Artificial ribonucleases
Teija Niittymäki and Harri Lönnberg*
Department of Chemistry, University of Turku, FIN-20014, Turku, Finland
First published on 28th October 2005
Abstract
Mimicking the action of enzymes by simpler and more robust man-made catalysts has long inspired bioorganic chemists.1 During the past decade, mimics for RNA-cleaving enzymes, ribonucleases,2 or, more precisely, mimics of ribozymes3 that cleave RNA in sequence-selective rather than base-selective manner, have received special attention. These artificial ribonucleases are typically oligonucleotides (or their structural analogs) that bear a catalytically active conjugate group and catalyze sequence-selective hydrolysis of RNA phosphodiester bonds.4
 Teija Niittymäki Teija Niittymäki | Teija Niittymäki was born in 1976 in Turku, Finland. She received her MSc from the University of Turku in 2000. She presently carries out her PhD studies under the supervision of Professor Harri Lönnberg at the same University. Her research is involved with the development of artificial ribonucleases. |
 Harri Lönnberg Harri Lönnberg | Harri Lönnberg was born in 1949 in Turku, Finland. He received his MSc degree from the University of Turku in 1972 and his PhD degree from the same university in 1976, having done postgraduate research on the kinetics and mechanisms of the hydrolysis of the O-glycosidic bond of aldofuranosides under the supervision of Professor Alpo Kankaanperä. From 1976 to 1992, he held various research posts at the Academy of Finland and was then appointed as the Professor of organic chemistry at the University of Turku. His research work has been involved with kinetics and mechanisms of the hydrolytic reactions of glycosides, nucleosides, nucleotides and oligonucleotides, solid-supported synthesis of oligonucleotide-, peptide- and glycoconjugates, fluorescence-based hybridisation diagnostics and DNA-base modifications. |
1. Introduction
Artificial ribonucleases have received interest for two reasons. Obviously, they may be used as artificial restriction enzymes for sequence-selective manipulation of large RNA molecules in vitro. Potential applications as catalytic antisense oligonucleotides in chemotherapy have, however, attracted more attention, in spite of the fact that numerous barriers still exist on the way to oligonucleotide-based drugs in general.5 The reason why gene silencing by antisense oligonucleotides could benefit from artificial ribonucleases is as follows. Oligodeoxyribonucleotides (ODN) and their phosphorothioate analogs activate an intracellular enzyme, RNase H, which degrades the RNA component of an RNA/ODN duplex, releasing the antisense ODN.6 In other words, mRNA hybridized with these kind of antisense oligonucleotides is destroyed by the cells' own machinery. Structurally more extensively modified antisense oligonucleotides do not usually trigger a similar activity.7 Accordingly, their antisense effect remains stoichiometric, unless they bear a catalyst that is able to destroy the target RNA and, hence, release the intact antisense oligomer.As mentioned above, artificial ribonucleases consist of two moieties, a catalytic group and a probe for sequence recognition. The role of the catalytic group is obvious; it cleaves the phosphodiester bond. The oligonucleotide moiety brings the sequence selectivity. Hybridization with the target RNA increases the effective concentration of the catalytic moiety in the vicinity of one particular phosphodiester bond, converting the otherwise random cleavage of the target to site-specific. The oligonucleotide moiety, however, also plays another role. Duplex formation between the artificial nuclease and the target may be exploited to shape the secondary structure and chain folding of the target optimal for cleavage. The site of tethering of the conjugate group within the artificial nuclease and the length of the complementary region(s) with the target, in turn affect the efficiency of turnover. The overall construct should be such that it binds the intact target chain more tightly than the cleavage products.
The predominant mechanism for cleavage of RNA under physiological conditions involves the formation of a marginally stable dianionic phosphorane by attack of the 2′-oxyanion on the phosphorus atom and the subsequent rate-limiting breakdown of this intermediate by departure of the 5′-linked nucleoside as an oxyanion (Scheme 1).8–10 This reaction may be accelerated by facilitating the proton transfer from the attacking nucleophile, 2′-OH, to the leaving nucleophile, 5′-O−. Accordingly, viable candidates for cleaving agents are molecules or ions that (i) enhance deprotonation of the 2′-OH, (ii) reduce the electron density at the phosphorus atom upon formation of the phosphorane intermediate, but allow it to be increased upon cleavage of the P–O5′-bond, or (iii) reduce the electron density at the departing 5′-oxygen atom upon cleavage of the P–O5′ bond.
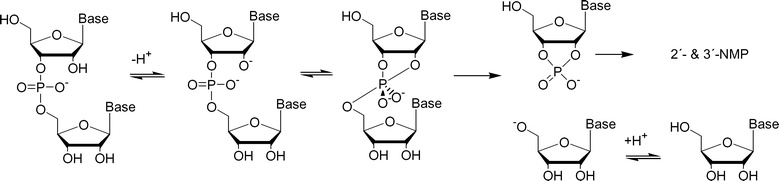 | ||
| Scheme 1 | ||
Another factor that affects the stability of RNA phosphodiester bonds is the secondary structure. Within double helical structures, intra-strand base-stacking around the cleavage site prevents the departing 5′-linked nucleoside (5′-O−) to adopt an apical position within the phosphorane intermediate,11–13 which is a prerequisite for the chain cleavage. According to Westheimer's rules on oxyphosphorane species,14 nucleophiles may enter and leave the phosphorane only through an apical position. Base-stacking forces the 5′O to an equatorial position (Fig. 1a) and the dianionic phosphorane, if ever formed, is too unstable to allow the 5′O to take an apical position (Fig 1b) via pseudorotation. Even within a single strand, base-stacking around the cleavage site is rate-retarding.15 Artificial nucleases should, therefore, be designed so that the phosphodiester bond aimed at being cleaved is not situated in a double helical stem and will not be engaged in a double helix upon hybridization with the nuclease. In addition, strongly stacked poly(purine) sequences should not be selected as target sites even when present in a single stranded region.
 | ||
| Fig. 1 Equatorial and apical orientation of the 5′-linked nucleoside within the phosphorane intermediate of hydroxide ion-catalyzed cleavage. | ||
The artificial ribonucleases developed so far fall in three different categories according to the catalytically active conjugate group, viz. cleaving agents based on (i) lanthanide ion chelates, (ii) Cu2+ and Zn2+ chelates, and (iii) metal ion-independent conjugate groups. The success within each category has been previously reviewed.4,16–23 The present discussion is aimed at giving a general overview of the present situation and perspectives of the development of artificial ribonucleases. Our attention is paid to studies where sequence-selectivity has really been achieved. In other words, reports on the catalytic activity of monomeric cleaving agents generally fall outside the scope of the present review, in particular when only aryl phosphoesters have been used as model compounds. Since the rate-limiting step for the intramolecular transesterification of aryl phosphodiesters is rather the formation than the breakdown of the phosphorane intermediate, in striking contrast to alkyl esters,10 efficient cleavage of an aryl ester does not necessarily indicate efficient cleavage of RNA. Furthermore, development of small molecule cleaving agents has been recently reviewed.23,24
In addition to the oligonucleotide conjugates described above, deoxyribozymes composed entirely of 2′-deoxyribonucleotide units constitute an interesting class of ribozyme mimics, which might well be called artificial ribonucleases. These ODNs bearing no conjugate group effectively cleave RNA phosphodiester bonds, as shown for the first time in 1994.25 Since then, many deoxyribozymes that catalyze not only RNA cleavage, but also ligation, have been identified by in vitro selection strategies.26 Owing to the marked similarity of the action of these DNzymes to ribozyme catalysis, they are not discussed in the present review.
2. Experimental techniques and conditions
The catalytic efficiency of artificial ribonucleases has usually been determined in excess of the nuclease compared to the target oligoribonucleotide (ORN). The assumption behind this experimental set-up is that the target is entirely engaged in a nuclease–target complex and the cleavage of the target follows simple first-order kinetics. In other words, the first-order rate constant and, hence the half-life, is independent of the concentration of both the target and the nuclease. Unfortunately, the validity of this assumption has seldom been experimentally verified. A few studies apart,27–30 the reported cleavage rates are based on densitometric traces of gel electrophoretic autoradiograms of only few samples, a method that at its best is semiquantitative. In addition, the results often refer to only one pH, temperature and ionic strength and the experimental conditions used by different groups differ somewhat from each other. For these reasons, accurate comparison of the efficiency of various artificial nucleases is impossible. Table 1 records illustrative examples of the data available. To facilitate the comparison, an estimation for half-lives at pH 7.5 and 37 °C (I = 0.1 M), the most frequently used experimental conditions, is given. These values should, however, be taken only as rough approximates, since the data available do not allow exact extrapolation to the reference conditions.| Nuclease | Base pairsa | c (nuclease)/µM | c (target)/µM | pH | I/mM | T (°C) | Time/h | Cleavage (%) | Ref. | Half-life/hl |
|---|---|---|---|---|---|---|---|---|---|---|
| a Number of complementary bases between the artificial nuclease and the RNA target.b The concentration of the target is not reported, but is obviously much lower than that of the nuclease.c The nuclease and target are fully complementary.d A dinucleotide bulge opposite to the cleaving agent.e A mononucleotide bulge opposite to the cleaving agent.f A trinucleotide bulge opposite to the cleaving agent.g A tetranucleotide bulge opposite to the cleaving agent.h A pentanucleotide bulge opposite to the cleaving agent.i [Zn2+] = 100 µM.j [Zn2+] = 10 µM.k [Zn2+] ≤50 µMl Estimate of the authors for the half-life at pH 7.5 and 37 °C (I = 0.1 M). | ||||||||||
| 1-Lu3+ | 15 | 10 | 0.3 | 8.0 | ca.10 | 37 | 8 | 17 | 35 | 100 |
| 2a-Eu3+ | 20 | 0.4 | 0.05 | 7.4 | ca.30 | 37 | 16 | 88 | 37 | 4 |
| 2b-Eu3+ | 20 | 0.4 | 0.05 | 7.4 | ca.30 | 37 | 16 | 51 | 37 | 10 |
| 3a-Eu3+ | 20 | 0.0025–2.5 | b | 7.5 | ca.130 | 37 | 28 | 30 | 38 | 50 |
| 3a-Dy3+ | 20 | 0.05 | 0.002 | 7.5 | ca.130 | 37 | 10 | 50 | 39 | 10 |
| 3b-Dy3+ | 20 | 0.05 | 0.002 | 7.5 | ca.130 | 37 | 2.2 | 50 | 39 | 2 |
| 3c-Dy3+ | 20 | 0.05 | 0.002 | 7.5 | ca.130 | 37 | 2.1 | 50 | 39 | 2 |
| 4-Eu3+ | 29c | 0.6 | 0.05 | 7.4 | ca.30 | 37 | 16 | 7 | 41 | 120 |
| 4-Eu3+ | 29e | 0.6 | 0.05 | 7.4 | ca.30 | 37 | 16 | 92 | 41 | 4 |
| 5-Dy3+ | 17h | 0.05 | 0.002 | 7.5 | ca.130 | 37 | 2.2 | 50 | 40 | 2 |
| 6-Cu2+ | 17c | 5.0 | 0.01 | 7.5 | ca.110 | 37 | 72 | 11 | 43 | 400 |
| 7-Cu2+ | 28f | 5.0 | 0.25 | 7.4 | ca.110 | 45 | 40 | 64 | 46 | 40 |
| 8-Cu2+ | 12 | 1.0 | 0.10 | 7.5 | ca.130 | 37 | 20 | 18 | 48 | 70 |
| 10-Cu2+ | 16e | 5.0 | 0.10 | 7.4 | ca.110 | 37 | 15 | 65 | 50 | 8 |
| 11-Zn2+ | 10g | 4.0i | 4.0 | 7.4 | 100 | 37 | 40 | 50 | 27 | 30 |
| 12-Zn2+ | 10g | 4.0i | 4.0 | 7.4 | 100 | 37 | 11 | 50 | 27 | 9 |
| 13-Zn2+ | 10g | 4.0i | 4.0 | 7.4 | 100 | 37 | 14 | 50 | 28 | 10 |
| 14-Zn2+ | 10 | 5.0j | 0.1 | 7.4 | 100 | 37 | 24 | 30 | 53 | 40 |
| 15-Zn2+ | 10 | 2.5k | 0.5 | 7.0 | 1000 | 25 | 19 | 2–5 | 49 | 100 |
| 16-Zn2+ | 13 | 18k | 18 | 7.3 | 100 | 35 | 130 | 50 | 30 | 80 |
| 17-Zn2+ | 13 | 4.5j | 9 | 7.3 | 100 | 35 | 20 | 50 | 29 | 13 |
| 18a-Zn2+ | 16h | 18k | 18 | 7.3 | 100 | 35 | 160 | 50 | 30 | 100 |
| 18b-Zn2+ | 16h | 18k | 18 | 7.3 | 100 | 35 | 180 | 50 | 30 | 120 |
| 18c-Zn2+ | 16h | 18k | 18 | 7.3 | 100 | 35 | 210 | 50 | 30 | 130 |
| 25 | 19 | 100 | 1.0 | 8.0 | ca.1 | 50 | 4 | 10 | 65 | 200 |
| 26 | 20e | 20 | 0.2 | 7.5 | ca.50 | 37 | 16 | 3 | 67 | 400 |
| 27 | 10 | 2.0 | 0.06 | 7.0 | ca.110 | 40 | 4 | 29 | 68 | 5 |
| 31a,b | 17e | 75 | b | 7.2 | ca.130 | 25 | 120 | 10 | 78 | 130 |
It is worth noting that the data in Table 1 refers merely to the efficiency of various sequence-selective cleaving agents as stoichiometric reagents. To be a real catalyst, an artificial nuclease should exhibit turnover, i.e. it should to be able to cleave the target ORN entirely, even when the concentration of the target is much higher than that of the nuclease. In many cases, the catalytic nature of the action of the nuclease has been studied separately by using the target in excess. The results of such studies are not included in Table 1, but are discussed below.
3. Artificial ribonucleases based on lanthanide ions
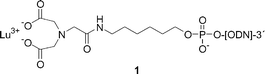
It has been known since the 1960s that metal ions catalyze the cleavage of RNA phosphodiester bonds at physiological pH.31,32 Among various metal ions, lanthanide ions are exceptionally effective in this respect.33,34 Accordingly, it is not surprising that conjugates of lanthanide ion complexes have received interest as artificial ribonucleases, and the most efficient nucleases so far described belong to this category.Komiyama et al.35 first showed that Lu3+ ion-promoted cleavage could be converted to sequence selective with the aid of a 15-mer ODN with iminodiacetic acid tethered to its 3′-terminus (1). The cleaving activity of this Lu3+ chelate conjugate was, however, quite modest. A 39-mer oligoribonucleotide (ORN) target was predominantly cleaved at a single phosphodiester bond two nucleotides towards the 3′-terminus from the last base pair, the half-life in excess of the conjugate about 30 h at pH 8 and 37 °C (Table 1). Since the metal ion-promoted cleavage usually shows first-order dependence of the rate on hydroxide ion concentration (higher order when multinuclear hydroxo complexes are formed),36 the half-life at pH 7.5 evidently is of the order of 100 h. The Eu3+ complex exhibited a comparable cleaving activity, while the La3+ complex was 50% less effective.
Although ligands containing negatively charged carboxylate groups, such as 1, exhibit high affinity to lanthanide ions, they markedly reduce the catalytic activity of the metal ion. For this reason, conjugates derived from neutral macrocyclic ligands appear more attractive and, indeed, such conjugates (when complexed with a lanthanide ion) have shown considerably improved cleaving activity. Two different types of macrocycles, viz. pyridine cyclophanes (cf.2a,b)37 and texaphyrin (cf.3a–c),38,39 have been used for this purpose. The highest cleavage rate has been obtained with the Dy3+ chelate conjugate, 3c-Dy3+, which cleave the target at the 3′-side of the first unpaired nucleotide, the half-life being 2.1 h at pH 7.5 and 37 °C.39 The conjugate has also been shown to exhibit turnover.403a-Dy3+39 and 3a-Eu3+38 exhibit half-lives of 10 and 50 h, respectively, under the same conditions and the cleavage site is shifted one nucleotide further from the last base pair. The pyridino cyclophane derived conjugates, 2a-Eu3+ and 2b-Eu3+, are almost as efficient as 3c-Dy3+. They cleave the target at the 3′-side of the third nucleotide from the last base pair, the half-lives at pH 7.5 and 37 °C being 4 and 10 h, respectively.37
Intra-chain conjugated versions of the most efficient cleaving agents, Eu3+-2a and Dy3+-3c, have additionally been prepared to ensure turnover.38,41 The underlying idea is that the affinity of the artificial ribonuclease to the target is reduced upon cleavage of the target within the double helical region and the release from the target is facilitated. As discussed above, metal ion chelates are not, however, able to cleave double-helical RNA,12,13 since strong base-stacking interactions between the base pairs prevent the 5′-linked nucleoside from taking an apical orientation within the phosphorane intermediate. For this reason, the base sequence of the artificial nuclease must be planned so that the target chain forms a bulge at the aimed cleavage site upon hybridization. Consistent with this, 4-Eu3+, bearing the pyridino cyclophane group at 2′-O of an intra-chain 5-methyluridine nucleoside,41 cleave a fully complementary ORN sequence only slowly (τ1/2 120 h at pH 7.5, 37 °C), but introduction of a two nucleotide bulge at the cleavage site accelerates the reaction by more than one order of magnitude (τ1/2 4 h). The site of attachment of the cleaving agent still plays a role, since the reaction is retarded by a factor of ten (τ1/2 50 h) when the chelate is tethered to C5 of an intra-chain 2′-deoxyuridine via the same linker. It has been argued that conjugates reaching the bulge across the minor groove are more efficient than those interacting across the major groove. As expected, these intra-chain conjugates exhibit efficient turnover.42 Up to 40 cleavage events have been shown to take place on using the 4-Eu3+ at a concentration of 1 µM and the ORN target in a 50-fold excess. Similarly, 5-Dy3+, bearing texaphyrin conjugated to a non-nucleosidic intra-chain unit, has been reported to show turnover.39 In this case, a one nucleotide bulge, viz. the unpaired nucleoside opposite to the cleaving agent, appears to be sufficient to allow cleavage of the target.

4. Artificial ribonucleases based on Cu2+ ion
The Cu2+-based artificial nucleases introduced so far are less efficient than their lanthanide ion counterparts. They are, however, of interest for the reason that Cu2+ is present in intracellular fluids. Ligands that bind Cu2+ very tightly may, hence, be expected to occur as Cu2+ complexes even in an intracellular environment.The history of Cu2+-based artificial nucleases is as long as that of lanthanide ion based agents. The first such ribozyme mimic, having terpyridine conjugated to C5 of an intra-chain uracil base of a 17-mer ODN (6), was reported by Bashkin in 1994.43 When a 159-mer segment of the gag-mRNA was used as a target, the cleavage took place at the 5′-side of adenosine just opposite to the cleaving agent. The half-life was, however, 400 h in excess of Cu2+-6 at pH 7.5 and 37 °C. The cleaving activity was later improved by tethering the cleaving agent to a serinol backbone unit (7)44,45 and inserting propane-1,3-diol spacers to both sides of the serinol unit.46 Accordingly, a trinucleotide internal loop was formed at the cleavage site, which facilitated the cleavage. The half-life was in this manner reduced to 30 h at pH 7.4 and 45 °C, corresponding a half-life of about 40 h at pH 7.5 and 37 °C. The reaction also showed turnover in excess of the target.

3′- and 5′-tethered Cu2+-chelate conjugates show marked cooperativity when hybridized to the target in such a manner that no gap remains between them. While the 5′-terminal 2′-O-methyl ORN conjugate (9-Cu2+) exhibited a half-life of 70 h at pH 7.4 and 45 °C and the 3′-conjugate (8-Cu2+) was inactive, the half-life was reduced to 5 h in the presence of both conjugates.47,48 When the two conjugates were covalently tethered to each other via a flexible linker (Fig. 2), the cleaving activity was twice as high as on using a mixture of 8-Cu2+ and 9-Cu2+.49 The cleavage was fastest at pH 7.5 and showed turnover.

 | ||
| Fig. 2 Structure of the linker used to tether the 3′-and 5′-terminal terpyridine conjugates of 2′–O-methyl oligoribonucleotides.49 Notation: n = 0–2. | ||
Another Cu2+ chelate that has been used for construction of artificial ribonucleases is 2,9-dimethyl-5-aminophenentroline. When tethered to an intra-chain serinol unit of an ODN (10), the cleaving activity is 5-fold compared to the corresponding terpyridine conjugate.50 The methyl substituents are essential, since they retard dimerization of the Cu2+ chelates,51 which markedly reduces the cleaving activity of phenanthroline conjugates.

5. Artificial ribonucleases based on Zn2+ ion
Zn2+ is another 3d transition metal ion that has received attention in design of artificial ribonucleases. The intracellular concentrations of Zn2+ fall in micromolar range, being sufficiently high to guarantee formation of complexes with formation constants in the nanomolar range. The Zn2+ chelate of 5-amino-2,9-dimethylphenanthroline is a somewhat less efficient cleaving agent than its Cu2+ counterpart; the cleaving activity of 7-Zn2+ has been reported to be 40% of that of 7-Cu2+.45 Rather extensive studies have been carried out with artificial ribonucleases derived from this Zn2+ chelate to elucidate the effects of the linker structure and site of tethering on the cleaving activity.27,28 For this purpose, three different types of conjugates of 11-mer 2′-O–Me ORNs (11–13) have been prepared. According to melting temperature measurements, all these conjugates hybridize with 80–90% efficiency to complementary ORN targets containing a bulge of 1–5 nucleosides opposite to the nucleoside that bears the cleaving agent. Conjugates 12 and 13, containing a base-moiety tethered cleaving agent, cleave these bulges somewhat faster than the 2′–O-tethered conjugate, 11. The targets containing a tri- or tetra-nucleotide bulge are usually cleaved more readily than those with a smaller or larger bulge. 12-Zn2+, the most efficient one among the nucleases tested, cleaves a tetranucleotide bulge 5 times as readily as a dinucleotide bulge.27 Usually the differences in hydrolytic stability are, however, smaller. All the bonds within a bulge are cleaved, those close to the double-helix somewhat less readily than the others.27,28 Since the stability constant for the Zn2+ complex of 2,9-dimethylphenanthroline is only of the order of 105 M−1,52 the maximal cleavage rate is achieved at Zn2+ concentrations as high as 100 µM. Under such conditions (pH 7.4, 37 °C), half-lives for the cleavage of a tetranucleotide bulge have been reported to be 40 11 and 14 h with 11, 12 and 13, respectively.27,28 The acceleration compared to a non-conjugated chelate is less than two orders of magnitude. At the 5′-terminal position, the cleaving activity of the base moiety tethered chelates is dropped to about one third. All the conjugates show turnover. Instead of 2′-O-Me ORN, a peptide nucleic acid oligomer (PNA) has been used as the sequence recognizing moiety (14).53 This has not brought in any marked change in the cleaving activity. Slow Zn2+-promoted cleavage of an ORN target (τ1/2 300 h at pH 7.0, 25 °C) has also been observed with a 3′-terminal imidazole conjugate of a 10-mer ODN (15) in excess of the metal ion (50 µmol dm−3).54

Zn2+-based artificial ribonucleases that bind the Zn2+ ion more tightly than those derived from 2,9-dimethylphenanthroline have been obtained by functionalization of 2′-O-methyl ORNs with 1,4,7-triazacyclododecane.29 The stability constant of the Zn2+ complex of this azacrown is 108.6 M−1.55 The cleaving activity of both 3′-terminal (16,17) and intrastrand (18a–c) conjugates have been tested at pH 7.3 and 35 °C in 1 : 1 mixtures of the conjugate and target and in excess of the target.29,30 Both types of conjugates show turnover, in spite of the fact that the cleavage with 16-Zn2+ and 17-Zn2+ takes place outside the complementary region of the ORN target, viz. one nucleotide from the last base pair towards the 5′-end of the target.30 The cleavage efficiency is surprisingly sensitive to the structure of 3′-terminal linker. Although the linkers in conjugates 16 and 17 are approximately as long (10 and 12 atoms from the 3′-terminal phosphate, respectively), the disulfide linker (17) affords an 8 times higher cleavage rate than the β-peptide linker (16). The reason for this difference remains obscure. As with 2,9-dimethylphenanthroline conjugates, the average cleavage rate is two orders of magnitude higher than that observed with the non-conjugated chelate. The intra-chain conjugates (18a–c), when targeted to a pentanucleotide (A5) bulge, exhibit cleavage rates from 60 to 80% of that of 16-Zn2+. The length of the linker plays only a minor role, the cleavage being slightly retarded with the increasing length. The cleavage rate of a trinucleotide bulge (A3) is about 60% of that of a pentanucleotide bulge. Interestingly, a U3-bulge has turned out to be virtually stable, possibly due to the well-established56 tendency of the Zn2+ azacrown chelates to coordinate to uracil (and thymine) bases.
The only dinuclear Zn2+-dependant nuclease described so far is the 5′-terminal ODN conjugate 19.57 This has been shown to cleave a complementary ORN approximately as readily as the 2,9-dimethylphenanthroline conjugates (11–13), but at a considerably lower Zn2+ concentration (10 µM).


6. Monomeric metal ion-dependent cleaving agents
Several metal ion-dependent cleaving agents have been introduced that effectively catalyze the cleavage of 2-hydroxypropyl-4-nitrophenylphosphate, which is generally used as a simple model of RNA. One should, however, bear in mind that efficiently promoted cleavage of an aryl phosphodiester is not necessarily an indication of an as markedly promoted cleavage of RNA. The rate-limiting step of the metal ion promoted cleavage of RNA phosphodiester bonds in all likelihood consists of intra-complex proton transfer from the aquo ligand of the phosphate-bound metal ion to the departing alkoxy ion. Consistent with this argument, Co(III) complexes have been shown to accelerate the cleavage of 3′,5′-ApA much more efficiently than the cleavage of adenosine 3′-phenylphosphate, in which formation of the phosphorane intermediate (rather than its breakdown) is rate limiting.58 In addition, the βlg value for the metal ion catalyzed cleavage of ribonucleoside 3′-alkylphosphates is close to zero (−0.32 ± 0.04), suggesting that the leaving group departs as an alcohol rather than an alkoxide ion.59 In other words, while factors facilitating the formation of the phosphorane intermediate play an important role in the cleavage of aryl esters, acceleration of the breakdown of the phosphorane intermediate is needed for RNA cleavage. With some metal ion complexes, efficient cleavage of RNA phosphodiester bonds has, however, been established. These include the trinuclear Zn2+ complex of N,N,N′,N′,N″,N″-hexakis(pyridin-2-ylmethyl)[tris(2-aminoethyl)amine] (20),60 the trinuclear Cu2+ complex of N,N,N′,N′,N″,N″-hexakis(pyridin-2-ylmethyl)[1,3,5-tris(2-aminomethyl)benzene] (21),61 the mononuclear Zn2+ complex of 2,6-bis[2-(guanidiniummethyl)pyrimidin-4-yl)]pyridine (22),62 the heterodinuclear Zn2+,Ni2+ complex of spiroazacrown 23,63 and the dinuclear Cu2+ complex of 1,8-bis[(1,4,7-triazacyclononan-1-yl)methyl]naphthalene (24).64 So far these cleaving agents have not been converted to sequence-selective nucleases.
7. Metal ion-independent artificial ribonucleases
The simplest example of organic sequence-selective nucleases is offered by diethylenetriamine conjugates of ODNs. Unfortunately, such simple conjugates are rather inefficient. A 19-mer ODN bearing a 5′-terminal diethylenetriamine tail (25) has been shown to cleave the target at the 3′-side of the first unpaired nucleoside, the half-life being 40 h at pH 8 and 50 °C.65,66 The half-life at pH 7.5 and 37 °C may be estimated to be at least 5-fold. The respective intra-chain conjugate (26) is approximately as efficient (τ1/2 400 h at pH 7.5, 37 °C).67 A considerably higher cleaving activity has been observed on using PNA for the sequence recognition and a urea linkage for tethering the diethylenetriamine moiety (27).68 A half-life of only 8 h has been reported at pH 7.0 and 40 °C, the cleavage taking predominantly place at the 3′-side of the third nucleoside from the last base pair. This corresponds to a half-life of 5 h at pH 7.5 and 37 °C. The efficiency compared to related ODN conjugates, 25 and 25, is surprisingly high.


Besides oligoamine conjugates, various imidazole containing constructs have been studied as artificial nucleases. These structures are aimed at mimicking the catalytic center of RNase A that contains two histidine residues, His-12 and His-119.69 The results obtained are to some extent controversial. Several ODN-conjugates bearing two imidazole residues at the 5′-terminus have been prepared and studied by the group of Vlassov.70–76 Among them, 28 has turned out to be most efficient.76 Usually tRNAPhe has been used as the target and the nuclease has been targeted towards a site known to exhibit inherent instability. For example, when 28 was targeted to the sequence C61–ACA–G65, known to be one of the most hydrolytically labile sites of tRNAPhe, the C63–A64 bond was cleaved very rapidly. The half-life in 50 mM imidazole buffer (pH 7.0, 37 °C, I = 0.2 M) was only 1.5 h. In addition, the duplex formation rather than cleavage was suggested to constitute the rate-limiting step of the overall process. Conjugates 2970,71 and 3073–75 are other diimidazole constructs shown to cleave tRNAPhe rather rapidly.


Most likely, the high cleavage rate observed on using conjugates 28–30 reflects the exceptional inherent hydrolytic instability of the scissile phosphodiester bond rather than the efficiency of the diimidazole constructs as a cleaving agent. Fast cleavage of RNA by imidazole-containing ODN conjugates has almost invariably been obtained by targeting the nuclease towards a 5′-CpA-3′ or 5′-UpA-3′ site within a single stranded region of tRNAPhe, i.e. towards sites known to exhibit high inherent cleavage rates.70–76 Targeting of the same ODN conjugates towards shorter linear ORNs would be of interest, especially since several closely related diimidazole conjugates planned to mimic the catalytic center of RNase A have turned out to be catalytically inactive towards simple ORN targets.77 Similarly, methanephosphonate ODNs bearing an diimidazole (31a) or imidazole/amino (31b) cleaving agent in an intra-chain position have been shown to cleave a complementary 22-mer ORN only very slowly (τ1/2 800 h at pH 7.2, 25 °C, corresponding τ1/2ca. 130 h at pH 7.5, 37 °C).78 Much faster cleavage has again be observed when a 5′-CpA-3′ bond of half-tRNAAsp has been used as a target: an imidazole/amino conjugate (32) exhibited a half-life of 18 h at pH 7.0 and 37 °C.79


Peptides composed of alternating basic and hydrophobic amino acids cleave RNA at a moderate rate. For example, the 5′-terminal ODN conjugate of H–Gly–(Arg–Leu)4 has been shown to display pronounced 5′-GpX-3′ cleaving activity, in addition to the frequently encountered 5′-CpA-3′ and 5-U′pA-3′ cleavages. Again inherent instability of the scissile bond, however, seems to play a dominant role, since the cleavage is not sequence-selective. In other words, even when the ODN moiety does not participate by Watson–Crick base pairing, it still enhances the cleaving activity of the peptide.80–82 However, a sequence-selective cleavage of tRNAPhe has also been reported. A conjugate bearing a 5′-terminal H–Leu–Arg–(Leu–Arg)3–Gly–NH2 sequence has been shown to cleave tRNALys3 predominantly at the C56–A57 site, i.e. 3 nucleotides towards the 3′-end from the last base pair with the conjugate.81 This cleavage has also been shown to be subject to competitive inhibition by the unconjugated ODN moiety.
Finally, the PNA conjugate of neamine (33) has been shown to cleave in 4-fold excess (0.5 µM) a considerable proportion of a 96-mer TAR RNA in 5 h at pH 7.4 and 25 °C.83 The cleavage rate is unaffected by Mg2+ when the concentration of this remains below 2 mmol dm−3, but at a higher concentration, metal ion inhibition takes place. Neomycin, the parent compound of neamine, is known to exhibit high affinity to the TAR region of HIV-1 RNA.84 Accordingly, it is not clear whether this cleaving agent could be targeted towards other sites and, hence, be of more general applicability.

Interestingly, tris{2-[(benzimidazol-2-yl)amino]ethyl}amine (34) has been shown to catalyze the cleavage of RNA phosphodiester bonds without the need for any special secondary or tertiary structure.85 A linear 29-mer ORN heterosequence labelled with a fluorescent dye at the 5′-terminus was cleaved, when incubated with this agents, to a mixture of labelled products containing all possible truncated sequences. The half-life for the overall disappearance of the intact ORN was 2 h at pH 6 and 37 °C (50 mM Tris-HCl buffer, I not adjusted). Accordingly, the half-life for the cleavage of a single bond may be estimated to be of the order of 60 h. This is a remarkable cleaving activity for a purely organic monomeric cleaving agent at such a low pH. Congeners of 34 may well form a solid basis for further development of purely organic sequence-selective artificial nucleases.

Some particular phosphodiester bond within RNA, usually a 5′-CpA-3′ or 5′-UpA-3′ bond, has been cleaved with several other organic small molecule compounds. These include an imidazole conjugate of dicationic bicyclo[2,2,2]-1,4-diazaoctane,86–88 diimidazole conjugates of phenazine,89 imidazole conjugates of oligoamines,90,91 a cyclen conjugate of an arginine rich peptide,92 bleomycin in the absence of metal ions,93 kanamycin A in the presence of Cu2+ ion,94,95 and a bis(guanidinium) conjugate of arginine.96

8. Hybridization directed cleavage by small molecules or metal ions
An alternative way to tailor RNA site-selectively in vitro is exploitation of an appropriate ODN probe and an exogenous catalyst for the cleavage. Hybridization with the ODN probe protects the ORN sequence from cleavage, leaving a bulge created at a desired site susceptible to the influence of the exogenous cleaving agent.97 Often, a non-nucleosidic unit is incorporated in the ODN to form a one nucleotide bulge. The first example of this approach was the study of Reynolds et al.78 A methanephosphonate ODN incorporating an abasic unit (35) cleaved in the presence of ethylenediamine the otherwise complementary ORN opposite to the non-nucleosidic unit. More recently, a similar approach has been applied by using an intra-chain acridine conjugate of ODN for creation of a bulge in the target RNA and Lu3+ ion as the cleaving agent.98 Cleavage at two sites within RNA, resulting in clipping of a designated RNA fragment, has also shown to be possible.99 The efficiency of the cleavage reaction depends on the structure of the linker used to tether it to the sugar–phosphate backbone100,101 and on substituents on the acridine ring.102,103 Introduction of N6-(N-phenylcarbamoyl) group into a dA residue within an ODN probe has been shown to result in site-selective cleavage of the complementary ORN opposite to the modified base in the presence of Mg2+.104 The structure of the modified adenine base is assumed to mimic an AT base pair and, hence, interrupt base-stacking interaction within the ORN strand.9. Concluding remarks
Several reasonably efficient artificial ribonucleases that contain a metal ion chelate as the catalytic moiety have been introduced. In principle, these could be used for tailoring of large RNA molecules in vitro. Nevertheless, only few examples of such applications are available. The structural analogs of conjugate 2b, with the Eu3+ chelate attached through the m-position (not para as in 2b) to the 5′-terminus of a 2′-O-(2-methoxyethoxy) substituted ORN, have been shown to cleave 571- and 2977-nucleotide long c-raf-1 RNA transcripts in a sequence specific manner.105 Up to 70% of the target was cleaved in 4 h at 37 °C on using a 2-fold excess of conjugate to the target. A 647-mer RNA related to the RNA component of human telomerase has been cleaved to 399- and 248-nucleotide long fragments with Zn2+-14.53 Finally, hybridization of RNA target with an ODN conjugate bearing two acridine groups at a distance of 12 nucleotides from each other, followed by cleavage with Lu3+ ion at the bulged nucleotides, has been used to cut a desired 13-mer fragment from the RNA.106 Mass spectrometric analysis of such a short fragment then allows precise detection of single nucleotide polymorphism at the site of interest.Applications of artificial ribonucleases in vivo have so far limited to the observation that the phosphorothioate analog of conjugate 33 is a more efficient antisense agent towards HIV-1 gag-mRNA than the corresponding unconjugated sequence.107 No real breakthrough has been described. The metal ion-based nucleases tend to suffer from time-dependent leakage and exchange reactions of metal ions, which may disturb their intracellular use. The most efficient lanthanide ion-based nucleases (2a, 3c) have, however, turned out to be so stable in vitro, that in vivo applications appear feasible.37–42 Another possibility is to use nucleases dependent on metal ions available at sufficient concentration in biological fluids. The stability constants of the Cu2+ and Zn2+ complexes of small azacrowns, for example, seem to be sufficiently high (>108–1012 M−1) to guarantee binding of these ions under intracellular conditions. Still, entirely covalent organic nucleases appear more attractive. A major hurdle on the way to oligonucleotide based drugs is poor cellular uptake of polyionic antisense oligomers. Metal ions chelates tethered to antisense oligomers may additionally retard their penetration to cells. Unfortunately, sufficiently efficient organic catalysts that cleave RNA phosphodiester bonds without any special demands for the base composition or chain folding have not been described. The initial results with diethylenetriamine–PNA conjugate (27)68 are promising, but no in vivo screening has been reported. It has only been observed that tethering of a cell-penetrating peptide to 27 reduces the catalytic activity.108 Recent studies of Zepik and Benner109 imply that development of purely organic cleaving agents may really be difficult. These authors, guided by the previous studies of Anslyn,110,111 Hamilton112,113 and Göbel,96,114 investigated the effect of numerous bisguanidinium compounds on the rate of transesterification of 3′,5′-UpU to 2′,3′-cUMP in aqueous triethanolamine buffer at pH 7.5. While some of the compounds moderately accelerated the cleavage, their congeners exhibiting quite similar ground state affinity to the starting material inhibited the transesterification. The recently reported sequence-independent cleaving activity of tris(guanidinium) 34 construct, however, lends some confidence to the feasibility of covalent organic catalysts and motivates to continue the development.
A factor that has not yet been properly studied is the attachment of the catalytic group to the sequence recognizing moiety. On using metal ion chelates as cleaving agents, the catalytic activity of the artificial nuclease is only 100-fold compared to the corresponding unconjugated chelate. This acceleration probably results from a proximity effect, i.e. from increased concentration of the cleaving agent in the vicinity of the scissile bond. One might, however, speculate that by a proper orientation and anchoring of the cleaving agent, a considerably greater acceleration could be achieved. This possibility may be more important with organic cleaving agents than with metal ion chelates, since the long-range electrostatic interaction between the metal ion and the negatively charged phosphodiester bond will eventually play an important role in the catalytic action of metal chelates. The efficiency of metal ion-based nucleases would most likely benefit from synergy of two appropriately immobilized metal ions. In an ideal case, two metal ions may provide double Lewis acid activation by coordination to both non-bridging phosphoryl oxygens, and simultaneously their aquo ligand can serve as an intra-complex general acid, protonating the leaving alkoxide concerted with rupture of the P–O bond.
Success in development of artificial nucleases does not guarantee that they also will find applications as chemotherapeutic agents. The overall future of antisense oligonucleotide drugs still depends on several other problems. The in vitro manipulation of RNA, however, definitely benefits from tools, with which pre-designed fragments could be cut from the RNA chains in such manner that enzymatic relegation of a novel sequence is possible.
References
- A. J. Kirby, Angew. Chem., Int. Ed. Engl., 1996, 35, 707 CrossRef CAS.
- For a general review on ribonucleases, see: H. M. Frankfort and H. D. Robertson, Cold Spring Harbor Monogr. Ser., 1982, 14, 359 Search PubMed.
- For a recent review on ribozyme mechanisms, see: Y. Takagi, Y. Ikeda and K. Taira, Top. Curr. Chem., 2004, 232, 213 Search PubMed.
- B. N. Trawick, A. T. Daniher and J. K. Bashkin, Chem. Rev., 1998, 98, 939 CrossRef CAS.
- For the current status of the development of antisense oligonucleotide-based chemotherapy, see: S. T. Crooke, Annu. Rev. Med., 2004, 55, 61 Search PubMed.
- H. Nakamura, Y. Oda, S. Iwai, H. Inoue, E. Ohtsuka, S. Kanaga, S. Kimura, C. Katsuda, K. Katayanagi, K. Morikawa, H. Miyashiro and M. Ikehara, Proc. Natl. Acad. Sci. U. S. A., 1991, 88, 11535 CAS.
- For antisense oligonucleotides that activate RNase H, see: M. M. Mangos, K.-L. Min, E. Viazovkina, A. Galarneau, M. I. Elzagheid, M. A. Parniak and M. J. Damha, J. Am. Chem. Soc., 2003, 125, 654 Search PubMed , and references therein.
- D. M. Perrault and E. V. Anslyn, Angew. Chem., Int. Ed., 1997, 36, 432 CrossRef.
- M. Oivanen, S. Kuusela and H. Lönnberg, Chem. Rev., 1998, 98, 961 CrossRef CAS.
- H. Lönnberg, R. Strömberg and A. Williams, Org. Biomol. Chem., 2004, 2, 2165 RSC.
- D. A. Usher and A. H. McHale, Proc. Natl. Acad. Sci. U. S. A., 1976, 73, 1149 CAS.
- U. Kaukinen, L. Bielecki, S. Mikkola, R. W. Adamiak and H. Lönnberg, J. Chem. Soc., Perkin Trans. 2, 2001, 1024 RSC.
- K. A. Kolasa, J. R. Morrow and A. P. Sharma, Inorg. Chem., 1993, 32, 3983 CrossRef CAS.
- F. H. Westheimer, Acc. Chem. Res., 1968, 1, 70 CrossRef CAS.
- U. Kaukinen, T. Venäläinen, H. Lönnberg and M. Peräkylä, Org. Biomol. Chem., 2003, 1, 2439 RSC.
- M. Komiyama, J. Biochem., 1995, 118, 665 CAS.
- R. Häner and J. Hall, Antisense Nucleic Acid Drug Dev., 1997, 7, 423 CAS.
- M. Komiyama and J. Sumaoka, Curr. Opin. Chem. Biol., 1998, 2, 751 CrossRef CAS.
- J. A. Cowan, Curr. Opin. Chem. Biol., 2001, 5, 634 CrossRef.
- R. Häner, Chimia, 2001, 55, 1035 CAS.
- M. Komiyama, J. Sumaoka, A. Kuzuya and Y. Yamamoto, Methods Enzymol., 2001, 341, 455 CrossRef CAS.
- M. Zenkova, N. Beloglazova, V. Silnikov, V. Vlassov and R. Giege, Methods Enzymol., 2001, 341, 468 CrossRef CAS.
- J. R. Morrow and O. Iranzo, Curr. Opin. Chem. Biol., 2004, 8, 192 CrossRef CAS.
- E. Kimura, Curr. Opin. Chem. Biol., 2000, 4, 207 CrossRef CAS.
- R. R. Breaker and G. F. Joyce, Chem. Biol., 1994, 1, 223 CrossRef CAS.
- S. K. Silverman, Org. Biomol. Chem., 2004, 2, 2701 RSC.
- H. Åström, N. H. Williams and R. Strömberg, Org. Biomol. Chem., 2003, 1, 1461 RSC.
- H. Åström and R. Strömberg, Org. Biomol. Chem., 2004, 2, 1901 RSC.
- T. Niittymäki, U. Kaukinen, P. Virta, S. Mikkola and H. Lönnberg, Bioconjugate Chem., 2004, 15, 174 CrossRef.
- T. Niittymäki and H. Lönnberg, Bioconjugate Chem., 2004, 15, 1275 CrossRef.
- J. J. Butzow and G. L. Eichhorn, Biopolymers, 1965, 3, 95 CAS.
- G. L. Eichhorn, E. Tarien and J. J. Buzow, Biochemistry, 1971, 10, 2014 CrossRef CAS.
- G. L. Eichhorn and J. J. Butzow, Biopolymers, 1965, 3, 79 CAS.
- J. R. Morrow, L. A. Buttrey, V. M. Shelton and K. A. Berback, J. Am. Chem. Soc., 1992, 114, 1903 CrossRef CAS.
- K. Matsumura, M. Endo and M. Komiyama, Chem. Commun., 1994, 2019 RSC.
- S. Kuusela and H. Lönnberg, H., J. Phys. Org. Chem., 1993, 6, 347 CrossRef CAS.
- J. Hall, D. Hüsken, U. Pieles, H. E. Moser and R. Häner, Chem. Biol., 1994, 1, 185 CrossRef CAS.
- D. Magda, R. A. Miller, J. L. Sessler and B. L. Iverson, J. Am. Chem. Soc., 1994, 116, 7439 CrossRef CAS.
- D. Magda, S. Crofts, A. Lin, D. Miles, M. Wright and J. L. Sessler, J. Am. Chem. Soc., 1997, 119, 2293 CrossRef CAS.
- D. Magda, M. Wright, S. Crofts, A. Lin and J. L. Sessler, J. Am. Chem. Soc., 1997, 119, 6947 CrossRef CAS.
- J. Hall, D. Hüsken and R. Häner, Nucleic Acids Res., 1996, 24, 3522 CrossRef CAS.
- R. Häner, J. Hall, A. Pfützer and D. Hüsken, Pure Appl. Chem., 1998, 70, 111 CrossRef CAS.
- J. K. Bashkin, E. I. Frolova and U. Sampath, J. Am. Chem. Soc., 1994, 116, 5981 CrossRef CAS.
- A. T. Daniher and J. K. Bashkin, Chem. Commun., 1998, 1077 RSC.
- W. C. Putnam and J. K. Bashkin, Chem. Commun., 2000, 767 RSC.
- B. N. Trawick, T. A. Osiek and J. K. Bashkin, Bioconjugate Chem., 2001, 12, 900 CrossRef CAS.
- H. Inoue, T. Furukawa, T. Tamura, A. Kamada and E. Ohtsuka, Nucleosides, Nucleotides Nucleic Acids, 2001, 20, 833 CrossRef CAS.
- H. Inoue, T. Furukawa, M. Shimizu, T. Tamura, M. Matsui and E. Ohtsuka, Chem. Commun., 1999, 45 RSC.
- S. Sakamoto, T. Tamura, T. Furukawa, Y. Komatsu, E. Ohtsuka, M. Kitamura and H. Inoue, Nucleic Acids Res., 2003, 31, 1416 CrossRef CAS.
- W. C. Putnam, A. T. Daniher, B. N. Trawick and J. K. Bashkin, Nucleic Acids Res., 2001, 29, 2199 CrossRef CAS.
- B. Linkletter and J. Chin, Angew. Chem., Int. Ed., 1995, 37, 472 CrossRef CAS.
- H. Irving and D. Mellor, J. Chem. Soc., 1962, 5222 RSC.
- A. Whitney, G. Gavory and S. Balasubramanian, Chem. Commun., 2003, 36 RSC.
- J. Hovinen, A. Guzaev, E. Azhayeva, A. Azhayev and H. Lönnberg, J. Org. Chem., 1995, 60, 2205 CrossRef CAS.
- M. Kodama and E. Kimura, J. Chem. Soc., Dalton Trans., 1977, 2269 RSC.
- E. Kimura, M. Kikuchi, H. Kitamura and T. Koike, Chem. Eur. J., 1999, 5, 3113 CrossRef CAS.
- S. Matsuda, A. Ishikubo, A. Kuzuya, M. Yashiro and M. Komiyama, Angew. Chem., Int. Ed., 1998, 37, 3284 CrossRef CAS.
- M. Komiyama, Y. Matsumoto, H. Takahashi, T. Shiiba, H. Tsuzuki, H. Yajima, M. Yashiro and J. Sumaoka, J. Chem. Soc., Perkin Trans. 2, 1998, 691 RSC.
- S. Mikkola, E. Stenman, K. Nurmi, E. Yousefi-Salakdeh, R. Strömberg and H. Lönnberg, J. Chem. Soc., Perkin Trans. 2, 1999, 1619 RSC.
- M. Yashiro, A. Ishikubo and M. Komiyama, Chem. Commun., 1997, 83 RSC.
- M. Komiyama, S. Kina, K. Matsumura, J. Sumaoka, S. Tobey, V. M. Lynch and E. Anslyn, J. Am. Chem. Soc., 2002, 124, 13731 CrossRef CAS.
- H. Ait-Haddou, J. Sumaoka, S. L. Wiskur, J. F. Folmer-Andersen and E. Anslyn, Angew. Chem., Int. Ed., 2002, 41, 4014 CAS.
- Q. Wang, S. Mikkola and H. Lönnberg, Chem. Biodiversity, 2004, 1, 1316 Search PubMed.
- M. J. Young and J. Chin, J. Am. Chem. Soc., 1995, 117, 10577 CrossRef CAS.
- M. Komiyama, T. Inokava and K. Yoshinari, Chem. Commun., 1995, 77 RSC.
- M. Komiyama and T. Inokava, J. Biochem., 1994, 116, 719 CAS.
- M. Endo, Y. Azuma, Y. Saga, A. Kuzuya, G. Kawai and M. Komiyama, J. Org. Chem., 1997, 62, 846 CrossRef CAS.
- J. C. Verheijen, B. A. L. M. Deiman, E. Yeheskiely, G. A. van der Marel and J. H. van Boom, Angew. Chem., Int. Ed., 2000, 39, 369 CrossRef CAS.
- R. T. Raines, Chem. Rev., 1998, 98, 1045 CrossRef CAS.
- V. Vlassov, T. Abramova, R. Giege and V. Silnikov, Antisense Nucleic Acid Drug Dev., 1997, 7, 39 CAS.
- L. Yurchenko, V. Silnikov, T. Godovikova, G. Shishkin, J.-J. Toulme and V. Vlassov, Nucleosides Nucleotides, 1997, 16, 1721 CAS.
- N. G. Beloglazova, N. N. Polushin, V. N. Silnikov, M. A. Zenkova and V. V. Vlassov, Dokl. Akad. Nauk, 1999, 369, 827 CAS.
- N. G. Beloglazova, V. N. Silnikov, M. A. Zenkova and V. V. Vlassov, FEBS Lett., 2000, 481, 277 CrossRef CAS.
- N. G. Beloglazova, A. Yu. Epanchintsev, V. N. Silnikov, M. A. Zenkova and V. V. Vlassov, Mol. Biol., Engl. Ed., 2002, 36, 731 Search PubMed.
- I. Y. Garipova and V. N. Silnikov, Russ. Chem. Bull., 2002, 51, 1112 CrossRef CAS.
- N. G. Beloglazova, M. M. Fabani, M. A. Zenkova, E. V. Bichenkova, N. N. Polushin, V. V. Silnikov, K. T. Douglas and V. V. Vlassov, Nucleic Acids Res., 2004, 32, 3887 CrossRef CAS.
- B. Verbeure, C. J. Lacey, M. Froeyen, J. Rozenski and P. Herdewijn, Bioconjugate Chem., 2002, 13, 333 CrossRef CAS.
- M. A. Reynolds, T. A. Beck, P. B. Say, D. A. Schwartz, B. P. Dwyer, W. J. Daily, M. M. Vaghefi, M. D. Metzler, R. E. Klem and L. J. Arnold, Jr., Nucleic Acids Res., 1996, 24, 760 CrossRef CAS.
- K. Ushijima and H. Takaku, Biochim. Biophys. Acta, 1998, 1379, 217 CrossRef CAS.
- N. L. Mironova, D. V. Pyshnyi, E. M. Ivanova, M. A. Zenkova, H. J. Gross and V. V. Vlassov, Nucleic Acids Res., 2004, 32, 1928 CrossRef CAS.
- N. L. Mironova, D. V. Pyshnyi, E. M. Ivanova, V. F. Zarytova, M. A. Zenkova, H. J. Gross and V. V. Vlassov, Russ. Chem. Bull., 2002, 51, 1177 CrossRef CAS.
- N. L. Mironova, Y. I. Boutorine, D. V. Pyshnyi, E. M. Ivanova, M. A. Zenkova and V. V. Vlassov, Nucleosides, Nucleotides Nucleic Acids, 2004, 23, 885 CrossRef CAS.
- E. Riguet, S. Tripathi, B. Chaubey, J. Desire, V. N. Pandey and J.-L. Decout, J. Med. Chem., 2004, 47, 4806 CrossRef CAS.
- H.-Y. Mei, A. A. Galan, N. S. Halim, D. P. Mack, D. W. Moreland, K. B. Sanders, H. N. Truong and A. W. Czarnik, Bioorg. Med. Chem. Lett., 1995, 5, 2755 CrossRef CAS.
- U. Scheffer, A. Strick, V. Ludwig, S. Peter, E. Kalden and M. Göbel, J. Am. Chem. Soc., 2005, 127, 2211 CrossRef CAS.
- D. A. Konevetz, I. E. Beck, N. G. Beloglazova, I. V. Sulimenkov, V. N. Silnikov, M. A. Zenkova, G. V. Shiskin and V. V. Vlassov, Tetrahedron, 1998, 55, 503.
- D. A. Konevetz, N. L. Mironova, I. E. Beck, M. A. Zenkova, G. V. Shishkin, V. V. Vlassov and V. N. Silnikov, Russ. J. Bioorg. Chem., 2002, 28, 331 Search PubMed.
- I. L. Kuznetsova, M. A. Zenkova, H. J. Gross and V. V. Vlassov, Nucleic Acids Res., 2005, 33, 1201 CrossRef CAS.
- M. A. Podyminogin, V. V. Vlassov and R. Giege, Nucleic Acids Res., 1993, 21, 5950 CrossRef CAS.
- S. Fouace, C. Gaudin, S. Picard, S. Corvaisier, J. Renault, B. Carboni and B. Felden, Nucleic Acids Res., 2004, 32, 151 CrossRef CAS.
- V. Silnikov, G. Zuber, J. P. Behr, R. Giege and V. Vlassov, Phosphorus, Sulfur Silicon Relat. Elem., 1996, 110, 277 Search PubMed.
- K. Michaelis and M. Kalesse, Angew. Chem., Int. Ed., 1999, 38, 2243 CrossRef CAS.
- M. V. Keck and S. M. Hecht, Biochemistry, 1995, 34, 12029 CrossRef CAS.
- A. Sreedhara, A. Patwardhan and J. A. Cowan, Chem. Commun., 1999, 1147 RSC.
- A. Sreedhara and J. A. Cowan, JBIC, J. Biol. Inorg. Chem., 2001, 6, 166 Search PubMed.
- K. Kurz and M. W. Göbel, Helv. Chim. Acta, 1996, 79, 1967 CAS.
- D. Husken, G. Goodall, M. J. J. Blommers, W. Jahnke, J. Hall, R. Häner and H. E. Moser, Biochemistry, 1996, 35, 16591 CrossRef CAS.
- A. Kuzuya, R. Mizoguchi, F. Morisawa, K. Machida and M. Komiyama, J. Am. Chem. Soc., 2002, 124, 6887 CrossRef CAS.
- A. Kuzuya, R. Mizoguchi, T. Sasayama, J.-M. Zhou and M. Komiyama, J. Am. Chem. Soc., 2004, 126, 1430 CrossRef CAS.
- Y. Shi, A. Kuzuya, K. Machida and M. Komiyama, Tetrahedron Lett., 2004, 45, 3703 CrossRef CAS.
- Y. Shi, K. Machida, A. Kuzuya and M. Komiyama, Bioconjugate Chem., 2005, 16, 306 CrossRef CAS.
- A. Kuzuya, K. Machida, R. Mizoguchi and M. Komiyama, Bioconjugate Chem., 2002, 13, 365 CrossRef CAS.
- A. Kuzuya, K. Machida and M. Komiyama, Tetrahedron Lett., 2002, 43, 8249 CrossRef CAS.
- S.-i. Nakano, Y. Uotani, K. Uenishi, M. Fujii and N. Sugimoto, J. Am. Chem. Soc., 2005, 127, 518 CrossRef CAS.
- L. Canaple, D. Hüsken and R. Häner, Bioconjugate Chem., 2002, 13, 945 CrossRef CAS.
- A. Kuzuya, R. Mizoguchi, F. Morisawa and M. Komiyama, Chem. Commun., 2003, 770 RSC.
- K. Ushijima, M. Shirakawa, K. Kagoshima, W.-S. Park, N. Miyano-Kurosaki and H. Takaku, Bioorg. Med. Chem., 2001, 9, 2165 CrossRef CAS.
- L. Petersen, M. C. de Koning, P. van Kuik-Romeijn, J. Weterings, C. J. Pol, G. Platenburg, M. Overhand, G. A. van der Marel and J. H. van Boom, Bioconjugate Chem., 2004, 15, 576 CrossRef CAS.
- H. H. Zepik and S. A. Benner, J. Org. Chem., 1999, 64, 8080 CrossRef CAS.
- J. Smith, K. Ariga and E. V. Anslyn, J. Am. Chem. Soc., 1993, 115, 362 CrossRef CAS.
- D. M. Kneeland, K. Ariga, V. M. Lynch, C. Y. Huang and E. V. Anslyn, J. Am. Chem. Soc., 1993, 115, 10042 CrossRef CAS.
- V. Jubian, R. P. Dixon and A. D. Hamilton, J. Am. Chem. Soc., 1992, 114, 1120 CrossRef CAS.
- V. Jubian, A. Veronese, R. P. Dixon and A. D. Hamilton, Angew. Chem., 1995, 34, 1237 CAS.
- M.-S. Muche and M. W. Göbel, Angew. Chem., Int. Ed., 1996, 35, 2126 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2006 |