Activity of synthetic ion channels is influenced by cation–π interactions with phospholipid headgroups
Michelle E.
Weber
a,
Elizabeth K.
Elliott
b and
George W.
Gokel
*ab
aDepartment of Molecular Biology & Pharmacology, Washington University School of Medicine, Campus Box 8103, 660 S. Euclid Ave., St. Louis, MO 63110, USA. E-mail: ggokel@wustl.edu; Fax: +1 314 362 9298; Tel: +1 314 362 9297
bDepartment of Chemistry, Washington University, 1 Brookings Drive, St. Louis, MO 63130, USA
First published on 30th November 2005
Abstract
A suite of synthetic hydraphile ion channels has been used to probe the possibility of cation–π interactions between the channel and the phospholipid bilayer. The hydraphiles selected for this study contained either no sidearm, aliphatic sidearms or aromatic sidearms that varied in electron-richness. An ion selective electrode (ISE) method was used to evaluate the ion transport ability of these hydraphiles across synthetic bilayers. Transport was dependent on sidearm identity. Ion transport activity for the aromatic sidechained compounds was greatest when the sidearms were electron rich and vesicles were prepared from 100% DOPC (trimethylammonium cation headgroup, overall neutral). When the lipid headgroups were made more negative by changing the composition from DOPC to 70 : 30 (w/w) DOPC : DOPA, transport by the aromatic-sidechained channels was reduced. Fluorescence studies showed that when the lipid composition changed, the headgroups experienced a different polarity, suggesting reorientation. The data are in accord with a stabilizing cation–π interaction between the aromatic sidearm of the hydraphile channel and the ammonium phospholipid headgroup.
Introduction
The phospholipid bilayer is a molecular cloister that defines the boundary of most cells and organelles. The plasma membranes of eukaryotic cells typically contain only about 50% by weight of phospholipids. The other half is a mixture of sterols, various other lipids, peptides, proteins, various anchor groups, and the like. Even within the group of phospholipids, there is remarkable structural variation in chain length, unsaturation, and head group identity. It is clear that all aspects of membrane structure contribute to the bilayer's ability to interact with various guest molecules, fuse, form lipid rafts, etc., although details of many such interactions remain elusive. We report here that the transport efficacy of hydraphile synthetic ion channels depends in part on the headgroup present on phospholipid monomers.Synthetic ion channels were essentially unknown two decades ago but a number of credible examples have been reported in recent years.1 We have developed structurally different families of cation-2 and anion-selective3 channels. The hydraphile family of cation channels is now well characterized and structure–activity studies have revealed details about ion transport mediated by simple, synthetic ion channels. The hydraphiles consist of two distal diaza-18-crown-6 macrocycles that serve as portals for ion entry and egress. The central relay serves the function4 of the “water and ion-filled capsule” recently identified in protein channels.5 We have shown that the central relay, typically a macrocycle, adopts an extended conformation and lies parallel to the lipid bilayer's acyl chains.6 The distal macrocycles are connected by polymethylene linkers to the central relay (macrocycle), both of which were dodecyl chains in the original design.
A method we used at an early stage to evaluate hydraphile-mediated ion transport relied on dynamic 23Na NMR in phospholipid vesicles.7 Although useful and reliable, this method is somewhat cumbersome and labor-intensive. We have developed and recently reported a methodology utilizing a Na+ selective microelectrode.8 The ion selective electrode (ISE) method is equally reliable but has proved to be faster and more reproducible. We have used the ISE method to record Na+ transport mediated by hydraphiles having systematically varied lengths as a means to dynamically assay liposomal membrane thickness. We found that shorter channels were active in the thinnest membranes but not thicker ones, and vice versa.9
Our confidence in the ion transport function of these synthetic channels has now permitted us to assay the effect of changes in various hydraphile subunits. In previous studies, we found that changes in the sidearms profoundly altered Na+ transport ability.6 A surprising observation was that Na+ transport was significantly higher when benzyl groups comprised the sidearms than when they were n-dodecyl groups. Another program underway in our laboratory has been to identify alkali metal cation–π interactions, both in solution and in the solid state.10 It occurred to us that the difference in activity of aromatic vs. alkyl-sidearmed hydraphiles might result, at least in part, from cation–π interactions between the channels' benzyl headgroups and the cationic phospholipid headgroups. We report here an effort to assay the interaction between hydraphile sidearms and the phospholipids in the bilayers where the former are active.
Results and discussion
We have studied hydraphile-mediated Na+ transport in liposomes prepared from several different phospholipid monomer mixtures. The transport results we obtained were reproducible for specific lipid mixtures, but sometimes quite different from other membrane compositions. In particular, we compared sodium cation transport in pure 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC) with results obtained in a 70 : 30 (w/w) mixture of DOPC and 1,2-dioleoyl-sn-glycero-3-phosphate (DOPA). Relatively little difference was observed when the hydraphile sidearms were aliphatic (see below) but significant when the sidearms were benzyl or substituted benzyl.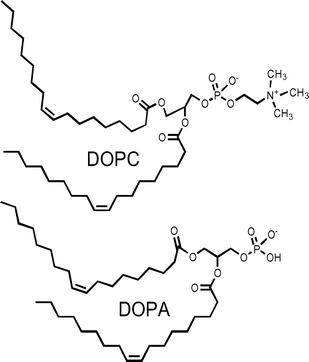
In earlier work, we found that sodium cation transport by hydraphiles having substituted benzyl sidearms correlated with the arene's electron richness (its Hammett parameter).11 The observed effect could have resulted from an electronic effect on the distal macrocycle nitrogen to which the benzyl group was attached. Alternately or additionally, it could have indicated a cation–π interaction, but how this might operate remained obscure. Moreover, cation–π interactions were poorly documented at the time of that earlier study. Extensive work in our10 and other laboratories12 has placed on firm footing the cation–π interaction for benzene,13 phenol,14 and indole15 with sodium or potassium cations.
Compounds studied
The compounds used in this study are shown as 1–8. The covalent spacer chains are 1,12-dodecyl and the macrocycles are 4,13-diaza-18-crown-6 in all of the compounds studied. The distal macrocycles are terminated by the groups indicated as “R”, and are aliphatic (1) or aromatic (2–7). Channel 8 does not contain a sidearm; rather it terminates in a secondary amine as the macrocycle nitrogen.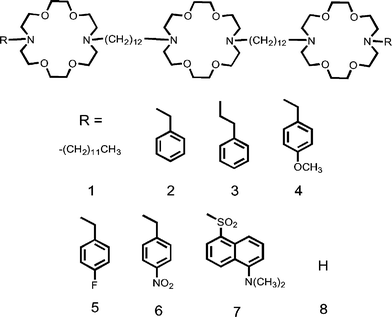
Compounds 2, 4, 5, and 6 are benzyl derivatives for which Hammett constants are available. Compound 3 is a phenethyl derivative that was chosen because previous studies showed that this sidechain is subject to alkali metal cation–π interactions.10a The dansyl sidechain (7) was used as a fluorescent probe to help assess sidechain environment. Two comments are appropriate concerning phenethyl-sidearmed 3. First, it is attached to nitrogen through a two-carbon, ethylene unit, so it is not a benzyl derivative. Second, we were able to prepare numerous cation–π complexes of diaza-18-crown-6 derivatives when the sidearm was 2-phenylethyl12–15 but not when it was benzyl. Thus, a direct comparison between 2 and 3 is problematic.
Sodium transport studies
Liposomes of approximate diameter 200 nm were prepared from an individual phospholipid or a mixture of lipids, as described in the Experimental section. The liposome's internal and external buffers were 750 mM NaCl and 750 mM choline chloride, respectively. Addition of an active hydraphile (1–8, 12 µM) led to Na+ efflux, which was monitored by ISE. The method used to assay sodium release from liposomes was reported recently in detail.9 The results obtained for compounds 2–7 in DOPC liposomes are shown graphically in Fig. 1. Reproducibility by this method is good: each curve shown is the average of at least three experiments.![Fractional sodium ion release from liposomes ([lipid] = 0.4 mM) mediated by 12 µM of compounds 2–7 as determined by ISE methods.](/image/article/2006/OB/b513179k/b513179k-f1.gif) | ||
| Fig. 1 Fractional sodium ion release from liposomes ([lipid] = 0.4 mM) mediated by 12 µM of compounds 2–7 as determined by ISE methods. | ||
The data show an activity order for Na+ release of 4 (4-methoxybenzyl) > 3 (2-phenylethyl) > 2 (benzyl) > 5 (4-fluorobenzyl) > 6 (4-nitrobenzyl). Compounds 2, 4, 5, and 6 can be directly compared. Sodium transport decreases with decreasing electron richness of the sidearm. Thus, 4-methoxybenzyl channel 4, which has the most electron-rich sidearms in the series, afforded 100% release by t = 1500 s. Hydraphiles 2 and 3 both have benzene-terminated sidearms, but compound 3 has an additional methylene spacer in the sidearm. The latter (3) was ∼25% more active than the former (2). The X-ray crystallographic studies of diaza-lariat ethers noted above showed clear evidence for cation–π complexation of alkali metal cations when the sidearm was phenylethyl but not when it was benzyl.16 This difference, observed originally in the solid state between lariat ethers having the sidearms of 2 and 3, was confirmed for the solution state by use of NMR.10a
Among compounds 2–6, those having electron-deficient arenes in the sidearms are the least active. Thus 5 (p-fluorobenzyl sidearm) mediates only 40% release at 1500 s, and the compound having p-nitrobenzyl (6) sidearms releases ∼20% of the total Na+.
As noted above, changing the benzyl group's para substituent predictably alters Na+ transport through liposomal bilayers.11 Variations in transport rates observed when the lipid composition was altered suggested an additional effect. It seemed possible that a cation–π interaction between the cationic headgroup of DOPC and the benzyl sidearm was enhancing channel function. The quaternary ammonium residue of DOPC is cationic even though the headgroup is overall neutral. If a cation–π interaction was involved, changing from a zwitterionic to an anionic lipid should diminish or eliminate the observed differences.
The activity of each hydraphile was determined under otherwise identical conditions in DOPC (zwitterionic headgroups) and a 70 : 30 (w/w) mixture of DOPC and 1,2-dioleoyl-sn-glycero-3-phosphate (DOPA). In each case, the hydraphile was present at 12 µM and the lipid concentration was 0.4 mM. Each line, graphed in Fig. 2, is the average of at least three replicates.
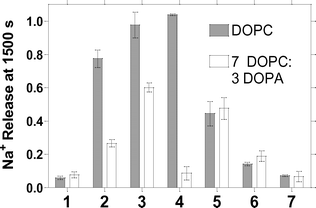 | ||
| Fig. 2 Dependence of Na+ release at 1500 s from liposomes mediated by compounds 1–7. Filled bars: liposomes prepared from pure DOPC. Open bars: liposomes prepared from 70 : 30 (w/w) DOPC : DOPA. | ||
The data in Fig. 2 reveal four interesting properties. First, the presence of a sidearm is required for significant Na+ transport in this assay system. The activity for channel 8 (not shown on graph), which lacks a sidearm, is <2% total Na+ release. We previously showed, by using an NMR assay, that no activity at all could be detected when the NH group of 8 was replaced by an oxygen atom.6 The present study confirmed that finding (data not shown). Second, the activity of otherwise identical hydraphiles is dependent on the stereoelectronic character of the sidearm residue. A clear correlation of substituted dibenzyldiaza-18-crown-6 compounds binding Na+ with Hammett parameters was reported some years ago.17 The effect is particularly quantifiable for benzyl-sidearmed compounds 2, 4, 5, and 6, which differ only in the arene substituent. Third, strikingly different transport activity was observed for certain individual compounds when the lipid composition changed. Finally, the transport by the aliphatic sidechained compound 1 is not significantly lipid dependent.
It is also apparent from the data of Fig. 2, that when the sidearms are aliphatic (1), changing the lipid composition makes little or no difference. Little effect is also apparent when the benzyl sidechains bear electron withdrawing groups. In marked contrast, however, more electron rich benzyl hydraphiles 2 and 3 show much higher Na+ transport in pure DOPC (filled bars in Fig. 2) than in the 70 : 30 DOPC : DOPA mixture. It is tempting to say that electron poor 5 and 6 are slightly more effective when the lipid headgroups are partly negative (DOPC : DOPA) than when wholly positive (DOPC), but the transport rates are within experimental error of each other (see error bars in Fig. 2). The data presented here comprise what is, to our knowledge, the first evidence of a cation–π interaction that affects ion transport through a phospholipid membrane.
A final observation is that the lipid dependence of 4 is the most dramatic of the group. The successful Hammett correlation discussed above makes the high transport value observed in 7 : 3 DOPC : DOPA seem reasonable. Why transport is so low for 4 in DOPA (open bars) is less clear. There are at least two possible explanations, both of which are speculative. First, the electron donating ability of the methoxy group, transmitted through the benzyl group to a distal macroring nitrogen, could enhance complexation and restrict ion transport. Alternately, the arene's increased electron richness could manifest itself in π-complexation of the transient cation and thus reduce flux. Of course, both effects could contribute to the poor transport.
Structural information
A search of the Cambridge Structural Database (CSD) revealed structural information for only six phospholipids. Two of these (LAPETM1018 and QACGAC19) are structures of phosphatidylethanolamines. The group includes a structure of a phosphatidic acid, namely sodium 2,3-dimyristoyl-D-glycero-1-phosphate (sodium 1,2-sn-dimyristoylphosphatididate, CSD: DAHJAX).20 One structure possesses a dimethylamine headgroup: 2,3-dilauroyl-rac-glycero-1-phospho-N,N-dimethylethanolamine (QACGAC).19 The two remaining structures are of phosphocholines: 1,2-dipalmitoyl-L-lecithin (PALLEC21) and 1,2-dimyristoyl-sn-glycero-3-phosphorylcholine dihydrate (WAHQAX22). The structure of sodium 1,2-sn-dimyristoylphosphatididate, (CSD: DAHJAX) is typical in that the fatty acyl chains are linear and aligned throughout the crystal as are the headgroups. The monomers crystallize in bilayers in which the fatty acid chains and headgroups are alternately in contact. Although these results are as expected, they shed no light on the interaction of the headgroups with aromatic residues.Ammonium ion interactions with arenes
The CSD reveals numerous contacts between tetramethylammonium ion and various arenes that are part of larger molecules such as metal complexes of phenol or catechol. An example is the tin complex (tetramethylammonium 2-chloro-5,5′-dimethyl-2,2′-spirobis(1,3,2-benzodithiastannole)) shown below and identified in the CSD as BTHSNA10.23 Using the crystal coordinates, we measured the distance24 from the quaternary nitrogen to the arene's centroid as 4.541 Å. In a previous study of ammonium ion–arene contacts,25 we identified relatively few structures in the CSD that exhibited contacts shorter than 4.7 Å.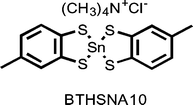
Lipid–hydraphile interactions
We infer from the results shown in Fig. 2 that the hydraphile's aromatic sidechains interact with the lipid headgroups. The solid state structures available for the phospholipids suggest that there is room between headgroups for interstitial placement of the hydraphile sidechains. It seems probable that when this interaction involves an arene and a quaternary ammonium cation, the hydraphile's active ion-conducting conformation is stabilized, resulting in increased ion transport. The distances involved are clearly critical, however, as interposition of a single methylene group (2 → 3) enhances transport activity. The ammonium–π interaction is not favorable when the hydraphile headgroup is electron poor. Thus, ion transport activity in DOPC is lower for 5 and 6 than for 2 and 4. When the lipids are changed from DOPC to a DOPC : DOPA mixture (filled bars in Fig. 2), the chance of such a favorable interaction is diminished. In the DOPC : DOPA case, neither 5 nor 6 is much affected.One might expect the electron poor hydraphiles to show improved transport when DOPA is present. The latter is a shorter lipid and it is unknown whether the distribution of DOPA is uniform throughout the bilayer. If the chain length sensitivity of 3 compared to 2 is extensible to the lipids, positioning alone may account for the differences in transport rates in the lipid mixture.
The Hammett relationship
The Hammett equation describes a linear free energy relationship that permits the correlation of stereoelectronic properties with structural variables. Compounds 2 and 4–6 are benzyl derivatives that differ only in the para-substituent. As noted above, we previously reported that Na+ cation transport obeys a Hammett relationship as determined by 23Na NMR transport studies.11 The experiments presented here assume that there is a predictable electronic effect on the distal macrocycles. The effect should be separate from the cation–π interaction with trimethylammonium cations. Indeed, the results presented here, obtained by use of the ISE method, confirm the previous 23Na-NMR studies for the DOPC system.Fig. 3 shows the Hammett plot of Na+ transport vs.σ° for 2 and 4–6, which are the only sidearms in our study for which Hammett coefficients are available.26 The data are correlated by a straight line, which shows that the variation in channel activity corresponds with the electronic effect of the sidearms in pure DOPC liposomes (r2 = 0.86, slope = −0.86). No correlation was observed when the liposomes were prepared from 70 : 30 (w/w) DOPC : DOPA. When a linear fit of the obviously poor data set was forced, the slope was ∼0 as was the correlation (i.e., r2). We note that in the previous study11 using the rather complex NMR method, we observed a similar correlation, albeit with fewer compounds and with a lipid mixture.
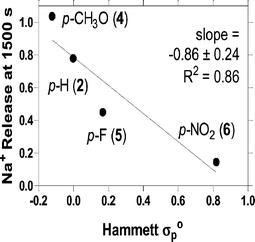 | ||
| Fig. 3 Hammett substituent constant dependence of Na+ release at 1500 s from liposomes mediated by compounds 2, 4, 5, and 6. The transport data show a reasonable correlation with the electronic effect. | ||
Dansyl fluorescence emission spectra
Dansyl channel (7) emission spectra were recorded in the absence or presence of DOPC or DOPC–DOPA vesicles. The results are presented in Fig. 4. Maximal fluorescence was observed for 7 at 532 nm in sodium-free external buffer (Fig. 4, bottom line). This absorption maximum is typical for a dansyl group in a polar environment. The emission spectrum for dansyl channel in DOPC vesicles (Fig. 4, middle trace) showed a small blue shift, to a fluorescence maximum of 527 nm and fluorescence intensity increased. The fluorescence maximum for dansyl typically occurs at shorter wavelength and is often more intense in less polar media.27 The largest blue shift was observed for 7 (532→515 nm), when it was inserted into liposomes prepared from a 70 : 30 mixture of DOPC–DOPA (Fig. 4, upper curve). The maximum blue shift (17 nm) was observed in the 70 : 30 system; no further shift was observed in a 50 : 50 mixture of these lipids (data not shown). We interpret the data of Fig. 4 to reflect movement of the dansyl group to a more apolar environment when the surface charge of the lipid becomes more negative.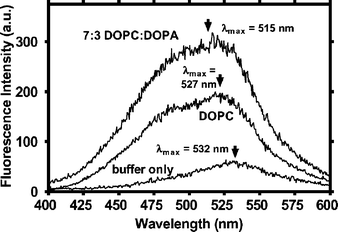 | ||
| Fig. 4 Fluorescence spectra for dansyl channel 7 in (bottom line) buffer, in pure DOPC liposomes, and (top line) 70 : 30 (w/w) DOPC : DOPA liposomes. | ||
We have previously used dansyl channel fluorescence to assess the position of the headgroup in the bilayer.28 We reported that the dansyl headgroup of 7 experienced a local dielectric similar to that expected for the midpolar regime of the bilayer.29 The latter measurements were confirmed using fluorescence depth quenching. We now consider the effect of lipid composition on the position of dansyl channel 7, as reported by its fluorescence maximum. The difference in the fluorescence maximum when 7 is in DOPC compared to DOPC : DOPA is apparent in Fig. 4. When the lipids have a neutral surface charge (DOPC), the sidearms are closer to the bilayer surface. Stabilizing cation–π interactions between the sidearm arenes and quaternary ammonium groups are likely. The presence of DOPA in DOPC makes the membrane surface more negative, obviating the favorable cation–π interaction.
When the liposome surface is more negative, the electron rich sidearms are repelled, they move closer to the hydrophobic core, and the headgroup fluorescence spectrum is blue-shifted (Fig. 4). The fluorescence data show that the sidearm of 7 is closer to the DOPC liposome surface than when DOPA is present in the bilayer. The ion transport data show greater efficacy for those hydraphiles that can interact favorably with an ammonium cation. When this stabilization is diminished, either by electronegative substitution on the sidearm arenes or inclusion of negative lipids in the liposomal bilayer, Na+ transport decreases.
Implications for proteins
Cation–π interactions are increasingly documented. In our own work, we showed using solution and solid state methods that sodium and potassium are π-complexed by benzene, phenol, and indole, the sidechain arenes of Phe, Tyr, and Trp.14–16,30 The preference of tryptophan residues for membrane interfaces is well documented.31 In some cases, cation–π interactions between Trp and positively charged Arg or Lys, have been observed.32 The possible roles of Phe residues have been less well investigated but were recently implicated in hydrophobic interactions in the neuropeptide Achatin-I.33 Molecular dynamics simulations in that case predicted the existence of close contacts between amino acid sidechains and lipid headgroups.34 Recent NMR studies have reported a cation–π interaction between the quaternary ammonium headgroup of phosphatidylcholine and Trp35 alone or Trp and Tyr.36 The crystal structure of human phosphatidylcholine transfer protein (PC-TP) shows that the lipid's quaternary ammonium headgroup is surrounded by an “aromatic cage” formed from a Trp and two Tyr residues.37 We note that so-called aromatic side chain “belts” have been recognized as stabilizing ensembles.38In other work from our lab, we noted that the indole residues of tryptophan could serve as amphiphilic headgroups39 at aqueous–bilayer boundaries.40 We illustrate this potential with the ribbon diagram drawn from the coordinates in reference 5 and shown as Fig. 5. It illustrates the position of the aromatic sidechains of the KcsA potassium channel from Streptomyces lividans. With the exception of four Phe residues in the ion pathway, every aromatic amino acid in the structure is found at the lipid interface. The hydraphile synthetic channels show that the electron richness of π-donor residues near the membrane interface correlates with increased ion transport activity.
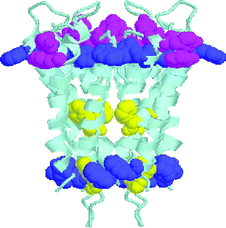 | ||
| Fig. 5 Ribbon structure of the KcsA potassium channel with Phe (yellow), Tyr (pink), Trp (blue) depicted as space filling models. This image was constructed using Protein Explorer from the crystallographic coordinates 1BL8 of the KcsA channel from Streptomyces lividans as determined by Doyle et al.5 | ||
Conclusion
We have used an ion selective electrode method in conjunction with fluorescence studies to show that hydraphile channels can be stabilized in the bilayer to afford Na+ transport activity. These compounds, which contain sidearms with varying degrees of π-density, demonstrate the importance of cation–π interactions in obtaining a stable, active conformation for cation transport by hydraphile channels. In a liposome system comprising neutral lipid headgroups, those compounds with π-dense sidearms exhibit higher levels of Na+ transport than those with electron-withdrawing groups on the arene system. When the lipid headgroup is negative, the activity of the latter channels is unaffected, while the channels with highly aromatic sidearms have decreased transport ability.The difference in activity is presumably due to hydraphile stabilization in the bilayer that is lost when the lipid surface is negatively charged. The fluorescence studies presented above confirm that the channel's sidearm is localized further into the hydrophobic core of the bilayer in the negative headgroup system, suggesting that this stabilization comes from the lipid headgroup. We presume this interaction to be a cation–π interaction between the quaternary amine of the phosphocholine headgroup and the arene-containing sidearms of the hydraphile channel.
Experimental
General
1H-NMR were recorded at 300 MHz in CDCl3 solvents and are reported in ppm (δ) downfield from internal (CH3)4Si. 13C-NMR were recorded at 75 MHz in CDCl3 unless otherwise stated. Infrared spectra were recorded on a Perkin-Elmer 1710 Fourier Transform Infrared Spectrophotometer and were calibrated against the 1601 cm−1 band of polystyrene. Melting points were determined on a Thomas Hoover apparatus in open capillaries and are uncorrected. Thin layer chromatographic (TLC) analyses were performed on aluminium oxide 60 F-254 neutral (Type E) with a 0.2 mm layer thickness or on silica gel 60 F-254 with a 0.2 mm layer thickness. Preparative chromatography columns were packed with activated aluminium oxide (MCB 80–325 mesh, chromatographic grade, AX 611) or with Kieselgel 60 (70–230 mesh).All reactions were conducted under dry N2 unless otherwise stated. All reagents were the best (non-LC) grade commercially available and were distilled, recrystallized, or used without further purification, as appropriate. Combustion analyses were performed by Atlantic Microlab, Inc., Atlanta, GA, and are reported as percents. Where water is factored into the analytical data, spectral evidence is presented for its presence.
Synthesis of hydraphiles
Compounds 1, 2, 4–8 have been reported previously.6,29,41Preparation of 3, PhCH2CH2<N18N>C12<N18N>C12<N18N>CH2CH2Ph
H<N18N>(CH2)12<N18N>(CH2)12<N18N>H (9) (149 mg, 0.134 mmol) and (2-bromoethyl)benzene (55 µL, 0.4 mmol), Na2CO3 (716 mg, 6.7 mmol), and KI (cat.) were dissolved in n-PrCN (30 mL). The reaction was stirred at reflux for 72 h and the solvent was removed. The residue was washed with 5% NaHCO3 (3 × 20 mL), dried and concentrated to afford a yellow residue which was crystallized from acetone to give 3 (128 mg, 73%) as white crystals (mp 65–66 °C). 1H-NMR: 1.25 (s, 32H), 1.43 (m, 8H), 2.47 (m, 8H), 2.77 (m, 24H), 2.855 (t, J = 6.0, 8H), 3.58–3.64 (m, 48H), 3.59 (s, 4H), 7.16–7.27 (m, 10H). 13C-NMR: 27.10, 27.50, 29.61, 33.74, 53.91, 56.01, 57.88, 69.96, 70.23, 70.73, 125.91, 128.32, 128.73, 140.52 ppm. Calculated for C76H138N6O12: C, 68.74; H, 10.47; N, 6.33%. Found: C, 68.52; H, 10.47; N, 6.32%.Vesicle preparation and sodium transport measurements
Details of this ion selective electrode (ISE) methodology were recently reported.9 Liposomes were formed (20 s sonication) from 1,2-dioleoyl-sn-glycero-3-phosphocholine and 0% or 30% (w/w) of 1,2-dioleoyl-sn-glycero-3-phosphate (Avanti Polar Lipids, AL). The internal buffer was 750 mM NaCl–15 mM N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid (HEPES), pH 7.0. The liposomes were filtered (0.2 µm membrane) and passed over a Sephadex G25 column equilibrated in sodium-free buffer (750 mM cholineCl–15 mM HEPES, pH 7.0). Lipid concentration was measured as previously reported42 and vesicle size was confirmed on a Coulter N4MD submicron particle analyzer to be ∼200 nm. Sodium efflux was measured by using a pH–Na+ combination microelectrode (Thermo-Orion) as previously described.9 Final values were determined after vesicular lysis and curve calibration.Data analysis
The data were acquired in mV units, which were converted to concentration by using the calibration curve for the electrode (recalibrated each ∼3 weeks). After converting from mV to concentration, the data were normalized to the total sodium concentration, as determined by lysing the vesicles with n-octylglucoside. Data manipulation was done using Origin Pro 7 software.Dansyl fluorescence emission spectra
Fluorescence was measured using a Perkin Elmer LS50B fluorimeter with continuous stirring. A stock solution of 0.67 mM dansyl channel (7) in 2-propanol was prepared and stored at 4 °C when not in use. Dansyl channel was added and stirred for ∼60 s before spectra were taken. The emission spectrum for 17 µM 7 was measured in 2 mL of buffer (750 mM cholineCl–15 mM HEPES, pH 7.0). The dansyl group was excited at 340 nm and the emission scanned from 350–700 nm. In each case, five scans were averaged to reduce baseline noise. For the measurements in vesicles, 7 was added to the liposome suspension (as prepared above) in buffer to achieve a total volume of 2 mL and overall lipid concentration of 0.4 mM.Acknowledgements
We thank the NIH (GM-36262) and NSF (CHE-0415586) for grants that supported this work and for a Training Grant Fellowship under grant T32-GM08785 to MEW.References
- (a) G. W. Gokel and A. Mukhopadhyay, Chem. Rev., 2001, 30, 274–286 CrossRef CAS; (b) G. W. Gokel, P. H. Schlesinger, N. K. Djedovic, R. Ferdani, E. C. Harder, J. Hu, W. M. Leevy, J. Pajewska, R. Pajewski and M. E. Weber, Bioorg. Med. Chem., 2004, 12, 1291–1304 CrossRef CAS.
- G. W. Gokel, Chem. Commun., 2000, 1–9 RSC.
- P. H. Schlesinger, R. Ferdani, J. Liu, J. Pajewska, R. Pajewski, M. Saito, H. Shabany and G. W. Gokel, J. Am. Chem. Soc., 2002, 124, 1848–1849 CrossRef CAS.
- C. L. Murray, H. Shabany and G. W. Gokel, Chem. Commun., 2000, 2371–2372 RSC.
- D. A. Doyle, J. M. Cabral, R. A. Pfuetzner, A. Kuo, G. J. M., S. L. Cohen, B. T. Chait and R. MacKinnon, Science, 1998, 280, 69–77 CrossRef CAS.
- O. Murillo, S. Watanabe, A. Nakano and G. W. Gokel, J. Am. Chem. Soc., 1995, 117, 7665–7679 CrossRef CAS.
- F. G. Riddell and S. J. Tompsett, Biochim. Biophys. Acta, 1990, 990, 193–197.
- W. M. Leevy, M. E. Weber, P. H. Schlesinger and G. W. Gokel, Chem. Commun., 2004, 89–91 Search PubMed.
- M. E. Weber, P. H. Schlesinger and G. W. Gokel, J. Am. Chem. Soc., 2005, 127, 636–642 CrossRef CAS.
- (a) E. S. Meadows, S. L. De Wall, L. J. Barbour and G. W. Gokel, J. Am. Chem. Soc., 2001, 123, 3092–3107 CrossRef CAS; (b) J. Hu, L. J. Barbour and G. W. Gokel, Proc. Natl. Acad. Sci. USA, 2002, 99, 5121–5126 CrossRef.
- O. Murillo, I. Suzuki, E. Abel and G. W. Gokel, J. Am. Chem. Soc., 1996, 118, 7628–7629 CrossRef CAS.
- (a) J. C. Ma and D. A. Dougherty, Chem. Rev., 1997, 97, 1303–1324 CrossRef CAS; (b) G. W. Gokel, S. L. D. Wall and E. S. Meadows, Eur. J. Org. Chem., 2000, 2967–2978 CrossRef CAS; (c) G. W. Gokel, L. J. Barbour, R. Ferdani and J. Hu, Acc. Chem. Res., 2002, 35, 878–86 CrossRef CAS.
- S. L. De Wall, E. S. Meadows, L. J. Barbour and G. W. Gokel, Proc. Natl. Acad. Sci. USA, 2000, 97, 6271–6276 CrossRef CAS.
- S. L. De Wall, L. J. Barbour and G. W. Gokel, J. Am. Chem. Soc., 1999, 121, 8405–8406 CrossRef.
- S. L. De Wall, E. S. Meadows, L. J. Barbour and G. W. Gokel, J. Am. Chem. Soc., 1999, 121, 5613–5614 CrossRef.
- J. Hu, L. J. Barbour and G. W. Gokel, Chem. Commun., 2002, 1808–1809 RSC.
- D. A. Gustowski, V. J. Gatto, J. Mallén, L. Echegoyen and G. W. Gokel, J. Org. Chem., 1987, 52, 5172–5176 CrossRef CAS.
- M. Elder, P. B. Hitchcock, R. Mason and G. G. Shipley, Proc. R. Soc. London, Ser. A, 1977, 354, 157–170 CrossRef CAS.
- I. Pascher and S. Sundell, Biochim. Biophys. Acta, 1986, 855, 68–78 CAS.
- K. Harlos, H. Eibl, I. Pascher and S. Sundell, Chem. Phys. Lipids, 1984, 34, 115–126 CrossRef.
- N. Albon, J. McAlister and M. Sundaralingam, Am. Crystallogr. Assoc., Ser. 2, 1977, 5, 69–70 Search PubMed.
- R. H. Pearson and I. Pascher, Nature (London), 1979, 281, 499–501 CAS.
- A. C. Sau, R. O. Day and R. R. Holmes, Inorg. Chem., 1981, 20, 3076–3081 CrossRef CAS.
- J. L. Atwood and L. J. Barbour, Cryst. Growth Des., 2003, 3, 3–8 CrossRef CAS.
- J. Hu, L. J. Barbour and G. W. Gokel, New J. Chem., 2004, 28, 907–911 RSC.
- R. W. Taft, J. Phys. Chem., 1960, 64, 1805–1815 CrossRef.
- E. Breukink, C. van Kraaij, A. van Dalen, R. A. Demel, R. J. Siezen, B. de Kruijff and O. P. Kuipers, Biochemistry, 1998, 37, 8153–8162 CrossRef CAS.
- E. Abel, G. E. M. Maguire, O. Murillo, I. Suzuki, S. L. De Wall and G. W. Gokel, J. Am. Chem. Soc., 1999, 121, 9043–9052 CrossRef CAS.
- S. Ohki and K. Arnold, J. Membr. Biol., 1990, 114, 195–203 Search PubMed.
- G. W. Gokel, L. J. Barbour, R. Ferdani and J. Hu, Acc. Chem. Res., 2002, 35, 878–886 CrossRef CAS.
- (a) M. R. R. De Planque, J. A. W. Kruijtzer, R. M. J. Liskamp, D. Marsh, D. V. Greathouse, R. E. Koeppe, II, B. de Kruijff and J. A. Killian, J. Biol. Chem., 1999, 274, 20839–20846 CrossRef CAS; (b) W. Hu and T. A. Cross, Biochemistry, 1995, 34, 14147–14155 CrossRef CAS; (c) W.-M. Yau, W. C. Wimley, K. Gawrisch and S. H. White, Biochemistry, 1998, 37, 14713–14718 CrossRef CAS.
- (a) F. Liu, N. A. H. R. Lewis, R. S. Hodges and R. N. McElhaney, Biochemistry, 2004, 43, 3679–3687 CrossRef CAS; (b) M. P. Aliste, J. L. MacCallum and D. P. Tieleman, Biochemistry, 2003, 42, 8976–8987 CrossRef CAS.
- T. Kimura, E. Okamura, N. Matubayasi, K. Asami and M. Nakahara, Biophys. J., 2004, 87, 375–385 CrossRef CAS.
- (a) C. Domene, P. J. Bond, S. S. Deol and M. S. P. Sansom, J. Am. Chem. Soc., 2003, 125, 12966–14967; (b) S. S. Deol, P. J. Bond, C. Domene and M. S. P. Sansom, Biophys. J., 2004, 87, 3737–3749 CrossRef CAS.
- D. A. Wah, C. Fernandez-Tornero, L. Sanz, A. Romero and J. J. Calvete, Structure, 2002, 10, 505–514 CrossRef CAS.
- J. M. Sanderson and E. J. Whelan, Phys. Chem. Chem. Phys., 2004, 6, 1012–1017 RSC.
- S. L. Roderick, W. W. Chan, D. S. Agate, L. R. Olsen, M. W. Vetting, K. R. Rajashankar and D. E. Cohen, Nat. Struct. Biol., 2002, 9, 507–511 CAS.
- W.-M. Yau, W. C. Wimley, K. Gawrisch and S. H. White, Biochemistry, 1998, 37, 14713–14718 CrossRef CAS.
- E. Abel, M. F. Fedders and G. W. Gokel, J. Am. Chem. Soc., 1995, 117, 1265–1270 CrossRef CAS.
- E. Abel, S. L. De Wall, W. B. Edwards, S. Lalitha, D. F. Covey and G. W. Gokel, Chem. Commun., 2000, 433–434 RSC.
- (a) O. Murillo, I. Suzuki, E. Abel, C. L. Murray, E. S. Meadows, T. Jin and G. W. Gokel, J. Am. Chem. Soc., 1997, 119, 5540–5549 CrossRef CAS; (b) E. Abel, G. E. M. Maguire, O. Murillo, I. Suzuki, S. L. De Wall and G. W. Gokel, J. Am. Chem. Soc., 1999, 121, 9043–9052 CrossRef CAS; (c) C. L. Murray, E. S. Meadows, O. Murillo and G. W. Gokel, J. Am. Chem. Soc., 1997, 119, 7887–7888 CrossRef CAS.
- J. C. Stewart, Anal. Biochem., 1980, 104, 10–4 CAS.
| This journal is © The Royal Society of Chemistry 2006 |