e) B. L. Chenard and C. M. Van Zyl, J. Org. Chem., 1986, 51, 3561–3566 CrossRef CAS; (f) D. P. Matthews, S. C. Miller, E. T. Jarvi, J. S. Sabol and J. R. McCarthy, Tetrahedron Lett., 1993, 34, 3057–3060 CrossRef CAS; (g) N. A. Petasis, A. K. Yudin, I. A. Zavialov, G. K. S. Prakash and G. A. Olah, Synlett, 1997, 6, 606–608 CrossRef.
- (a) S. Rozen and D. Hebel, J. Org. Chem., 1987, 52, 2588–2590 CrossRef; (b) P. Nussbaumer, G. Petranyi and A. Stutz, J. Med. Chem., 1991, 34, 65–73 CrossRef CAS; (c) J. DeYoung, H. Kawa and R. J. Lagow, J. Chem. Soc., Chem. Commun., 1992, 11, 811–812 RSC; (d) M. Schlosser and G. Heinz, Chem. Ber., 1969, 102, 1944–1953 CrossRef CAS; (e) M. A. McClinton and V. Sik, J. Chem. Soc., Perkin Trans. 1, 1992, 15, 1891–1895 RSC.
- A. C. Spivey and C. M. Diaper, Sci. Synth., 2003, 5, 149–157 Search PubMed.
- V. Gouverneur and B. Greedy, Chem.–Eur. J., 2002, 8, 766–772 CrossRef CAS.
- (a) A. M. Stuart, P. L. Coe and D. J. Moody, J. Fluorine Chem., 1998, 88, 179–184 CrossRef CAS; (b) L. J. Diorazio, D. A. Widdowson and J. M. Clough, Tetrahedron, 1992, 48, 8073–8088 CrossRef CAS; (c) A. P. Lothian and C. A. Ramsden, Synlett, 1993, 10, 753–755 CrossRef.
- (a) P. Di Raddo, M. Diksic and D. Jolly, J. Chem. Soc., Chem. Commun., 1984, 3, 159–160 RSC; (b) M. Speranza, C.-Y. Shiue, A. P. Wolf, D. S. Wilbur and G. Angelini, J. Fluorine Chem., 1985, 30, 97–107 CrossRef.
- V. Gouverneur and B. Greedy, unpublished results.
- B. Greedy and V. Gouverneur, Chem. Commun., 2001, 3, 233–234 RSC.
- L. Chabaud, P. James and Y. Landais, Eur. J. Org. Chem., 2004, 15, 3173–3199 CrossRef.
- S. Thibaudeau and V. Gouverneur, Org. Lett., 2003, 5, 4891–4893 CrossRef CAS.
- M Tredwell, K. Tenza, Mc Pacheco and V. Gouverneur, Org. Lett., 2005, 7, 4495–4497 CrossRef CAS.
- B. Greedy, J.-M. Paris, T. Vidal and V. Gouverneur, Angew. Chem., Int. Ed., 2003, 42, 3291–3294 CrossRef CAS.
- M. C. Pacheco and V. Gouverneur, Org. Lett., 2005, 7, 1267–1270 CrossRef CAS.
- M. C. Pacheco, L. Garcia and V. Gouverneur, Abstracts of Papers, 230th ACS National Meeting, Washington, DC, United States, Aug. 28–Sept. 1, 2005 ORGN-134.
| This journal is © The Royal Society of Chemistry 2006 |