A promiscuous glutathione transferase transformed into a selective thiolester hydrolase†
Sofia
Hederos
a,
Lotta
Tegler
b,
Jonas
Carlsson
c,
Bengt
Persson
c,
Johan
Viljanen
a and
Kerstin S.
Broo
*a
aIFM Chemistry, Division of Organic Chemistry, Linköping University, SE-581 83, Linköping, Sweden. E-mail: kerbr@ifm.liu.se; Fax: +46 13 122587; Tel: +46 13 286679
bDivision of Organic Chemistry, Biomedical Centre, Uppsala University, Box 599, SE-751 24, Uppsala, Sweden
cIFM Theory and Modelling, Division of Bioinformatics, Linköping University, SE-581 83, Linköping, Sweden
First published on 1st December 2005
Abstract
Human glutathione transferase A1-1 (hGST A1-1) can be reengineered by rational design into a catalyst for thiolester hydrolysis with a catalytic proficiency of 1.4 × 107 M−1. The thiolester hydrolase, A216H that was obtained by the introduction of a single histidine residue at position 216 catalyzed the hydrolysis of a substrate termed GSB, a thiolester of glutathione and benzoic acid. Here we investigate the substrate requirements of this designed enzyme by screening a thiolester library. We found that only two thiolesters out of 18 were substrates for A216H. The A216H-catalyzed hydrolysis of GS-2 (thiolester of glutathione and naphthalenecarboxylic acid) exhibits a kcat of 0.0032 min−1 and a KM of 41 µM. The previously reported catalysis of GSB has a kcat of 0.00078 min−1 and KM of 5 µM. The kcat for A216H-catalyzed hydrolysis of GS-2 is thus 4.1 times higher than for GSB. The catalytic proficiency (kcat/KM)/kuncat for GS-2 is 3 × 106 M−1. The promiscuous feature of the wt protein towards a range of different substrates has not been conserved in A216H but we have obtained a selective enzyme with high demands on the substrate.
Introduction
The design of novel catalysts is a challenging task that, in addition to extending our knowledge of proteins, eventually may lead to the development of selective and environmentally friendly catalysts1 for reactions not catalyzed by Nature. Progress in this field has been made by rational design,2–5 computational design6–8 and combinatorial approaches.9–14 Other studies have been performed with enzyme models5 and through covalent modification of natural proteins to introduce artificial functional groups.15–17A prerequisite in the redesign of proteins is the use of an appropriate scaffold and the glutathione transferases (GSTs EC 2.5.1.18)‡§ are good candidates because of their stability, ease of purification18 and the wealth of knowledge concerning structure–activity relationships.19 The GSTs belong to a large family of detoxication enzymes that catalyze the conjugation of the tripeptide glutathione (GSH) (Scheme 1) to a broad range of different hydrophobic electrophiles.20–23 The proteins exist as homo- or heterodimers22,24 and the active site consists of a G-site, where GSH binds, and a promiscuous hydrophobic substrate-binding site, the H-site, where the electrophilic molecules bind.25,26 Catalysis is mediated through a conserved tyrosine, serine, or cysteine residue that activates GSH for nucleophilic attack22 and the G-site is essentially conserved throughout the classes.20,26 The differences that provide the substrate specificities of the isozymes are mainly located in the H-site.25 In the alpha class, the H-site is shielded from the solvent by a flexible helical segment.26
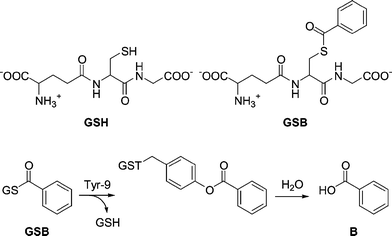 | ||
| Scheme 1 | ||
We have previously, through rational design, constructed a thiolester hydrolase by introducing a single histidine residue in position 216 in hGST A1-1 (Fig. 1).27 The reaction catalyzed by the designed enzyme, A216H, is an example of catalytic promiscuity28–30 where an alternative function is revealed by a minimal change of active site residues. The A216H-catalyzed hydrolysis of the synthetic substrate GSB (Scheme 1) to form GSH and benzoic acid showed a catalytic proficiency31 (kcat/KM)/kuncat of more than 107 M−1.27 The wt protein is instead covalently modified with the acyl moiety on residue Y9.32 This acylated Y9 is an intermediate on the reaction pathway in the A216H-catalyzed hydrolysis reaction of GSB.27
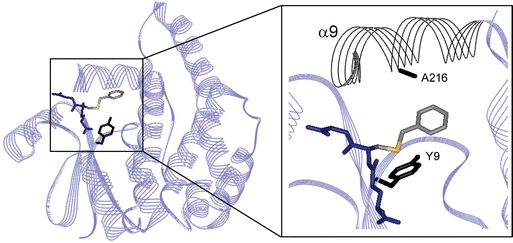 | ||
| Fig. 1 A close-up of the crystal structure (1GUH) of hGST A1-1 in a complex with S-benzyl-glutathione.26 The amino acids Y9 and A216 are shown in “stick” representation and helix 9 (α9) is also indicated. The protein is dimeric but shown here as a monomer for reasons of clarity of presentation. | ||
Since the natural detoxication activity of wt hGST A1-1 includes a wide range of substrates,25 we were interested in investigating the substrate requirements of the novel thiolester hydrolase A216H. A panel of 17 thiolesters of glutathione (GS-thiolesters) was used as a library in screening experiments. The library contains GS-thiolesters that vary in size, shape, reactivity and hydrophobicity.33 Two non-aromatic GS-thiolesters were included since GST A1-1 from rat (rGST A1-1), a protein highly homologous to hGST A1-1, has been shown to hydrolyze non-aromatic thiolesters.34 This library has previously been used to investigate the requirements for the site-specific Y9 modification of wt hGST A1-1.33 In that study 78% of the GS-thiolesters were able to site-specifically modify Y9 and since the same library was used in this study, we hoped to explore the requirements for catalysis versus modification.
Results
To analyze all GS-thiolesters (18 including GSB) with five time points per sample, would generate more than 300 injections when using reversed phase HPLC (samples incubated with protein and reference samples). This is a time-consuming process (approximately 15 000 min of HPLC time) and, in addition, the measurements would have been performed only once. We have instead used an alternative method with a screening approach where the selection criteria have been sharpened in every new step. In total, four screening experiments (A–D) were performed to verify the results and to elucidate the substrate requirements of the designed enzyme A216H.The reaction products of the hydrolysis reaction would be GSH and the corresponding acid (Scheme 1) but we have only tried to detect either the decrease of substrate (GS-thiolester) or formation of acid. This is because GSH was very difficult to detect quantitatively in our pilot experiments using reversed phase HPLC due to its high solubility in water and thus poor and unreliable retention times.35 In addition, the thiol is prone to oxidation and this also makes quantitative HPLC analyses difficult.
Prior to starting the screening experiment, we determined that a common detection limit for the acids was approximately a few µM using our HPLC method with detection at 250 nm. This experiment also provided the retention time of each acid. To maximize the yields of the reactions, the samples were incubated for an extended period of time (26 h), something that is possible due to the stability of A216H as measured by the CDNB activity that is constant over at least 72 hours incubation at 25 °C (ESI†). As discussed in our previous study, the uncatalyzed hydrolysis reactions of the substrates were almost negligible.27,33 All compounds in the library were incubated separately in all experiments either with or without protein. The reaction mixtures were quenched with trifluoroacetic acid and analyzed by reversed phase HPLC following addition of internal standard.
Selection criteria
The selection criteria of the screening procedure were initially generous and a stepwise sharpening of the analytical processing filtered out false positives. This procedure also automatically included repeated measurements of potential substrates even though a screening process was used. We had previously determined27 that 5.6 µM benzoic acid was produced after 24 h using 5 µM A216H and 75 µM GSB. With the detection limits taken into account, a higher concentration (15 µM) of A216H was used in screening experiment A to allow the identification of substrates that displayed at least 25% of the rate constant of A216H-catalyzed hydrolysis of GSB. The selection criteria in screening experiment A were generous and samples that showed any trace of acid with a signal-to-noise ratio larger than 10 were further analyzed in screening experiment B.In screening experiment B the concentrations of A216H and GS-thiolester were lowered to match previous studies performed with A216H27 since we intended to analyze the reactions with UV spectroscopy. The lowered concentration of A216H from 15 µM to 5 µM placed higher demands on the substrate. To elucidate the importance of the histidine residue in position 216, samples of GS-thiolesters incubated with wt protein were included in screening experiments B–D. Background samples of the GS-thiolesters incubated without protein were also examined. A threshold was set in experiment B so that there had to be a difference of 65% or more in relative concentration for A216H compared to the wt sample in either GS-thiolester consumption or acid production. This threshold was set to allow for the fact that the wt protein reacts covalently in a stoichiometric fashion with 13 of the 17 GS-thiolesters in the panel.33 In addition, it has previously been shown that the wt protein hydrolyzes some non-aromatic GS-thiolesters36 and the threshold was also set to filter out such GS-thiolesters.
The stringency in experiment C was increased by the use of a kinetic analysis of the samples. The observed rate constants for both GS-thiolester consumption and acid production had to display a difference of 65% or more for the A216H-incubated sample compared to the sample incubated with wt protein.
The final screening experiment, D, was mainly performed to resolve the ambiguity concerning one of the GS-thiolesters (GS-3).
Results from the screening experiments
A summary of the outcome of the screening experiments is presented in Table 1. In screening experiment A, nine of the GS-thiolesters (Table 1) and the control substrate GSB displayed traces of acid in the presence of A216H (Figs. 2A and B). The previously thoroughly investigated substrate GSB27 was included as a control (Fig. 2A) to verify that the reaction conditions used were satisfactory and that the protein was in good shape. The analyses were done at various wavelengths based on the UV spectrum of each acid and GS-thiolester (typically 229 and 250 nm) since the HPLC was equipped with a photo diode detector. However, a peak from the protein was seen in close proximity to the internal standard at 229 nm (Fig. 2A) so analysis was made at 250 nm when allowed by the UV spectrum of each compound. The remaining eight GS-thiolesters that did not show any trace of acid were excluded from further experiments.![[hair space]](https://www.rsc.org/images/entities/char_200a.gif)
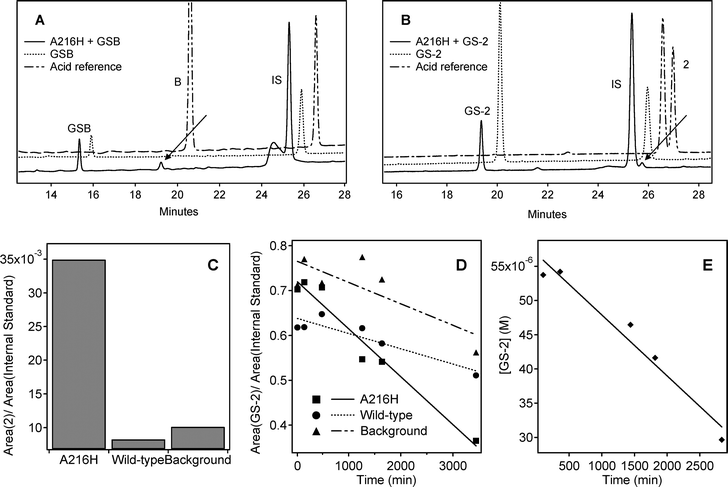 | ||
| Fig. 2 Representative data from the screening experiments A–D. In screening experiment A, 15 µM A216H was incubated with 100 µM GS-thiolester, and analyzed by reversed phase HPLC. Traces are shown from (A) the positive control GSB (λ = 229 nm) and (B) GS-2 (λ = 250 nm). The GS-thiolester and the acid reference are shown as well. The observed peak from the product (A) benzoic acid and (B) naphthalenecarboxylic acid (2) is indicated with an arrow. (C) Data from GS-2 obtained in screening experiment B. The thiolester GS-2 (75 µM) was incubated with 5 µM A216H and wt protein respectively. A background sample with no protein present was also collected. Late time points (after 46 hours of incubation) were analyzed by reversed phase HPLC and the peak areas were derived from integration of the peaks in the chromatograms. The increased production of naphthalenecarboxylic acid in the A216H sample compared to the wt and reference samples is seen. (D) In screening experiment C, a kinetic analysis was performed with both early and late time points that were analyzed by reversed phase HPLC. Here, the peak area of remaining GS-2 divided by the peak area of the internal standard is plotted as a function of time. (E) Data from screening experiment D that was derived from HPLC analysis of 5 µM A216H incubated with 75 µM GS-2. The concentration of GS-2 was obtained by using relative responses and is plotted as a function of time. | ||
The reactions in screening experiment B were followed by UV spectroscopy but due to the complexity of the resulting UV traces no effort was put into the interpretation of the data. Instead, late time points were collected after approximately two days and analyzed by reversed phase HPLC. The peaks in the chromatograms were integrated and the relative concentration was determined by dividing the area of the GS-thiolester or acid peak respectively by the area of the internal standard (Fig. 2C). Five of the GS-thiolesters (Table 1) fulfilled the selection criteria and were further analyzed in experiment C. The remaining four GS-thiolesters were rejected.
The kinetic analysis in screening experiment C is illustrated in Fig. 2D. Only one GS-thiolester (GS-2) fulfilled the selection criteria. The data from GS-3 were irreproducible but GS-3 showed interesting results and was further analyzed by conducting a fourth screening experiment (D).
The final screening experiment (D) was carried out with GS-2 and GS-3. The concentration of GS-3 was varied in an attempt to resolve the ambiguities concerning the irreproducible results. For all samples, five time points were withdrawn and analyzed by reversed phase HPLC. The concentration of remaining GS-thiolester was calculated, using relative responses, and plotted as a function of time (Fig. 2E) to determine the rate constants. Due to the low signal-to-noise ratio of the acids 2 and 3, only the decrease in GS-thiolester concentration was used. Again, experiments performed with GS-3 were not reproducible and GS-3 was therefore excluded as a possible substrate. Hence, only GS-2 was found to be a substrate for A216H.
Saturation kinetics of A216H
The Michaelis–Menten behavior of A216H towards GS-2 was analyzed by incubating various concentrations of GS-2 with 5 µM A216H at pH 7 and 25 °C. The hydrolytic reaction to form naphthalenecarboxylic acid (2) and GSH from GS-2 was followed by monitoring the consumption of GS-2 by reversed phase HPLC at five time points (ESI†). The reaction followed saturation kinetics under turnover conditions and the kinetic profile is seen in Fig. 3. The kinetic parameters were determined to be kcat = 0.0032 min−1 and KM = 41 µM (Table 2). The wt protein is acylated at Y9 by GS-233 but the reaction stops after addition of one equivalent of reagent and hGST A1-1 is thus not a catalyst for hydrolysis of GS-2 as shown by the inset in Fig. 3.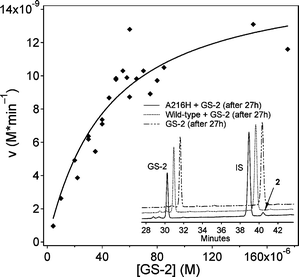 | ||
| Fig. 3 A216H-catalyzed hydrolysis of GS-2 to form GSH and naphthalenecarboxylic acid. The kinetic studies were performed with 5 µM A216H at pH 7 and 25 °C and the reaction rates were determined by analyzing five time points by reversed phase HPLC. The peaks of GS-2 and the internal standard (IS) were integrated to determine the remanining concentration of GS-2. HPLC traces (inset) show the decrease of GS-2 and formation of naphthalenecarboxylic acid (2) in the prescence of A216H. The decrease of GS-2 in the wt sample is due to acylation of Y9.33 | ||
The background hydrolysis of GS-2 was determined using N-acetylated GS-2. This is due to a previous observation with GSB where a competing intramolecular acyl transfer reaction to form N-acylated GSH proceeds with a rate of 5.8 × 10−9 M min−1 when no protein is present. The first-order rate constant of the uncatalyzed hydrolysis of N-acetylated GS-2 was determined by reversed phase HPLC to be 3 × 10−5 min−1 at pH 7 (100 mM NaPi) and 25 °C. The rate constant for hydrolysis of N-acetylated GSB has previously been determined to be of the same order of magnitude (1.1 × 10−5 min−1).27
Computer simulation studies
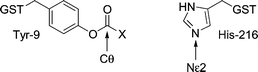
The distance between the epsilon nitrogen of H216 in A216H and the carbonyl carbon of the substrate was investigated by molecular modeling to obtain supporting information about why catalysis occurs for only two out of 18 GS-thiolesters tested. The acyl groups of nine representative GS-thiolesters (Table 3) were attached covalently to the side chain of Y9 using the crystal structure of A216H27 (Fig. 4) as a starting point. Energy minimizations of the modified structures were then done by Monte Carlo methods and the observed distances between the epsilon nitrogen (Nε2) of H216 and the carbonyl carbon (Cθ) of the Y9 ester are shown in Table 3. The only GS-thiolesters in this subset that show distances less than 5 Å between Nε2 and Cθ are GSB and GS-2. It should be noted that acyl groups that do not display reoccurring structures are probably not in the lowest energy conformation since several different structures were found when repeating the simulations. Out of the nine acids modeled at Y9, five show reoccurring structures, where typically half the simulations have very similar conformations and energies.|
|
||
|---|---|---|
| Estera | Distance Nε2–Cθ/Å | Reoccurring structureb,c |
| a B corresponds to benzoic acid and the numbers to the acids in Chart 1. b Several simulations have given the same conformation (rmsd < 0.6 Å) with similar energies. c + corresponds to yes and − to no. | ||
| Tyr-B | 4.6 | + |
| Tyr-2 | 4.0 | − |
| Tyr-3 | 5.1 | − |
| Tyr-4 | 6.6 | + |
| Tyr-8 | 6.1 | + |
| Tyr-10 | 5.9 | − |
| Tyr-11 | 8.2 | + |
| Tyr-12 | 5.2 | − |
| Tyr-13 | 6.2 | + |
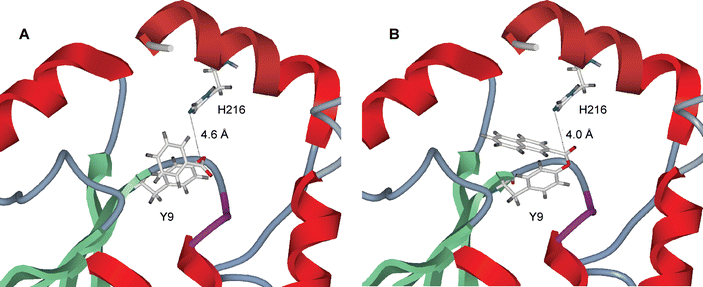 | ||
| Fig. 4 Close-up of the active site of the crystal structure (1USB) of A216H27 with (A) benzoic acid and (B) naphthalenecarboxylic acid introduced by computer modeling at the side chain of Y9. The distance between H216 and the carbonyl carbon of the Y9 ester is (A) 4.6 Å and (B) 4.0 Å. | ||
Discussion
We have previously reported that hGST A1-1 can be reengineered into a thiolester hydrolase by a single point mutation, A216H.27 This new enzyme catalyzed the hydrolysis of GSB to form GSH and benzoic acid via an Y9-acylated intermediate. The wt protein is also acylated at Y9 by GSB but the reaction stops at that stage. The introduced histidine thus opens up a new reaction pathway.27 In a separate line of investigation we found that Y9 in the wt protein could be acylated with a range of different GS-thiolesters33 and we were thus encouraged to explore the substrate requirements of A216H in order to investigate if the introduction of a histidine residue could open up the hydrolytic pathway for GS-thiolesters other than GSB.Only two out of 18 GS-thiolesters are substrates for A216H
Four successive screening experiments filtered out GS-2 as the only new substrate in the library. In each experiment a number of potential substrates were eliminated but false positives survived each round. The reasons behind the false positives are complex but the worst problems are probably low S/N ratios of the acid products, and acylation of Y9 without subsequent hydrolysis. Using this screening procedure we were able to analyze all the potential substrates in the library with a mere 160 injections instead of >300 as outlined in the Results section.Recently, the same GS-thiolesters (Chart 1) were also used to obtain information about the requirements for site-specific modification of Y9 in wt hGST A1-1, a step that corresponds to the first step in the catalytic cycle of A216H-catalyzed hydrolysis of GSB. We found that 14 of the 18 GS-thiolesters (78%) in the library (including GSB) were able to acylate Y9.33 The generality was perhaps not so surprising given that the natural function of the GSTs is conjugation of glutathione to a wide range of electrophiles and the H-site is thus accordingly promiscuous.20–23 Interestingly, we now observe that A216H is only able to hydrolyze two out of the 18 GS-thiolesters tested (10%). Even though several of the GS-thiolesters were able to acylate Y9 as demonstrated by decreases in the concentration of starting material, only in the case of GS-2 (and GSB) is this a true intermediate that allows H216 to complete the catalytic cycle. This demonstrates that correct orientation of the catalytic residues is necessary during the whole cycle. In retrospect, we were indeed fortunate when we designed and synthesized GSB and not a similar substrate. This further illustrates the difficulties encountered when trying to design novel enzymes; it is not only the protein but also the substrate that needs to be taken into account. Perhaps the best starting point would be to investigate a whole panel of substrates and not just one or a few to evaluate the effect of a certain mutation.
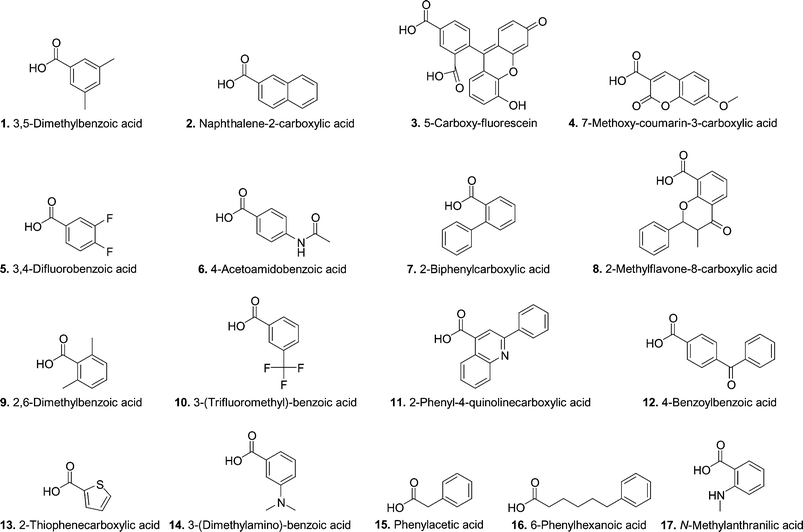 | ||
| Chart 1 | ||
The library contains a range of different GS-thiolesters with respect to size, shape, hydrophobicity and pKa of the acid.33 The non-aromatic GS-thiolesters (GS-15 and GS-16) were also included in the library since the wt rat GST A1-1 (closely homologous to hGST A1-1) is capable of hydrolysis of non-aromatic GS-thiolesters.34 Even though the library contains this variance, only two GS-thiolesters are substrates for A216H and it is thus not possible to draw any relevant conclusions about the substrate requirements. For example, the difference between GS-1 and GS-2 is very small and the substitution in position 5 of GS-1 may perhaps possess a sterical hindrance. It would be interesting to use a more refined library based on GSB where methyl groups are introduced in various positions in the aromatic ring. A primary choice would, for example, be 3,4-dimethylbenzoic acid that closely resembles GS-2.
The distance between acylated Y9 and H216 is the shortest for the two successful substrates
The computer simulations support our experimental finding that only GSB and GS-2 are substrates for A216H. The carbonyl carbons of the esters formed from Y9 and benzoic acid and naphthalenecarboxylic acid (2), respectively, are the only ones that are within a distance of 5 Å with respect to the epsilon nitrogen of H216 (Table 3). In the case of GSB a reoccurring structure appeared (Fig. 4A) with a distance of 4.6 Å between the epsilon nitrogen of H216 and the carbonyl carbon of the ester formed from benzoic acid and Y9. In the case of GS-2 (Fig. 4B) no reoccurring structure was obtained, however, the structure with lowest energy (out of 14 simulations) has been used for a qualitative comparison. The corresponding distance in the case of GS-2 was 4.0 Å. This suggests that only for GSB and GS-2 is the histidine residue in the correct orientation to aid in catalysis and it again stresses that precise positioning of the catalytic amino acids is of utmost importance in the design of novel enzymes. However, other factors may also contribute to the discrimination mechanism. For example, the positioning of the acyl intermediate may render some of the intermediates to be more stabilized than others. The naphthylated side chain of Y9 may, for example, bind better in the active site than the benzoylated one and thus give rise to a higher activation energy.The kcat(GS-2)/kcat(GSB) is 4.1
The kinetic parameters derived from the saturation kinetics studies show that the ratio kcat(GS-2)/kcat(GSB) is 4.1. A higher kcat indicates that the introduced histidine 216 possibly is more favorably oriented for catalysis of hydrolysis of GS-2 than GSB. That the KM for GS-2 should be higher (41 µM) than that for GSB (5 µM) is somewhat surprising since naphthalenecarboxylic acid (2) seemingly has the potential to interact more favorably with the hydrophobic H-site of A216H due to its larger aromatic system. However, it should be noted that KM cannot be treated as a pure dissociation constant (KD) in a multistep reaction. Again, the conclusion is that it is not easy to a priori model the outcome of the dynamic process of catalysis.The background hydrolysis rates of GSB and GS-2 were obtained using N-acetylated GS-thiolesters since we previously found that GSB does not hydrolyze even after nine days at pH 7 and 25 °C but rather undergoes a rearrangement reaction where the acyl group migrates to the α-amino group of GSH.27 Thiolester hydrolysis is not expected to be affected by N-acetylation. The catalytic proficiency (kcat/KM)/kuncat of A216H-catalyzed hydrolysis of GS-2 was calculated to be 3 × 106 M−1. Thus, the higher kcat compensates for the higher KM.
The suggested mechanism for A216H-catalyzed hydrolysis of GS-2
Through detailed mechanistic studies27 we determined that an acylated Y9 is an intermediate in the A216H-mediated hydrolysis of GSB. It is highly likely that the mechanism of hydrolysis of GS-2 by A216H also proceeds via an acylated Y9-intermediate. The basis of this assumption is that the wt protein becomes modified at Y9 by both GS-2 and GSB as shown by HPLC and MALDI-MS.32,33 The introduced histidine residue in A216H is crucial for the catalytic cycle to proceed and in the case of GSB it almost certainly functions as a general base and not a nucleophile.27 We find it highly likely that the same type of mechanism operates in the case of GS-2, especially since this is the only new substrate found during the screening process and it is not likely that the amino acids are positioned such that the mechanism would switch to nucleophilic catalysis by the histidine.Conclusion
The substrate requirements of the designed enzyme A216H have been investigated through a combinatorial screening process of 17 GS-thiolesters. Only one new substrate, GS-2, was found and the ratio kcat(GS-2)/kcat(GSB) is 4.1. The ratio of the catalytic proficiencies (GS-2/GSB) is 0.2. The promiscuous feature of the wt protein towards a range of different natural substrates has not been conserved in the designed enzyme A216H but we have obtained a selective enzyme with high demands on the substrate. We believe that our findings illustrate the challenges encountered in rational design of novel protein function and may be of importance for future biotechnological applications using GSTs.Experimental
All chemicals and reagents used were of the highest purity available. The synthesis of the GS-thiolesters has previously been reported33 and in this study the same stock solutions were used. The HPLC experiments were performed with a Varian system (ProStar 410 autosampler together with a ProStar 230 delivery system and a ProStar 330 photodiode array detector) using a Kromasil C8 column (4.6 mm × 250 mm, Supelco Inc, Bellefonte, USA) and shallow acetonitrile gradients with aqueous trifluoroacetic acid (0.1%) (ESI†). The HPLC samples were mixed with the internal standard 2-chloro-4-nitrophenol and trifluoroacetic acid to a final volume of 30 µL and stored at −20 °C until analysis. The chromatograms in screening experiment B–D and in the saturation kinetic studies were integrated using the Varian software. The UV measurements were carried out with a Varian Cary-100 UV-visible spectrophotometer and submicrocells (Varian, Palo Alto, USA).Site-directed mutagenesis and protein expression
The A216H mutant was prepared by inverse PCR using the wt vector pGNdeA137 and custom-synthesized primers (DNA Technology, Aarhus, Denmark) as described previously.27 The wt protein and the mutant A216H were expressed in Escherichia coli BL21(DE3) (Novagen, San Diego, USA) and purified by cation-exchange chromatography using a HiTrap SP column (Amersham Biosciences, Piscataway, USA).38 SDS-PAGE was used to confirm the purity and concentrations were determined spectrophotometrically using ε280(hGST A1-1) = 24700 M−1 cm−1).38 A standard assay39 using 1 mM 1-chloro-2,4-dinitrobenzene (CDNB) and 1 mM GSH in 100 mM NaPi pH 6.5 at 30 °C was conducted to check the activity of the proteins.Screening procedures
The screening procedures are illustrated in Fig. 5.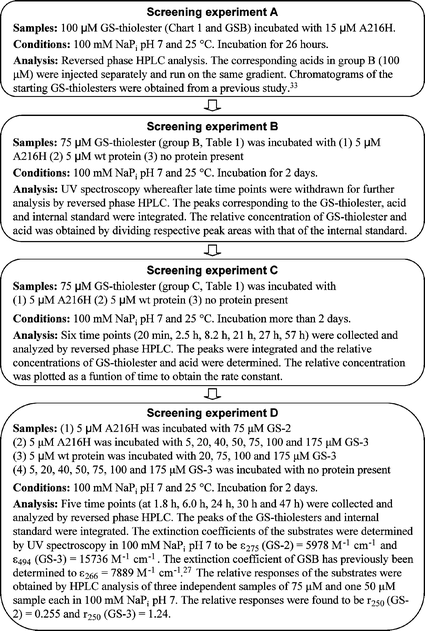 | ||
| Fig. 5 A flow chart of the experimental procedures used for screening experiments A–D. | ||
Saturation kinetics
The saturation behavior of A216H towards GS-2 was analyzed by mixing 5 µM A216H with different concentrations of GS-2 in 100 mM NaPi pH 7 and 25 °C. Time points (4–6 per sample) were collected and analyzed by reversed phase HPLC. Due to the high concentration of enzyme relative to substrate, the Michaelis–Menten equation in the form in which no assumption of [E] « [S] has been made was used to analyze the results, eqn (1).40 The kinetic parameters, kcat and KM, were determined using eqns (2) and (3).| ν = kcat/2(KM + [E]0 + [S]0 − √[(KM + [E]0 + [S]0)2 − 4[E]0[S]0]) | (1) |
| kcat = Vmax/[E]0 | (2) |
| KM = Vmax/2 | (3) |
The saturation kinetics in the case of GSB was previously determined by monitoring the disappearance of the thiolester functionality of GSB at 266 nm by UV spectroscopy.27
Background hydrolysis rate
N-Acetylated GS-2 was synthesized by adding four portions of 10 equivalents of sulfo-N-hydroxysuccinimide acetate during a period of four hours to GS-2 (final concentration 75 µM) in 100 mM NaPi pH 7. HPLC was used to confirm the acetylation of GS-2. The background hydrolysis sample was incubated at 25 °C and time points were collected over 11 days and mixed with internal standard and trifluoroacetic acid to quench the reaction whereupon the samples were stored frozen until analysis. Reversed phase HPLC was used to determine the remaining concentration of N-acetylated GS-2.Computer simulations
The PDB file 1USB was used as a starting structure for the simulations and a subset of acids were covalently attached to produce an ester at the side chain of Y9 before the computer simulations were started. The energy of the modified structure was then minimized by a Monte Carlo-based method as implemented in the ICM program (Molsoft LLC, La Jolla, CA). The method uses the ECEPP/3 forcefield41 and a form of implicit solvation called atomic solvation.42 For non-bonded interactions the energy function is a combination of van der Waals interaction,43 hydrogen bonding,43 torsion energy43 and electrostatic interactions.44 Amino acid residues included in the simulation where located within a sphere of 5 Å around Y9. Remaining residues of the molecule were fixed in order to speed up the calculations. Thus, 18 residues (7–16, 20, 33–35, 55–57 and 216) were included in the energy minimization, but in order for the Monte Carlo method to work there must be at least four amino acid residues in sequence. Therefore, only ten of the residues (7–16) could be run through the Monte Carlo minimization and the other eight residues were minimized with localized methods in the Monte Carlo simulation. Between four and fourteen simulations with one million Monte Carlo iterations per simulation were performed for each thiolester substrate.Acknowledgements
We thank Professor B. H. Jonsson (Linköping University) for critical reading of the manuscript. This work was financially supported by The Wenner-Gren Foundation, The Carl-Trygger Foundation, The Knut and Alice Wallenberg Foundation, and the Swedish Research Council. S.H. is enrolled in the graduate school Forum Scientium, supported by the Swedish Foundation for Strategic Research (SSF).References
- A. Schmid, J. S. Dordick, B. Hauer, A. Kiener, M. Wubbolts and B. Witholt, Nature (London), 2001, 409, 258–268 CrossRef CAS.
- T. M. Penning and J. M. Jez, Chem. Rev., 2001, 101, 3027–3046 CrossRef CAS.
- D. N. Bolon, C. A. Voigt and S. L. Mayo, Curr. Opin. Chem. Biol., 2002, 6, 125–129 CrossRef CAS.
- F. Cedrone, A. Menez and E. Quemeneur, Curr. Opin. Struct. Biol., 2000, 10, 405–410 CrossRef CAS.
- S. J. Rowan and J. K. M. Sanders, Curr. Opin. Chem. Biol., 1997, 1, 483–490 CrossRef CAS.
- D. N. Bolon and S. L. Mayo, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 14274–14279 CrossRef CAS.
- M. A. Dwyer, L. L. Looger and H. W. Hellinga, Science, 2004, 304, 1967–1971 CrossRef CAS.
- D. E. Benson, M. S. Wisz and H. W. Hellinga, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 6292–6297 CrossRef CAS.
- Y. Wei and M. H. Hecht, Protein Eng., Des. Sel., 2004, 17, 67–75 Search PubMed.
- A. Tramontano, K. D. Janda and R. A. Lerner, Science, 1986, 234, 1566–1570 CrossRef CAS.
- S. J. Pollack, J. J. W. and P. G. Schultz, Science, 1986, 234, 1570–1573 CrossRef CAS.
- D. Hilvert, Annu. Rev. Biochem., 2000, 69, 751–793 CrossRef CAS.
- E. T. Farinas, T. Bulter and F. H. Arnold, Curr. Opin. Biotechnol., 2001, 12, 545–551 CrossRef CAS.
- D. Y. Jackson, J. R. Prudent, E. P. Baldwin and P. G. Schultz, Proc. Natl. Acad. Sci. U. S. A., 1991, 88, 58–62 CAS.
- D. F. Qi, C. M. Tann, D. Haring and M. D. Distefano, Chem. Rev., 2001, 101, 3081–3111 CrossRef CAS.
- I. Hamachi, T. Nagase, Y. Tajiri and S. Shinkai, Bioconjugate Chem., 1997, 8, 862–868 CrossRef CAS.
- G. DeSantis and J. B. Jones, Curr. Opin. Biotechnol., 1999, 10, 324–330 CrossRef CAS.
- K. Lim, J. X. Ho, K. Keeling, G. L. Gilliland, X. H. Ji, F. Ruker and D. C. Carter, Protein Sci., 1994, 3, 2233–2244 CrossRef CAS.
- B. Ketterer, Chem.-Biol. Interact., 2001, 138, 27–42 CrossRef CAS.
- D. Sheehan, G. Meade, V. M. Foley and C. A. Dowd, Biochem. J., 2001, 360, 1–16 CrossRef CAS.
- R. C. Strange, M. A. Spiteri, S. Ramachandran and A. A. Fryer, Mutat. Res., 2001, 482, 21–26 CrossRef CAS.
- R. N. Armstrong, Chem. Res. Toxicol., 1997, 10, 2–18 CrossRef CAS.
- B. Mannervik, Biochem. Soc. Trans., 1996, 24, 878–880 CAS.
- N. E. Pettigrew and R. F. Colman, Arch. Biochem. Biophys., 2001, 396, 225–230 CrossRef CAS.
- J. D. Hayes and D. J. Pulford, Crit. Rev. Biochem. Mol. Biol., 1995, 30, 445–600 CrossRef CAS.
- I. Sinning, G. J. Kleywegt, S. W. Cowan, P. Reinemer, H. W. Dirr, R. Huber, G. L. Gilliland, R. N. Armstrong, X. H. Ji, P. G. Board, B. Olin, B. Mannervik and T. A. Jones, J. Mol. Biol., 1993, 232, 192–212 CrossRef CAS.
- S. Hederos, K. S. Broo, E. Jakobsson, G. J. Kleywegt, B. Mannervik and L. Baltzer, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 13163–13167 CrossRef CAS.
- S. D. Copley, Curr. Opin. Chem. Biol., 2003, 7, 265–272 CrossRef CAS.
- R. J. Kazlauskas, Curr. Opin. Chem. Biol., 2005, 9, 195–201 CrossRef CAS.
- U. T. Bornscheuer and R. J. Kazlauskas, Angew. Chem., Int. Ed., 2004, 43, 6032–6040 CrossRef CAS.
- B. G. Miller and R. Wolfenden, Annu. Rev. Biochem., 2002, 71, 847–885 CrossRef CAS.
- S. Hakansson, J. Viljanen and K. S. Broo, Biochemistry, 2003, 42, 10260–10268 CrossRef.
- J. Viljanen, L. Tegler and K. S. Broo, Bioconjugate Chem., 2004, 15, 718–727 CrossRef CAS.
- E. C. Dietze, M. P. Grillo, T. Kalhorn, B. S. Nieslanik, C. M. Jochheim and W. M. Atkins, Biochemistry, 1998, 37, 14948–14957 CrossRef CAS.
- S. Hederos and K. S. Broo, unpublished results.
- C. Ibarra, M. P. Grillo, M. Lo Bello, M. Nucettelli, T. K. Bammler and W. M. Atkins, Arch. Biochem. Biophys., 2003, 414, 303–311 CrossRef CAS.
- A. Gustafsson, P. L. Pettersson, L. Grehn, P. Jemth and B. Mannervik, Biochemistry, 2001, 40, 15835–15845 CrossRef CAS.
- A. Gustafsson and B. Mannervik, J. Mol. Biol., 1999, 288, 787–800 CrossRef CAS.
- W. H. Habig and W. B. Jakoby, Methods Enzymol., 1981, 77 Search PubMed.
- K. S. Broo, H. Nilsson, J. Nilsson and L. Baltzer, J. Am. Chem. Soc., 1998, 120, 10287–10295 CrossRef CAS.
- G. Nemethy, K. D. Gibson, K. A. Palmer, C. N. Yoon, G. Paterlini, A. Zagari, S. Rumsey and H. A. Scheraga, J. Phys. Chem., 1992, 96, 6472–6484 CrossRef CAS.
- L. Wesson and D. Eisenberg, Protein Sci., 1992, 1, 227–235 CAS.
- F. A. Momany, R. F. McGuire, A. W. Burgess and H. A. Scheraga, J. Phys. Chem., 1975, 79, 2361–2381 CrossRef CAS.
- J. Fernandez-Recio, M. Totrov and R. Abagyan, Protein Sci., 2002, 11, 280–291 CrossRef CAS.
- B. Mannervik, Y. C. Awasthi, P. G. Board, J. D. Hayes, C. Di Ilio, B. Ketterer, I. Listowsky, R. Morgenstern, M. Muramatsu, W. R. Pearson, C. B. Pickett, K. Sato, M. Widersten and C. R. Wolf, Biochem. J., 1992, 282, 305–306 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Stability of A216H as measured by CDNB activity; the acetonitrile gradients used in the HPLC analyses; diagrams and time points from saturation kinetics HPLC measurements. See DOI: 10.1039/b510115h |
| ‡ Abbreviations: CDNB, 1-chloro-2,4-dinitrobenzene; GSH, glutathione, γ-Glu-Cys-Gly; GST, glutathione transferase; hGST A1-1, GST A1-1 isoform from human; G-site, glutathione-binding site; GS-thiolester, thiolester of glutathione; HPLC, high performance liquid chromatography; H-site, hydrophobic electrophile binding site; MALDI-MS, matrix assisted laser desorption mass spectrometry; NaPi, sodium phosphate; TFA, trifluoroacetic acid; UV, ultraviolet; wt, wild-type. |
| § This nomenclature is derived from that recommended by Mannervik.45 |
| This journal is © The Royal Society of Chemistry 2006 |