Borrowing hydrogen: iridium-catalysed reactions for the formation of C–C bonds from alcohols
Phillip. J. Black, Gerta Cami-Kobeci, Michael G. Edwards, Paul A. Slatford, Michael K. Whittlesey and Jonathan M. J. Williams*
Department of Chemistry, University of Bath, Claverton Down, Bath, BA2 7AY, UK. E-mail: chsjmjw@bath.ac.uk; Fax: +44 (0)1225 386231; Tel: +44 (0)1225 383942
First published on 25th November 2005
Abstract
Alcohols have been employed as substrates for C–C bond-forming reactions which involve initial activation by the temporary removal of hydrogen to form an aldehyde. The intermediate aldehyde is converted into an alkene via a Horner–Wadsworth–Emmons reaction, nitroaldol and aldol reactions. The ‘borrowed hydrogen’ is then returned to the alkene to form a C–C bond.
Introduction
The interconversion of alcohols and carbonyl compounds can be readily achieved by transfer hydrogenation reactions.1 We have recently reported that the temporary, reversible interconversion of alcohols and carbonyl compounds can lead to interesting transformations. Thus, the indirect nucleophilic addition to allylic alcohols has been achieved by ‘borrowing hydrogen’ from the substrate to provide an α,β-unsaturated ketone which readily undergoes a conjugate addition reaction.2 The alcohol functionality is then restored by returning hydrogen to the temporarily formed carbonyl group. We have employed a similar strategy for the indirect bromination of alcohols, which proceeds via α-bromination of a temporarily formed ketone.3The construction of C–C bonds is a fundamental reaction in organic synthesis, although alcohols are not generally used as starting materials despite their wide availability. Herein, we report a strategy involving ‘borrowing hydrogen’ as a method for the formation of C–C bonds from alcohols. As outlined in Scheme 1, this strategy involves borrowing hydrogen from the substrate alcohol to generate an intermediate carbonyl compound. Conversion of the carbonyl compound into an alkene can then be achieved under the reaction conditions using a suitable method. The hydrogen is then returned to the intermediate alkene to provide the corresponding C–C bond.
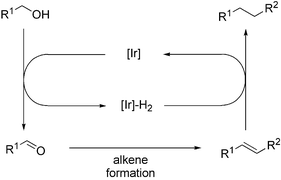 | ||
| Scheme 1 C–C bond formation from alcohols. | ||
We have reported our preliminary findings in an earlier communication on indirect Wittig and Wadsworth–Emmons reactions.4 Herein, we wish to report further details of the indirect Wadsworth–Emmons reaction, as well as previously unreported variants of the aldol condensation.
Results and discussion
Initially, we wished to identify a suitable catalyst which would be able to oxidise the alcohol and reduce the alkene via crossover transfer hydrogenation (Scheme 2).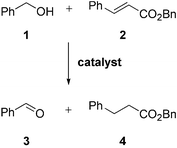 | ||
| Scheme 2 Crossover transfer hydrogenation. | ||
Both steps one and three in the domino process identified in Scheme 1 could be examined simultaneously by performing a catalytic crossover transfer hydrogenation reaction between suitable alcohol donors and alkene acceptors. The benefit of such a system was that it was easy to determine whether a catalyst was suitable or not.
A range of transfer hydrogenation catalysts was examined for their performance in the model reaction between benzyl alcohol 1 and benzyl cinnamate 2. The data reported in Table 1 indicate that although Meerwein–Ponndorf–Verley/Oppenauer type catalysts were unsuccessful (entry 3) both heterogeneous (entries 1–2) and homogeneous transition metal catalysts were successful. In 2001 Ishii and co-workers5 reported an iridium-based transfer hydrogenation system for the reduction of α,β-unsaturated carbonyl compounds with isopropanol. Using an [Ir(COD)Cl]2/dppp/Cs2CO3 system the selective reduction of α,β-unsaturated aldehydes or ketones into the saturated carbonyl derivative could be accomplished, although under extended reaction times reduction of the carbonyl group also occurred. Our results demonstrated that the reduction of α,β-unsaturated esters was more troublesome than the reduction of α,β-unsaturated ketones; longer reaction times and higher temperatures were necessary (entries 4–7). However, judicious choice of reaction conditions allowed complete conversion of benzyl alcohol 1 and benzyl cinnamate 2 into benzaldehyde 3 and benzyl dihydrocinnamate 4 respectively (entry 7).
| Entry | Catalyst (mol%) | Temp/°C | t/h | Conversion (%)b |
|---|---|---|---|---|
| a The reactions were carried out on a 0.5 mmol scale in toluene (1.5 mL).b Measured by 1H NMR.c Reaction run in THF. | ||||
| 1 | Ra–Ni (25) | 110 | 24 | 33 |
| 2 | Pd–C (25) | 110 | 48 | 65c |
| 3 | Al(OtBu)3 (100) | 110 | 48 | <5 |
| 4 | [Ru(η6-(p-cymene)(S,S-TsDPEN)] (5) | 110 | 48 | 25 |
| 5 | [Ir(COD)Cl]2/dppp/Cs2CO3 (2) | 80 | 24 | 35 |
| 6 | [Ir(COD)Cl]2/dppp/Cs2CO3 (2) | 150 | 4 | 25 |
| 7 | [Ir(COD)Cl]2/dppp/Cs2CO3 (5) | 150 | 72 | 100 |
The catalytic crossover transfer hydrogenation of malonate- and nitroaldol-derived alkenes was also demonstrated using these conditions. In both cases, the Ishii catalyst was effective for the crossover transfer hydrogenation reaction (Scheme 3).
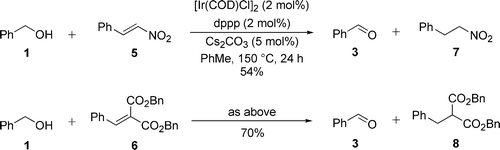 | ||
| Scheme 3 Initial crossover transfer hydrogenation reactions. | ||
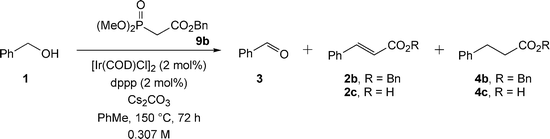
Initially, we decided to investigate the indirect Wadsworth–Emmons reaction according to Scheme 4, using benzyl alcohol 1 and phosphonates 9a and 9b (Table 2). Since caesium carbonate was already present in the Ishii crossover transfer hydrogenation catalyst system, it appeared sensible to use this base to deprotonate the phosphonate. In fact, the literature contains several reports of the use of caesium carbonate in Wadsworth–Emmons reactions; in particular Mouloungui and co-workers have exploited the use of this base.6
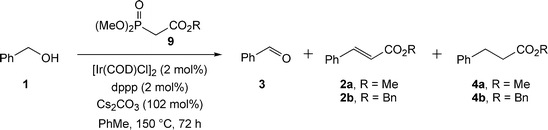 | ||
| Scheme 4 Initial domino indirect Wadsworth–Emmons reactions. | ||
To our delight, the crude product mixture contained the required dihydrocinnamate ester in addition to alkene and aldehyde intermediates; the composition of the crude mixtures was however surprising. The initial reaction with trimethylphosphonoacetate produced not the expected methyl ester products, but 15% of benzyl cinnamate 2b and 31% of benzyl dihydrocinnamate 4b (entry 1). When this reaction was reproduced under more dilute conditions (0.307 M solution), a mixture of benzyl and methyl esters was obtained (entry 2). Both these results are indicative of considerable transesterification under the reaction conditions. When the problem of transesterification was removed via the use of benzyl dimethylphosphonoacetate (entry 3) only a 14% conversion into benzyl dihydrocinnamate 4b was obtained. Nevertheless, these results were significant, since a three-step domino process had created a C–C bond from an alcohol substrate. We had also achieved a proof of principle that the project could succeed, and this spurred future development. These data also indicate that concentration is clearly an important variable in the reaction, since under greater dilution the process of transesterification was less evident, though not completely eradicated.
Such a phenomenon has been noted previously with phosphonate esters. Takano and co-workers discovered that catalytic amounts of DMAP effected an ester exchange reaction with phosphonoacetates and an alcohol substrate,7 whilst in a study of the Wadsworth–Emmons reaction in alcoholic solvents, Mouloungui and co-workers8 found that transesterification was catalysed by potassium carbonate.
The difficulty in separating the mixture of these three components relates mainly to the alkene and alkane products; in all solvent systems examined these two components co-ran on silica. Perhaps the easiest solution to this problem would be to convert all the alkene present into alkane by addition of hydrogen at the conclusion of the reaction. This was unsatisfactory from several perspectives; if hydrogen is added it is impossible to establish the real amount of product formed, and furthermore it removes the element of proof of principle from the system. The separation of alkenes from alkanes (and other alkenes) has been achieved frequently by use of silver-doped silica.9 Initially this method appeared ideal, though in reality it proved impossible to identify the two components on silver-doped TLC plates, and was therefore discounted. Following considerable experimentation, we discovered that the catalytic potassium permanganate/sodium metaperiodate oxidative system reported by von Rudloff10 was the most effective method of removing the unwanted aldehyde and alkene intermediates via chemical separation, leaving the product dihydrocinnamate untouched.
The initial experiments showed that the domino reaction process had the potential to succeed, but also revealed problems, most notably the presence of large amounts of the intermediate aldehyde and alkene in the reaction mixture. In theory, these should both progress through to the final alkane product. To enable a better understanding of the reaction process all the possible reaction variables were examined thoroughly in attempts to optimise the reaction conditions. Thus the catalyst, base, solvent, phosphonate, reaction time and concentration were all probed in a series of experiments.
The presence of intermediate benzaldehyde 3 in the product mixtures indicated that there were problems with the formation of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond, initially thought to be facile. In an attempt to enhance conversion of aldehyde 3 into benzyl dihydrocinnamate 4bvia benzyl cinnamate 2b, variation of the loading of benzyl phosphonoacetate 9b and caesium carbonate used was examined (Scheme 5, Table 3). These data indicate that increasing the amount of either the base or phosphonate decreased the amount of reaction, consistent with both of these components inhibiting the reaction. The major effect of increasing the number of equivalents of phosphonate was to promote hydrolysis of 2b and 4b, to cinnamic acid 2c and dihydrocinnamic acid 4c respectively. Presumably the water required is formed from carbonic acid decomposition, which itself is formed by deprotonation of benzyl dimethylphosphonoacetate 9b. Unsurprisingly, when no base or catalytic base was used (entries 4 and 5) the reactions were unsuccessful, indicating that any aldehyde formed by oxidation cannot be olefinated.
C bond, initially thought to be facile. In an attempt to enhance conversion of aldehyde 3 into benzyl dihydrocinnamate 4bvia benzyl cinnamate 2b, variation of the loading of benzyl phosphonoacetate 9b and caesium carbonate used was examined (Scheme 5, Table 3). These data indicate that increasing the amount of either the base or phosphonate decreased the amount of reaction, consistent with both of these components inhibiting the reaction. The major effect of increasing the number of equivalents of phosphonate was to promote hydrolysis of 2b and 4b, to cinnamic acid 2c and dihydrocinnamic acid 4c respectively. Presumably the water required is formed from carbonic acid decomposition, which itself is formed by deprotonation of benzyl dimethylphosphonoacetate 9b. Unsurprisingly, when no base or catalytic base was used (entries 4 and 5) the reactions were unsuccessful, indicating that any aldehyde formed by oxidation cannot be olefinated.
| Entry | 9b (equiv.) | Cs2CO3b (equiv.) | Conversionc (%) | 3 (%) | 2b/2c (%) | 4b/4c (%) |
|---|---|---|---|---|---|---|
| a Reactions were carried out on a 0.46 mmol scale in toluene (1.5 mL).b Includes 2 mol% of the catalyst combination (except for entry 4).c Total conversion of benzyl alcohol 1 into compounds 3, 2b/2c and 4b/4c as determined by 1H NMR. | ||||||
| 1 | 2 | 1.02 | 57 | 10 | 19 | 27 |
| 2 | 2 | 2.02 | 67 | 18 | 15 | 34 |
| 3 | 5 | 5.02 | 20 | 4 | 6 | 10 |
| 4 | 1 | 0 | <1 | <1 | 0 | 0 |
| 5 | 1 | 0.02 | 45 | 39 | 3 | 4 |
| 6 | 1 | 2 | 31 | 14 | 6 | 11 |
| 7 | 1 | 5 | 27 | 9 | 3 | 15 |
 | ||
| Scheme 5 Variation of base and phosphonoacetate quantities. | ||
The finding that both base and phosphonate could inhibit the reaction prompted examination of pre-formed phosphonoacetate enolates. In theory, pre-formation of the enolate would remove the base from the system, leaving only the phosphonoacetate anion and the catalytic caesium carbonate. Thus pre-formation of the enolate of benzyl dimethylphosphonoacetate 9b with n-butyllithium, sodium hydride and potassium hydride was attempted (Table 4). Although these systems did appear to be more reactive, the reactions afforded far more by-products than any observed previously. In particular, large amounts of hydrolysis were evident. Both n-butyllithium (entry 1) and sodium hydride (entry 3) led to the formation of more significant amounts of alkane product (45–48%); a 23% yield of benzyl dihydrocinnamate 4b was obtained from the reaction with n-butyllithium following oxidative workup. Even in these cases though, significant amounts of aldehyde and alkene remained. Sodium hydride appeared to be the best base for these reactions; no hydrolysis was observed when one equivalent was used. Therefore the reaction with five equivalents of the pre-formed sodium enolate was disappointing (entry 4). The results were ultimately the same as those obtained with one equivalent of phosphonate, except that significant hydrolysis occurred. The 26% yield of the product obtained from this reaction was ultimately disappointing. In addition a 14% yield of dibenzyl ether was obtained. Presumably this by-product arises from a base-catalysed reaction at the high reaction temperatures.
| Entry | Base (equiv.) | Conversionb (%) | 3 (%) | 2b/2c (%) | 4b/4c (%) |
|---|---|---|---|---|---|
| a Reactions were carried out on 0.92 mmol scale in toluene (3 mL).b Total conversion of benzyl alcohol 1 into compounds 3, 2b/2c and 4b/4c as determined by 1H NMR.c Yield of isolated 4b after flash column chromatography and von Rudloff oxidative workup in parentheses.d Reaction conducted using 5 equivalents of phosphonate 9b.e 14% of dibenzyl ether was also obtained. | |||||
| 1 | nBuLi (1) | 85 | 29 | 9 | 48 (23c) |
| 2 | KH (1) | 52 | 18 | 12 | 21 |
| 3 | NaH (1) | 65 | 10 | 10 | 45 |
| 4 | NaH (5)d | 65 | 8 | 11 | 46 (26c),e |
Removing the base from the system had not removed the issue of reaction inhibition, and thus the search for an alternative base was conducted (Table 5). These results did not deviate substantially from those observed previously; it was evident that large amounts of benzaldehyde 3 and benzyl cinnamate 2b still remained in the reaction mixture. A range of bases could successfully deprotonate the phosphonate, the best being the strong organic bases DBU and MTBD (entries 2 and 3). There is in fact precedent for this, since both MTBD and DBU proved to be the most successful bases in Taylor and co-workers’ domino oxidation–Wadsworth–Emmons system.11 With these two bases the progression through the reaction cycle appeared more complete than for alternative bases. Thus 58% (MTBD) and 45% (DBU) conversions to benzyl dihydrocinnamate 4b were observed for these two reactions. In the case of MTBD, an isolated 17% yield of benzyl dihydrocinnamate 4b was obtained. This is substantially lower than the observed 58% conversion and probably indicative of the messy nature of the reaction mixture and the associated purification difficulties.
| Entry | Base | Conversion b (%) | 3 (%) | 2b (%) | 4b (%) |
|---|---|---|---|---|---|
| a Reactions were carried out on 0.46 mmol scale in toluene (1.5 mL).b Total conversion of benzyl alcohol 1 into compounds 3, 2b and 4b as determined by 1H NMR analysis.c 1-Methyl-1,3,4,6,7,8-hexahydro-2H-pyrimido[1,2-a]pyrimidine.d Yield of isolated product after flash column chromatography and von Rudloff oxidative workup. | |||||
| 1 | Rb2CO3 | 48 | 13 | 20 | 15 |
| 2 | MTBDc | 89 | 18 | 14 | 58 (17d) |
| 3 | DBU | 65 | 10 | 10 | 45 |
| 4 | Phosphazene-P1-tBu | 82 | 12 | 52 | 17 |
| 5 | CsF | 71 | 16 | 28 | 27 |
| 6 | KOtBu | 77 | 39 | 15 | 22 |
An examination of alternative solvent systems also proved futile. As expected, the use of co-ordinating solvents (e.g. THF) inhibited the reaction, and amongst non-coordinating solvents toluene proved to be the solvent of choice. The use of other phosphine ligands, including PPh3, PCy3 and BINAP did not provide any significant change in product distribution, although the use of excess (20 mol%) dppp completely inhibited the reaction.
Whilst we were pleased that some product was formed in the indirect Wadsworth–Emmons reaction from alcohols, there were problems in obtaining a satisfactory yield. Significant amounts of aldehyde and alkene consistently remained, and progression through the domino sequence appeared to be troublesome. This seemed to indicate that a common inhibitor was present in the reactions. From these data the conclusion that the phosphonate was responsible for the poor performance of the system was reached, which led to the conclusion that the Wadsworth–Emmons phosphonate itself inhibits the reaction from proceeding. We reasoned that the structural similarity of Wadsworth–Emmons phosphonoacetates to acetoacetone may result in chelation to the iridium catalyst centre. Thus, the sites required for the oxidation/reduction steps to proceed are consumed. If the system cannot proceed to completion then hydrogen is presumably lost via alternative pathways such as aldehyde decarbonylation, and hence the significant amounts of aldehyde and alkene remaining. A search of the literature revealed that the complexation of β-carbonylphosphonates to metal centres is well known. The crystal structures of several of these species have been reported.12
Given the shortcomings of the indirect Wadsworth–Emmons reaction, our attention next turned to the indirect nitroaldol reaction, since we had already determined that the crossover transfer hydrogenation is feasible (vide supra). Using benzyl alcohol 1 as the substrate, we examined conditions for the reaction with nitromethane 10. Whilst we were able to achieve a modest conversion (44%) to the desired product 7, the reaction also contained intermediates from the catalytic cycle, benzaldehyde 3 and nitrostyrene 5. The use of 10 mol% Cs2CO3 led to an increased consumption of starting material, but afforded the dinitrocompound 11 as the main product,13 presumably by conjugate addition of nitromethane 10 onto the intermediate nitrostyrene 5. The results are summarised in Scheme 6 and Table 6.
| Entry | MeNO2 (equiv.) | Cs2CO3 (equiv.) | dppp (mol%) | Conversionb (%) | 3 (%) | 5 (%) | 7 (%) | 11 (%) |
|---|---|---|---|---|---|---|---|---|
| a Reactions were carried out on a 4 mmol scale in toluene (1.5 mL).b Total conversion of benzyl alcohol 1 into compounds 3, 5, 7, and 11 as determined by 1H NMR analysis. | ||||||||
| 1 | 1.5 | 2.5 | 2.5 | 74 | 7 | 22 | 44 | — |
| 2 | 1.5 | 5 | 2.5 | 60 | 10 | 10 | 40 | — |
| 3 | 1.5 | 5 | 5 | 70 | — | 20 | 40 | 10 |
| 4 | 1.5 | 10 | 2.5 | 100 | 7 | 16 | 5 | 72 |
| 5 | 3 | 2.5 | 5 | 57 | — | — | 31 | 26 |
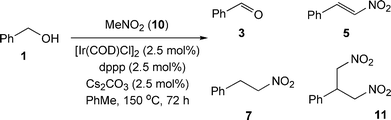 | ||
| Scheme 6 Indirect nitroaldol reaction with nitromethane and benzyl alcohol. | ||
Encouraged by these results we decided to test p-hydroxybenzyl alcohol 12 as an alternative substrate in this reaction. Quite pleasingly the reaction afforded the desired nitroalkane 13 in reasonable conversion, along with the dinitro compound 14 (Scheme 7).14
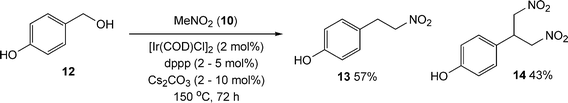 | ||
| Scheme 7 Use of p-hydroxybenzyl alcohol in the indirect nitroaldol reaction. | ||
The production of the undesired dinitroalkanes, by conjugate addition to the nitrostyrene, is in competition with the hydrogenation of the nitrostyrene to give the nitroalkane. Thus increasing the nitromethane concentration hinders the desired reaction (hydrogenation) by increasing the competing conjugate addition (Table 6, entry 5). To avoid this problem we changed the nucleophile to nitroethane, since a search in the literature revealed that increasing the carbon chain length markedly affects the reactivity.15
To our delight, the use of nitroethane 15 in the indirect nitroaldol reaction afforded the corresponding nitroalkanes in 14–70% conversion along with intermediate aldehyde and nitroalkene. In these cases, isoxazoles were identified as byproducts, which should be expected since it is known that 1,3-dinitro compounds are able to form isoxazoles.15,16 The results are summarised in Scheme 8 and Table 7. Unsubstituted benzyl alcohol (entry 1) gave the best conversion, whereas substrates containing an electron-withdrawing group (entry 3) gave lowest yields of the desired nitroalkane.
| Entry | Substrate | EtNO2 (equiv.) | Cs2CO3 (equiv.) | Conversionb (%) | 3 (%) | 16 (%) | 17 (%) | 18 (%) |
|---|---|---|---|---|---|---|---|---|
| a Reactions were carried out on a 4 mmol scale in toluene (1.5 mL).b Total conversion of benzyl alcohol 1 into compounds 3, 16, 17, and 18 as determined by 1H NMR analysis. | ||||||||
| 1 | Ph, 1a | 1.5 | 2.5 | 98 | 14 | — | 70 | 14 |
| 2 | p-OH-C6H4, 1b | 1.5 | 5 | 100 | — | — | 49 | 51 |
| 3 | p-Cl-C6H4, 1c | 1.5 | 2 | 93 | 42 | 28 | 14 | 9 |
| 4 | p-OMe-C6H4, 1d | 1.5 | 2 | 100 | — | 55 | 33 | 11 |
| 5 | m-NH2-C6H4, 1e | 1.5 | 2 | 100 | — | — | 57 | 43 |
| 6 | 3-Indolylmethanol 1f | 1.5 | 2 | 100 | — | — | 40 | 60 |
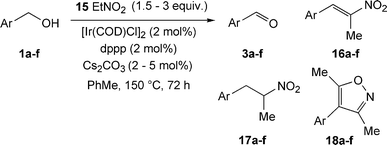 | ||
| Scheme 8 Indirect nitroaldol reaction using nitroethane as the nucleophile. | ||
We have also investigated the use of dibenzyl malonate 19 in an indirect crossed aldol (Knoevenagel) reaction. Thus, reaction of benzyl alcohol 1 with dibenzyl malonate 19 under the iridium-catalysed crossover transfer hydrogenation conditions provided some of the expected product 22, along with decarboxylated product 23. We presume that decarboxylation is caused by hydrolysis of one ester group of the malonate followed by a thermal decarboxylation, the water being formed during the aldol condensation process, and this also explains the presence of benzyl acetate 21, the decarboxylation product of the parent dibenzyl malonate 19. It is important to note that the loading of piperidinium actetate 20 plays an essential role in these reactions, since little or no reaction was observed when using 5 mol%. However, 25 mol% of 20 was found to give optimum results (Scheme 9).
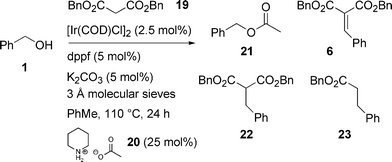 | ||
| Scheme 9 C–C Bond formation with dibenzyl malonate. | ||
A series of reactions was performed to determine whether the base influences the ratio of products (cf. Wadsworth–Emmons reactions, Table 3). As can be seen in Table 8, there is little difference between the bases when employing 5 mol%, however a noticeable change occurs when using higher loadings. Indeed, almost complete decarboxylation was observed when employing 50 mol% caesium carbonate, although only a moderate overall yield was achieved.
| Entry | Base (mol%) | Conversionb (%) | 3 (%) | 6 (%) | 21 (%) | 22 (%) | 23 (%) |
|---|---|---|---|---|---|---|---|
| a Reactions carried out on a 1 mmol scale in toluene (1.0 mL).b Total conversion of benzyl alcohol 1 into compounds 3, 6, 21, 22, and 23 as determined by 1H NMR analysis. | |||||||
| 1 | K2CO3 (5) | 54 | 9 | 10 | 4 | 31 | 2 |
| 2 | K2CO3 (50) | 55 | 9 | 4 | 5 | 34 | 3 |
| 3 | Cs2CO3 (5) | 59 | 14 | 14 | 8 | 22 | 1 |
| 4 | Cs2CO3 (50) | 36 | 5 | 1 | 15 | 3 | 13 |
| 5 | KOH (5) | 58 | 11 | 25 | 7 | 12 | 2 |
| 6 | KOH (50) | 51 | 17 | 3 | 5 | 24 | 2 |
Unfortunately, when employing dimethyl malonate as the nucleophile, substantial transesterification occured with benzyl alcohol to give an unpredictable mixture of methyl and benzyl esters in the starting material, product, and side-products. Disappointingly, this problem hindered the investigation on the scope of the alcohol that can be employed, since any but the most hindered of primary alcohols will undergo facile transesterification. To avoid this problem our attention switched to 1,3-diketones, which are also known to undergo Knoevenagel reactions with aldehydes.17
Initially we focused on the reaction between benzyl alcohol 1 and pentane-2,4-dione (acetyl acetone) 24, and were pleased to find that the reaction proceeded in 61% total conversion giving 27 as the major product (36%) (Scheme 10). Analysis of the product mixture is exacerbated by the keto-enol tautomerism, and closer examination determined that decarbonylation of the product also occurred to give 25 (24%) (cf. decarboxylation with dibenzyl malonate). Thus, a nucleophile was sought that eliminates the problems we have observed (metal co-ordination, transesterification, decarboxylation, decarbonylation). The commercially available mixed ketone-nitrile 28 appeared to meet these criteria, and was submitted to the standard reaction conditions with benzyl alcohol 1a (Scheme 11, Ar = Ph). We were delighted to find that the reaction proceeded to 55% conversion under these conditions (Table 9, entry 1), and could be driven to completion by increasing the reaction time (72 h) at an elevated temperature (150 °C) (entry 2). Having found a viable system for testing substrate specificity, various alcohols (1a–b,d,g–k) were employed to test the scope of the reaction. The results are summarised in Table 9.
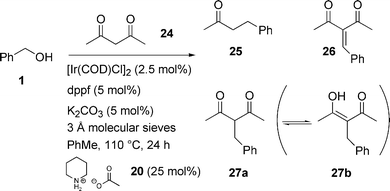 | ||
| Scheme 10 C–C Bond formation with 2,4-pentanedione. | ||
| Entry | ROH | Product | Conversion (%)b | Isolated yield (%)c |
|---|---|---|---|---|
| a Reactions were carried out on a 3 mmol scale in toluene (3 mL).b Total conversion of alcohol 1 into compound 29 as determined by 1H NMR analysis.c Yield of isolated 29 after flash column chromatography.d 150 °C, 72 h. | ||||
| 1 | PhCH2OH, 1a | 29a | 55 | 46 |
| 2d | PhCH2OH, 1a | 29a | 100 | 89 |
| 3 | p-OH-C6H4CH2OH, 1b | 29b | 79 | 44 |
| 4 | p-OMe-C6H4CH2OH, 1d | 29d | 48 | 48 |
| 5 | p-F-C6H4CH2OH, 1g | 29g | 40 | 30 |
| 6 | p-NO2-C6H4CH2OH, 1h | — | 18 | — |
| 7 | 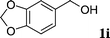 | 29i | 72 | 46 |
| 8 | PhCH2CH2OH, 1j | — | <1 | — |
| 9 | 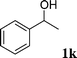 | — | <1 | — |
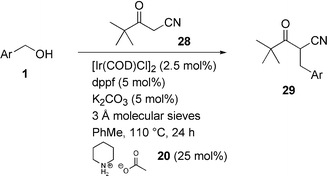 | ||
| Scheme 11 C–C Bond formation with 4,4-dimethyl-3-oxopentanenitrile. | ||
The results were somewhat mixed. Although the reaction worked moderately well for benzylic alcohols (except p-NO2), we were disappointed that in the case of n-phenethyl alcohol 1j and sec-phenethyl alcohol 1k (entries 8 and 9 respectively) only starting materials were recovered, though in the latter case it is not surprising since more forcing conditions involving TiCl4 are usually required for the condensation of ketones.18
In summary, we have demonstrated the concept of ‘borrowing hydrogen’ as a strategy for C–C bond formation. Whilst we were able to form the anticipated products in each case, the reactions suffered from the formation of by-products in many cases. The high temperature required for the crossover transfer hydrogenation process is almost certainly responsible for the lack of selectivity. We are now trying to develop more efficient catalysts to allow the reactions to proceed under milder conditions.
Experimental
General
Anhydrous toluene was distilled from sodium wire or obtained from an Anhydrous Engineering drying column. NMR: Bruker Avance 300, Bruker Avance 400, 1H and 13C{1H} NMR spectra were measured at 25 °C unless reported otherwise. 1H NMR signals are reported relative to TMS (0.0 ppm) or alternatively to the residual solvent peak. 13C{1H} NMR signals are reported relative to CDCl3 (77.0 ppm). 19F{1H} NMR signals were externally referenced to the 2H lock signal. Coupling constants (J) are quoted in Hertz (Hz) to the nearest 0.1 Hz. Mass spectra, including high-resolution spectra, were recorded on a Fisons Micromass Autospec mass spectrometer using EI, CI, and/or FAB sources, or sent to the EPSRC National Mass Spectrometry Service Centre, University of Wales Swansea, for analysis. FT-IR: Perkin–Elmer 1600 series as either liquid films, KBr discs or CDCl3 solutions. Flash column chromatography: Davisil LC 60A silica gel. TLC: 0.25 mm, Macherey–Nagel silica gel G/UV254 nm visualising at 254 nm, or with acidic (H2SO4) aq. KMnO4 solution, or p-anisaldehyde stain.General procedure 1: Iridium-catalysed crossover transfer hydrogenation reactions
To a nitrogen-purged pressure tube containing the required amounts of [Ir(COD)Cl]2, dppp and caesium carbonate, and benzyl cinnamate 2a (100 mg, 0.42 mmol) was added benzyl alcohol 1 (42 µL, 0.42 mmol) followed by anhydrous toluene. The tube was sealed, stirred vigorously and then heated at the required temperature for the time in Table 1. Upon completion, the reaction was quenched with wet diethyl ether (5 mL), filtered to remove the insoluble residues and then concentrated in vacuo to yield the crude product. Conversion was determined by analysis of the 1H NMR spectrum.General procedure 2: Iridium-catalysed indirect Wadsworth–Emmons reactions
To a nitrogen-purged ACE™ pressure tube containing [Ir(COD)Cl]2 (12.4 mg, 0.0184 mmol), dppp (7.6 mg, 0.0184 mmol), caesium carbonate (307 mg, 0.94 mmol) and benzyl dimethylphosphonoacetate 9b (238 mg, 0.92 mmol) was added via syringe benzyl alcohol 1 (92 µL, 0.92 mmol) followed by anhydrous toluene (3 mL). The tube was sealed and then heated at 150 °C for 72 h. Following cooling to room temperature the reaction was quenched by the addition of wet diethyl ether (5 mL), and then poured into a mixture of water (30 mL) and diethyl ether (50 mL). The ether layer was separated and the remaining aqueous layer further extracted with diethyl ether (3 × 20 mL). The combined organic extracts were washed with saturated brine (50 mL), dried (MgSO4), filtered and then concentrated in vacuo to afford the crude product. Conversion was determined by analysis of the 1H NMR spectrum. The crude reaction mixture was purified by flash column chromatography on silica using 40 : 1 petroleum ether (bp 40–60 °C)–diethyl ether as the eluent to afford an inseparable mixture of benzyl cinnamate 2b and benzyl dihydrocinnamate 4b.General procedure 3: von Rudloff oxidative workup procedure for the removal of alkenes
The alkene/alkane mixture was suspended in 50 mL of a 3 : 2 water–tert-butanol solution and treated with potassium permanganate (20 mg), sodium metaperiodate (1.625 g) and potassium carbonate (125 mg) in a single portion. This created a 0.0025 M/0.15 M/0.018 M potassium permanganate/sodium metaperiodate/potassium carbonate solution, which was allowed to stir for 2 h at room temperature and then diluted with diethyl ether (50 mL) and water (50 mL). The ether layer was separated and the remaining aqueous layer further extracted with diethyl ether (3 × 20 mL). The combined organic extracts were washed with 1 M sodium hydroxide (2 × 25 mL), saturated brine (50 mL), dried (MgSO4), filtered and then concentrated in vacuo to yield the desired product, benzyl dihydrocinnamate 4b. νmax (liquid film)/cm−1 3061, 3026, 2948, 1735, 1603, 1495, 1453, 1159, 1077, 767, 685; δH (400 MHz; CDCl3; Me4Si) 2.67 (2H, t, JHH 7.4, CH2CO2Bn), 2.96 (2H, t, JHH 7.4, PhCH2), 5.10 (2H, s, OCH2Ph), 7.16–7.21 (3H, m, Ph-H), 7.24–7.37 (7H, m, Ph-H); δC (100 MHz; CDCl3; CDCl3) 31.4 (PhCH2), 36.3 (CH2CO2Bn), 66.7 (OCH2Ph), 126.5 (Ph-C), 128.5 (4 × Ph-C), 128.7 (Ph-C), 128.8 (Ph-C), 136.1 (i-Ph-C), 140.6 (i-Ph-C), 172.8 (CO); m/z (FAB+) 240.1139 (M˙+, C16H16O2 requires 240.1150).Typical procedure for crossover transfer hydrogenation Wadsworth–Emmons reactions with pre-formed enolates
To a solution of benzyl dimethylphosphonoacetate 9b (238 mg, 0.92 mmol) in anhydrous toluene (3 mL) cooled to −78 °C was added dropwise n-butyllithium (2.5 M, 367 µL, 0.92 mmol). The mixture was stirred at −78 °C for 1 h and then warmed to room temperature. The phosphonate enolate solution thus obtained was treated with the other reagents and the reaction conducted as in General procedure 2. Purification of the crude reaction mixture was achieved according to General procedure 3 to afford benzyl dihydrocinnamate 4b (51 mg, 23%) as the isolated product.O), 135.6 (i-Ph-C), 129.0 (Ph-C), 128.8 (Ph-C), 128.7 (Ph-C), 67.8 (s, OCH2Ph), 53.5 (d, JPC 6.0, OCH3), 33.8 (d, JP,C 135, CH2CO2Bn); δP (121.4 MHz, CDCl3) 23.46 (s); m/z (EI+) 258 (8%), 151 (42), 124 (97), 109 (64), 108 (38), 107 (27), 94 (82), 93 (27), 91 (38), 79 (100), 77 (27), 63 (11), 47 (15); m/z (HRMS) 258.0664 (M˙+, C11H15O5P requires 258.0657).C), 1481(CCAr), 1434 (C–H2); δH (400 MHz; CDCl3; CHCl3) 7.45–7.58 (5H, m, Ph-H), 7.59 (1H, d, JHH 13.3, PhCHCHNO2), 8.02 (1H, d, JHH 13.3, PhCHCHNO2); m/z (EI) 149 (M˙+, 48%), 102 ([M − (HNO2˙)]+, 77), 91 (C7H7+, 77), 77 (C6H5+, 100).); m/z (EI) 372.14 (M+, 40%), 321 (10), 221 (28), 194 (100), 167.1 (23), 149.1 (37), 118.1 (36), 105.1 (48), 91.1 (80).Reaction of benzyl alcohol with dibenzyl malonate
This procedure is typical: Toluene (1 mL) was added to a mixture of benzyl alcohol (103 µL, 1 mmol), dibenzyl malonate (250 µL, 1 mmol), [Ir(COD)Cl]2 (16.8 mg, 0.025 mmol), dppf (28 mg, 0.05 mmol), potassium carbonate (6.9 mg, 0.05 mmol), piperidinium acetate (36.3 mg, 0.25 mmol), and activated 3 Å molecular sieves in a carousel reaction tube. The reaction mixture was then heated to 110 °C in a pre-warmed carousel reaction station, and stirred for 24 h. Upon cooling, the reaction mixture was filtered through a plug of silica (approx. 1 cm × 2 cm) washing with CH2Cl2 (5 × 5 mL). The solvent was removed in vacuo to give the crude product as a black oil; 1H NMR analysis showed that 6, 21, 22, and 23 were present. Purification of the crude reaction mixture was achieved by flash column chromatography on silica using 9 : 1 petroleum ether (bp 40–60 °C)–EtOAc as the eluent.O).O).O).); δC (75.5 MHz; CDCl3; CDCl3) 26.5 (CH3), 31.6 (CH3), 129.0 (Ph-C), 129.6 (Ph-C), 130.6 (Ph-C), 132.9 (Ph-C), 139.8 (PhCC), 142.8 (PhCC), 196.4 (CO), 205.5 (CO).![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N), 1715 (CO), 1498, 1248, 1041; δH (300 MHz; CDCl3; Me4Si) 1.00 (9H, s, 3 × CH3), 3.03 (1H, dd, JHH 13.6 and 7.6, CHAHB), 3.11 (1H, dd, JHH 13.6 and 7.6, CHAHB), 3.96 (1H, dd, JHH 7.6 and 7.6, CH(CN)CH2Ph), 7.1–7.3 (5H, m, Ph-H); δC (75.5 MHz; CDCl3; CDCl3) 24.5 (3 × CH3), 34.9 (CHCH2Ph), 37.7 (CHCH2Ph), 44.4 ((CH3)3CC(O)), 116.1 (CN), 126.6 (Ph-C), 127.8 (Ph-C), 128.1 (Ph-C), 135.2 (Ph-C), 203.9 (CO); m/z (CI + NH3) 233 ([M + NH4]+, 100%), 208 (5), 85 (2), 52 (10); m/z (ESI) 233.1649 ([M + NH4]+, C14H21N2O requires 233.1648).N), 1725 (CO), 1514, 1255, 1170; δH (300 MHz; CDCl3; Me4Si) 1.11 (9H, s, 3 × CH3), 3.06 (1H, dd, JHH 13.7 and 7.6, CHAHB), 3.13 (1H, dd, JHH 13.7 and 7.6, CHAHB), 4.00 (1H, dd, JHH 7.6 and 7.6, CH(CN)CH2Ph), 5.63 (1H, br s, OH), 6.77 (2H, d, JHH 8.5, m-Ph-H), 7.06 (2H, d, JHH 8.5, o-Ph-H); δC (75.5 MHz; CDCl3; CDCl3) 25.6 (3 × CH3), 35.3 (CHCH2Ph), 39.2 (CHCH2Ph), 45.6 ((CH3)3CC(O)), 115.8 (m-Ph-C), 117.2 (CN), 128.0 (i-Ph-C), 130.4 (o-Ph-C), 155.3 (p-Ph-C), 205.3 (CO); m/z (CI + NH3) 263 ([M + NH4]+, 100%), 245 (4), 121 (2), 52 (5); m/z (ESI) 263.1752 ([M + NH4]+, C15H23N2O2 requires 263.1754).N), 1722 (CO), 1613, 1514, 1251, 1179, 1035; δH (300 MHz; CDCl3; Me4Si) 1.01 (9H, s, 3 × CH3), 2.98 (1H, dd, JHH 13.7 and 7.6, CHAHB), 3.07 (1H, dd, JHH 13.7 and 7.6, CHAHB), 3.69 (3H, s, OCH3), 3.92 (1H, dd, JHH 7.6 and 7.6, CH(CN)CH2Ph), 6.75 (2H, d, JHH 8.7, m-Ph-H), 7.04 (2H, d, JHH 8.7, o-Ph-H); δC (75.5 MHz; CDCl3; CDCl3) 24.6 (3 × CH3), 34.2 (CHCH2Ph), 38.0 (CHCH2Ph), 44.5 ((CH3)3CC(O)), 54.2 (OCH3), 113.2 (m-Ph-C), 116.2 (CN), 127.2 (i-Ph-C), 129.2 (o-Ph-C), 158.0 (p-Ph-C), 204.1 (CO); m/z (CI + NH3) 263 ([M + NH4]+, 100%), 245 (4), 121 (2), 52 (5); m/z (ESI) 263.1752 ([M + NH4]+, C15H23N2O2 requires 263.1754).N), 1723 (CO), 1511, 1226; δH (300 MHz; CDCl3; Me4Si) 1.02 (9H, s, 3 × CH3), 3.02 (1H, dd, JHH 13.7 and 7.6, CHAHB), 3.10 (1H, dd, JHH 13.7 and 7.6, CHAHB), 3.94 (1H, dd, JHH 7.6 and 7.6, CH(CN)CH2Ph), 6.92 (2H, m, m-Ph-H), 7.11 (2H, m, o-Ph-H); δC (75.5 MHz; CDCl3; CDCl3) 24.8 (3 × CH3), 34.0 (CHCH2Ph), 37.8 (CHCH2Ph), 44.5 ((CH3)3CC(O)), 114.4 (2C, d, JCF 21.5, m-Ph-C), 115.9 (CN), 129.8 (2C, d, JCF 8.1, o-Ph-C), 131.0 (1C, d, JCF 3.3, i-Ph-C), 161.2 (1C, d, JCF 246.3, p-Ph-C), 203.8 (CO); δF(376.5 MHz; CDCl3) −114.6 (1F, s, Ph-F); m/z (CI + NH3) 251 ([M + NH4]+, 100%), 233 (5), 226 (33), 208 (3), 186 (6), 171 (2); m/z (ESI) 251.1553 ([M + NH4]+, C14H20N2OF requires 251.1554).N), 1719 (CO), 1507, 1501, 1448, 1369, 1247, 1040; δH (300 MHz; CDCl3; Me4Si) 1.12 (9H, s, 3 × CH3), 3.03 (1H, dd, JHH 13.7 and 7.6, CHAHB), 3.11 (1H, dd, JHH 13.7 and 7.6, CHAHB), 3.99 (1H, dd, JHH 7.6 and 7.6, CH(CN)CH2Ph), 5. 3 (2H, s, OCH2O), 6.63–6.75 (3H, m, Ph-H); δC (75.5 MHz; CDCl3; CDCl3) 26.0 (3 × CH3), 36.1 (CHCH2Ar), 39.4 (CHCH2Ar), 45.9 ((CH3)3CC(O)), 101.5 (OCH2O), 108.9 (Ar-CH), 109.8 (Ar-CH), 117.4 (CN), 122.8 (Ar-CH), 130.2 (i-Ar-C), 147.4 (Ar-C(OCH2)), 148.3 (Ar-C(OCH2)), 205.4 (CO); m/z (CI + NH3) 277 ([M + NH4]+, 100%), 252 (5), 52 (15); m/z (ESI) 277.1546 ([M + NH4]+, C15H21N2O3 requires 277.1547).
N), 1715 (CO), 1498, 1248, 1041; δH (300 MHz; CDCl3; Me4Si) 1.00 (9H, s, 3 × CH3), 3.03 (1H, dd, JHH 13.6 and 7.6, CHAHB), 3.11 (1H, dd, JHH 13.6 and 7.6, CHAHB), 3.96 (1H, dd, JHH 7.6 and 7.6, CH(CN)CH2Ph), 7.1–7.3 (5H, m, Ph-H); δC (75.5 MHz; CDCl3; CDCl3) 24.5 (3 × CH3), 34.9 (CHCH2Ph), 37.7 (CHCH2Ph), 44.4 ((CH3)3CC(O)), 116.1 (CN), 126.6 (Ph-C), 127.8 (Ph-C), 128.1 (Ph-C), 135.2 (Ph-C), 203.9 (CO); m/z (CI + NH3) 233 ([M + NH4]+, 100%), 208 (5), 85 (2), 52 (10); m/z (ESI) 233.1649 ([M + NH4]+, C14H21N2O requires 233.1648).N), 1725 (CO), 1514, 1255, 1170; δH (300 MHz; CDCl3; Me4Si) 1.11 (9H, s, 3 × CH3), 3.06 (1H, dd, JHH 13.7 and 7.6, CHAHB), 3.13 (1H, dd, JHH 13.7 and 7.6, CHAHB), 4.00 (1H, dd, JHH 7.6 and 7.6, CH(CN)CH2Ph), 5.63 (1H, br s, OH), 6.77 (2H, d, JHH 8.5, m-Ph-H), 7.06 (2H, d, JHH 8.5, o-Ph-H); δC (75.5 MHz; CDCl3; CDCl3) 25.6 (3 × CH3), 35.3 (CHCH2Ph), 39.2 (CHCH2Ph), 45.6 ((CH3)3CC(O)), 115.8 (m-Ph-C), 117.2 (CN), 128.0 (i-Ph-C), 130.4 (o-Ph-C), 155.3 (p-Ph-C), 205.3 (CO); m/z (CI + NH3) 263 ([M + NH4]+, 100%), 245 (4), 121 (2), 52 (5); m/z (ESI) 263.1752 ([M + NH4]+, C15H23N2O2 requires 263.1754).N), 1722 (CO), 1613, 1514, 1251, 1179, 1035; δH (300 MHz; CDCl3; Me4Si) 1.01 (9H, s, 3 × CH3), 2.98 (1H, dd, JHH 13.7 and 7.6, CHAHB), 3.07 (1H, dd, JHH 13.7 and 7.6, CHAHB), 3.69 (3H, s, OCH3), 3.92 (1H, dd, JHH 7.6 and 7.6, CH(CN)CH2Ph), 6.75 (2H, d, JHH 8.7, m-Ph-H), 7.04 (2H, d, JHH 8.7, o-Ph-H); δC (75.5 MHz; CDCl3; CDCl3) 24.6 (3 × CH3), 34.2 (CHCH2Ph), 38.0 (CHCH2Ph), 44.5 ((CH3)3CC(O)), 54.2 (OCH3), 113.2 (m-Ph-C), 116.2 (CN), 127.2 (i-Ph-C), 129.2 (o-Ph-C), 158.0 (p-Ph-C), 204.1 (CO); m/z (CI + NH3) 263 ([M + NH4]+, 100%), 245 (4), 121 (2), 52 (5); m/z (ESI) 263.1752 ([M + NH4]+, C15H23N2O2 requires 263.1754).N), 1723 (CO), 1511, 1226; δH (300 MHz; CDCl3; Me4Si) 1.02 (9H, s, 3 × CH3), 3.02 (1H, dd, JHH 13.7 and 7.6, CHAHB), 3.10 (1H, dd, JHH 13.7 and 7.6, CHAHB), 3.94 (1H, dd, JHH 7.6 and 7.6, CH(CN)CH2Ph), 6.92 (2H, m, m-Ph-H), 7.11 (2H, m, o-Ph-H); δC (75.5 MHz; CDCl3; CDCl3) 24.8 (3 × CH3), 34.0 (CHCH2Ph), 37.8 (CHCH2Ph), 44.5 ((CH3)3CC(O)), 114.4 (2C, d, JCF 21.5, m-Ph-C), 115.9 (CN), 129.8 (2C, d, JCF 8.1, o-Ph-C), 131.0 (1C, d, JCF 3.3, i-Ph-C), 161.2 (1C, d, JCF 246.3, p-Ph-C), 203.8 (CO); δF(376.5 MHz; CDCl3) −114.6 (1F, s, Ph-F); m/z (CI + NH3) 251 ([M + NH4]+, 100%), 233 (5), 226 (33), 208 (3), 186 (6), 171 (2); m/z (ESI) 251.1553 ([M + NH4]+, C14H20N2OF requires 251.1554).N), 1719 (CO), 1507, 1501, 1448, 1369, 1247, 1040; δH (300 MHz; CDCl3; Me4Si) 1.12 (9H, s, 3 × CH3), 3.03 (1H, dd, JHH 13.7 and 7.6, CHAHB), 3.11 (1H, dd, JHH 13.7 and 7.6, CHAHB), 3.99 (1H, dd, JHH 7.6 and 7.6, CH(CN)CH2Ph), 5. 3 (2H, s, OCH2O), 6.63–6.75 (3H, m, Ph-H); δC (75.5 MHz; CDCl3; CDCl3) 26.0 (3 × CH3), 36.1 (CHCH2Ar), 39.4 (CHCH2Ar), 45.9 ((CH3)3CC(O)), 101.5 (OCH2O), 108.9 (Ar-CH), 109.8 (Ar-CH), 117.4 (CN), 122.8 (Ar-CH), 130.2 (i-Ar-C), 147.4 (Ar-C(OCH2)), 148.3 (Ar-C(OCH2)), 205.4 (CO); m/z (CI + NH3) 277 ([M + NH4]+, 100%), 252 (5), 52 (15); m/z (ESI) 277.1546 ([M + NH4]+, C15H21N2O3 requires 277.1547).Acknowledgements
We would like to thank the EPSRC and the British Council for support of this work.References
- For reviews see: (a) G. Zassinovich and G. Mestroni, Chem. Rev., 1992, 1051 CrossRef CAS; (b) R. A. W. Johnstone and A. H. Wilby, Chem. Rev., 1985, 129 CrossRef CAS; (c) J.-E. Bäckvall, J. Organomet. Chem., 2002, 105 CrossRef CAS.
- P. J. Black, W. Harris and J. M. J. Williams, Angew. Chem., Int. Ed., 2001, 40, 4475 CrossRef CAS.
- G. Cami-Kobeci and J. M. J. Williams, Synlett, 2003, 1, 124.
- M. G. Edwards, R. F. R. Jazzar, B. M. Paine, D. J. Shermer, M. K. Whittlesey, J. M. J. Williams and D. D. Edney, Chem. Commun., 2004, 90 RSC.
- (a) S. Sakaguchi, T. Yamaga and Y. Ishii, J. Org. Chem., 2001, 66, 4710 CrossRef CAS; (b) K. Taguchi, H. Nakagawa, T. Hirabayashi, S. Sakaguchi and Y. Ishii, J. Am. Chem. Soc., 2004, 126, 72 CrossRef CAS.
- Z. Mouloungui, I. Murengezi, M. Delmas and A. Gaset, Synth. Commun., 1988, 18, 1241 CrossRef CAS.
- S. Hataeyma, K. Satoh, K. Sakurai and S. Takano, Tetrahedron Lett., 1987, 28, 2713 CrossRef CAS.
- Z. Mouloungui, R. Elmestour, M. Delmas and A. Gaset, Tetrahedron, 1992, 48, 1219 CrossRef CAS.
- C. M. Williams and L. N. Mander, Tetrahedron, 2001, 57, 425 CrossRef CAS.
- E. von Rudloff, Can. J. Chem., 1956, 34, 1413.
- L. Blackburn, C. X. Pei and R. J. K. Taylor, Synlett, 2002, 215 CrossRef CAS.
- (a) M. Gahagan, R. K. Mackie, D. J. Cole-Hamilton, D. C. Cupertino, M. Harman and M. B. Hursthouse, J. Chem. Soc., Dalton Trans., 1990, 2195 RSC; (b) T. H. Siddall, J. Inorg. Nucl. Chem., 1963, 25, 883 CrossRef CAS; (c) S. M. Bowen, E. N. Duesler and R. T. Paine, Inorg. Chem., 1983, 22, 286 CrossRef CAS.
- R. Ballini, G. Bosica, D. Fiorini and A. Palmieri, Synthesis, 2004, 1938 CrossRef CAS.
- B. K. Cassels and S. Sepulveda-Boza, Rev. Latinoam. Quim., 1988, 19(1), 25 Search PubMed.
- R. Ballini, F. Bigi, E. Gogni, R. Maggi and G. Sartori, J. Catal., 2000, 191, 348 CrossRef CAS.
- (a) M. D. Alcantara, F. C. Escribano, A. Gomez-Sanchez and M. J. Dianez, Synthesis, 1996, 64 CrossRef CAS; (b) A. T. Neilsen and T. G. Archibald, J. Org. Chem., 1969, 34, 984 CrossRef.
- (a) G. Jones, Org. React., 1967, 15, 204 CAS; (b) I. P. Sword, J. Chem. Soc. C, 1970, 1916 RSC; (c) K. Uehara, M. Ito and M. Tanaka, Bull. Chem. Soc. Jpn., 1973, 46, 1566 CAS; (d) M. Yamashita, Y. Watanabe, T. A. Mitsudo and Y. Takegami, Tetrahedron Lett., 1975, 1867 CrossRef CAS.
- W. Lehnert, Synthesis, 1974, 667 CrossRef CAS.
- J. C. Bradley and G. Buchi, J. Org. Chem., 1976, 41, 699 CrossRef CAS.
- (a) J. E. Baldwin and D. S. Johnson, J. Org. Chem., 1973, 38, 1973; (b) C. Wang, W. S., Synth. Commun., 2002, 32, 3481 CrossRef.
- A. F. McAnda, K. D. Roberts, A. J. Smallridge, A. Ten and M. A. Trewhella, J. Chem. Soc., Perkin Trans. 1, 1998, 3, 501 RSC.
- H. Chikashita, S. Nishida, M. Miyazaki, Y. Morita and K. Itoh, Bull. Chem. Soc. Jpn., 1987, 60, 737 CAS.
- Y. Kawai, Y. Inaba and N. Tokitoh, Tetrahedron: Asymmetry, 2001, 12, 309 CrossRef CAS.
- M. Takeshita, S. Yoshida and Y. Kohno, Heterocycles, 1994, 1, 1994.
- J. J. Lalonde, D. E. Bergbreiter and C. H. Wong, J. Org. Chem., 1988, 53, 2323 CrossRef CAS.
- R. A. Aitken, J. M. Armstrong, M. J. Drysdale, F. C. Ross and B. M. Ryan, J. Chem. Soc., Perkin Trans. 1, 1999, 593 RSC.
- S. L. Harbeson and D. H. Rich, J. Med. Chem., 1989, 1378 CrossRef CAS.
- I. Shiina, Tetrahedron, 2004, 1587 CrossRef CAS.
- J.-Y. Liu, Y.-J. Jang, W.-W. Lin, J.-T. Liu and C.-F. Yao, J. Org. Chem., 2003, 68, 4030 CrossRef CAS.
- L. Ridvan and J. Závada, Tetrahedron, 1997, 14793 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2006 |