Marine natural products: synthetic aspects
Gillian M.
Nicholas
a and
Andrew J.
Phillips
*b
aRoche Colorado Corporation, 2075 North 55th St., Boulder, Colorado 80301
bDepartment of Chemistry and Biochemistry, University of Colorado, Boulder, Colorado 80309, USA. E-mail: Andrew.Phillips@colorado.edu; Fax: 1 303 492 0439; Tel: 1 303 735 2049
First published on 5th January 2006
Abstract
Covering: January to December 2004. Previous review: Nat. Prod. Rep., 2005, 22, 144
An overview of marine natural products synthesis during 2004 is provided. As with the previous installment of this series, the emphasis is on total syntheses of molecules of contemporary interest, new total syntheses, and syntheses that have resulted in structure confirmation or stereochemical assignments.
 Gillian M. Nicholas | Gillian Nicholas received her B.Sc., M.Sc., and Ph.D. degrees from the University of Canterbury, where she worked on the isolation and structure elucidation of biologically active compounds from fungal sources with John Blunt and Murray Munro. Postdoctoral research with Ted Molinski at UC Davis and Carole Bewley at the National Institutes of Health was followed by a 2 year stay at Hauser Inc. in Denver, CO. She is currently a Principal Analytical Research Chemist at Roche Colorado Corporation in Boulder, CO. |
 Andrew J. Phillips | Andy Phillips obtained his B.Sc. (Hons) and Ph.D. degrees from the University of Canterbury in Christchurch, New Zealand. After a postdoctoral appointment with Peter Wipf at the University of Pittsburgh, he joined the faculty at the University of Colorado. His research interests focus around the development of new methods and strategies for the synthesis of complex natural products. |
1 Introduction
This review is designed to provide an overview of key features of the 2004 literature covering the synthesis of marine natural products and should act as a companion to the Marine Natural Products review published in this journal.1 The emphasis is on total syntheses of molecules of contemporary interest, as well as syntheses that have resulted in structure reassignments. Tabulated data for other syntheses are also provided.2 Review articles
A number of reviews that cover various aspects of marine natural products synthesis have appeared: “Chemistry and biology of curacin A”,2 “Progress in studies of novel marine bis(indole) alkaloids”,3 “Total synthesis of polycyclic ether natural products based on Suzuki–Miyaura cross-coupling”,4 “The chemistry of vicinal tricarbonyls and related systems”,5 “Synthesis of polyheterocyclic nitrogen-containing marine natural products”,6 “Bioorganic studies on marine natural products with bioactivity, such as antitumor activity and feeding attractance”,7 “Recent advances in the synthesis of trans-fused polycyclic ethers by hydroxy-epoxide-cyclization and ether-ring-expansion reactions”,8 “Elisabethin A: A marine diterpenoid yet to surrender to total synthesis”,9 “(+)-Discodermolide: a marine natural product against cancer”,10 “Lamellarins, from A to Z: A family of anticancer marine pyrrole alkaloids”,11 “Amphidinolides, bioactive macrolides from symbiotic marine dinoflagellates”,12 “Marine natural products”,13 “Recent advances in the total syntheses of oxazole-containing natural products”,14 and “Evolution of dithiane-based strategies for the construction of architecturally complex natural products”.15 Other reviews of relevance are cited in the text.3 Azaspiracid-1
In one of the significant achievements in the review period, the Nicolaou group has employed a combination of degradative studies and synthesis to revise the structure of azaspiracid-1.16 In work described in 2003, Nicolaou and co-workers established by synthesis17,18 that the initially proposed structure19 for azaspiracid-1 was incorrect (1, Scheme 1). Upon realizing that there were structural questions remaining to be resolved, the first path was to degrade natural azaspiracid-1 to smaller fragments and then locate the positions of error by synthesis. This approach was also expected to allow the determination of the relative stereochemistry between the ABCDE and FGHI domains. To this end, an authentic sample of azaspiracid was reacted with TMSCHN2 and the methyl ester obtained was treated with NaIO4, which resulted in cleavage of the C20–C21 bond. This provided lactone 3 and aldehyde 2, which were subjected to short sequences of common transformations to yield alcohol 6 and acid 7 (the stereochemistry shown for these compounds corresponds to that originally proposed). | ||
| Scheme 1 Chemical degradation and derivatization of azaspiracid-1 (originally proposed structure) to C1–C20 alcohol 6 and C26–C40 carboxylic acid 7. Reagents and conditions: (1) TMSCHN2, MeOH, 25 °C; (2) NaIO4, MeOH–H2O (4 : 1), 25 °C, ∼100% over 2 steps; (3) NaBH4, MeOH, 25 °C, ∼90%; (4) 5% Pd/C, H2, MeOH, 25 °C, ∼90%; (5) O3, Me2S, MeOH, −78 °C; (6) NaOH (0.1 N), MeOH–H2O (4 : 1), 25 °C, 2 h; (7) TMSCHN2, MeOH, 25 °C, ∼90% over 3 steps; (8) NaIO4, MeOH–H2O (4 : 1), 25 °C, ∼90%. | ||
Key steps of the synthesis of the EFGHI ring containing system 8 are shown in Scheme 2. The primary alcohol of dithiane 9 was acetylated, and subsequent dithiane deprotection with PhI(TFA)2 followed by oxidation of the lactol to the lactone with N-iodosuccinimide produced 10 in 51% over the three steps. Stille coupling with stannane 11 in the presence of Pd(0) resulted in the formation of dihydropyran 12. Removal of the TES ether with HF·pyridine, followed by treatment with N-iodosuccinimide to induce iodoetherification, produced iodoether 13 in 38% yield (for the three steps). Two further steps (removal of the iodide and cleavage of the Teoc carbamate) gave the structure corresponding to degradation product 3 and showed the structure of the compound obtained by degradation is in fact epimeric, in terms of the stereochemistry around the E ring, to that which was originally proposed. A synthesis of the compound in which the FGHI rings were enantiomeric was also completed by this route, but it did not match the data for 3. These, and related studies that involved the synthesis of acid 7, also established the absolute stereochemistry for this domain.
![Synthesis of the EFGHI ring containing lactone. Reagents and conditions: (1) Ac2O, 2,4,6-collidine, CH2Cl2, 85%; (2) PhI(TFA)2, MeCN–pH 7 buffer, 81%; (3) NIS, TBAI, CH2Cl2, 74%; (4) 3.0 equiv. lactone 10, 90 mol% [Pd2dba3], LiCl, AsPh3, DIPEA, syringe pump additon of stannane; (5) HF·py, THF–py; (6) NIS, NaHCO3, THF, 38% (3 steps).](/image/article/2006/NP/b501014b/b501014b-s2.gif) | ||
| Scheme 2 Synthesis of the EFGHI ring containing lactone. Reagents and conditions: (1) Ac2O, 2,4,6-collidine, CH2Cl2, 85%; (2) PhI(TFA)2, MeCN–pH 7 buffer, 81%; (3) NIS, TBAI, CH2Cl2, 74%; (4) 3.0 equiv. lactone 10, 90 mol% [Pd2dba3], LiCl, AsPh3, DIPEA, syringe pump additon of stannane; (5) HF·py, THF–py; (6) NIS, NaHCO3, THF, 38% (3 steps). | ||
At this juncture, attention turned to questions regarding the connectivity and stereochemistry of the ABCD ring containing domain. Desilylation of previously synthesized compound 14 to give 15 provided material that could be compared with degradation product 4 (Schemes 1 and 3). The spectroscopic data for these two samples differed substantially, particularly in the A ring. Progress towards a corrected structure was assisted by comparison of NMR data with a related compound, lissoketal (16).20 Based on this comparison, a new structure in which the A ring double bond has been relocated was proposed; however, final resolution of the problem did not come until the synthesis of several closely related structures had been completed. Based on this work the stereochemistry and connectivity shown in compound 17 was secured as corresponding to degradation compound 4.
 | ||
| Scheme 3 Comparison of synthetic and proposed ABCD ring containing alcohols. Reagents and conditions: (1) TBAF, THF, 88%. | ||
Armed with the information gleaned from these studies, and the earlier synthesis, plans could be laid to complete the total synthesis by employing the key couplings shown in Scheme 4.
 | ||
| Scheme 4 Key carbon–carbon bond-forming reactions in the Nicolaou azaspiracid-1 synthesis. | ||
Key steps of the route to the ABCDE domain are shown in Scheme 5. Malic acid derived tetrahydrofuran 18 was treated with TMSOTf in CH2Cl2 at low temperature to induce the desired deprotection–spirocyclization sequence to give 19 in 89% as a single stereoisomer. A sequence of 6 steps led to allylic carbonate 20, and deoxygenation of this compound to give 21 was achieved by employing a modification of an earlier-described Pd-catalyzed method21 which produced the desired compound in 82% yield (with 7 : 1 selectivity for the Δ7,8 olefin). After advancement to pentafluorophenyl ester 22, introduction of the C21–C27 domain involved acylation of the dithiane anion derived from 23 to give 24 in 50% yield. Six further steps provided compound 25 (see Scheme 6) which served as the key precursor to the A→E domain for the final steps of the synthesis.
![Synthesis of the ABCDE domain. Reagents and conditions: (1) TMSOTf (4.0 equiv.), CH2Cl2, −90 °C, 30 min, 89%; (2) [Pd2dba3]·CHCl3 (0.125 equiv.), nBu3P (0.48 equiv.), LiBH4 (10.0 equiv.), DME, 0 °C, 1 h, 82%, Δ7,8/Δ8,9 = 7 : 1; (3) 23 (7.0 equiv.), nBuLi–nBu2Mg (4.7 equiv.), THF, 0 → 25 °C, 1.5 h, then −90 °C, then 22, 30 min, 50%. PFP = pentafluorophenyl.](/image/article/2006/NP/b501014b/b501014b-s5.gif) | ||
| Scheme 5 Synthesis of the ABCDE domain. Reagents and conditions: (1) TMSOTf (4.0 equiv.), CH2Cl2, −90 °C, 30 min, 89%; (2) [Pd2dba3]·CHCl3 (0.125 equiv.), nBu3P (0.48 equiv.), LiBH4 (10.0 equiv.), DME, 0 °C, 1 h, 82%, Δ7,8/Δ8,9 = 7 : 1; (3) 23 (7.0 equiv.), nBuLi–nBu2Mg (4.7 equiv.), THF, 0 → 25 °C, 1.5 h, then −90 °C, then 22, 30 min, 50%. PFP = pentafluorophenyl. | ||
![The closing stages of the Nicolaou synthesis of azaspiracid-1. Reagents and conditions: (1) 25, [Pd2dba3] (0.3 equiv.), AsPh3 (0.3 equiv.), LiCl (6.0 equiv.), DIPEA, then 26 (3.0 equiv. in THF by syringe pump addition), NMP, 40 °C, 1 h, 55%; (2) TBAF (1.2 equiv.), THF, 0 °C, 80%; (3) NIS (2.0 equiv.), NaHCO3, THF, 0 °C, 12 h, 62%; (4) Et3B (0.2 equiv.), nBu3SnH–PhMe (1 : 2), 0 °C, 5 min, 86%.](/image/article/2006/NP/b501014b/b501014b-s6.gif) | ||
| Scheme 6 The closing stages of the Nicolaou synthesis of azaspiracid-1. Reagents and conditions: (1) 25, [Pd2dba3] (0.3 equiv.), AsPh3 (0.3 equiv.), LiCl (6.0 equiv.), DIPEA, then 26 (3.0 equiv. in THF by syringe pump addition), NMP, 40 °C, 1 h, 55%; (2) TBAF (1.2 equiv.), THF, 0 °C, 80%; (3) NIS (2.0 equiv.), NaHCO3, THF, 0 °C, 12 h, 62%; (4) Et3B (0.2 equiv.), nBu3SnH–PhMe (1 : 2), 0 °C, 5 min, 86%. | ||
The closing stages of the synthesis are illustrated in Scheme 6. The crucial coupling of the ABCD and FHI subunits occurred by Pd-mediated Stille-type reaction between allylic acetate 25 and dihydropyranyl stannane 26 to give 27 in 55% yield. Removal of the C34 TES ether (TBAF, 80%) and iodoetherification with N-iodosuccinimide produced 28 in an impressive 62% yield given the complexity of the substrate. Subsequent deiodination with Bu3SnH and Et3B give the fully elaborated A→I ring system (29) of the natural product, and the synthesis was completed by a short sequence of 5 steps that consisted of redox chemistry and protecting group manipulations.
In other studies on azaspiracid-1, Forsyth has descibed a concise approach to the trioxadispiroketal ABCD ring system 31 (the stereochemistry at C6 is epimeric to the final stereochemistry determined by Nicolaou). The key step consists of treatment of the ynone 30 with p-TsOH in toluene at room temperature for 1–2 days. Under these conditions the C6 and C17 TES ethers are removed and the ensuing sequence of hemiacetal formations and conjugate additions leads to 31 in 55% over two steps.22

Carter and Zhou have described a concise 23-step synthesis of the ABCDE ring system 32 from N-propionyl-(S)-benzyloxazolidinone.23 Ishikaway and Nishiyama have described a synthesis of compound 33, which contains the ABCD ring system, as well as related studies.24,25
4 Polyethers
Structurally complex polyethers continue to draw significant attention from the synthesis community, and a total synthesis of brevetoxin B26 has been reported by Nakata and co-workers (Scheme 7). Key features of this synthesis include the coupling of two large fragments by a Wittig reaction and then formation of the H ring by reductive etherification of an O,S-acetal. Advanced intermediate 36 (prepared in 17 steps from tri-O-acetyl-D-glucal) was advanced by a nice two-directional strategy that involves reductive cyclizations and selective Wittig reactions (Scheme 7). The intial reaction with ethyl propiolate gave β-alkoxyacrylate 37 in 95% yield. SmI2-induced reductive cyclization, followed by treatment with acidic ethanol, gave 38 in 79% yield over the two steps. Silylation of the tertiary alcohol and double reduction to the lactol-aldehyde 39 with DIBAL-H set the stage for the Wittig reaction sequence. Reaction, first with Ph3P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C(Me)CO2Et at 0 °C to rt, followed by reaction with Ph3PCHC(O)Me at 100 °C, gave 41 in 67%. Silylation of the tertiary alcohol followed by a sequence of 28 steps then yielded advanced intermediate 40 that contains the A→G rings. Subunit coupling with aldehyde 35, followed by TMS ether hydrolysis and formation of the O,S-acetal gave 42. Reduction of the O,S-acetal was achieved with Ph3SnH under radical conditions to yield 43. A short sequence of three steps that mirrored the Nicolaou synthesis provided brevetoxin B.
C(Me)CO2Et at 0 °C to rt, followed by reaction with Ph3PCHC(O)Me at 100 °C, gave 41 in 67%. Silylation of the tertiary alcohol followed by a sequence of 28 steps then yielded advanced intermediate 40 that contains the A→G rings. Subunit coupling with aldehyde 35, followed by TMS ether hydrolysis and formation of the O,S-acetal gave 42. Reduction of the O,S-acetal was achieved with Ph3SnH under radical conditions to yield 43. A short sequence of three steps that mirrored the Nicolaou synthesis provided brevetoxin B.
 | ||
| Scheme 7 Key steps of Nakata's total synthesis of brevetoxin B. Reagents and conditions: (1) ethyl propiolate, N-methylmorpholine, CH2Cl2, rt, 95%; (2) SmI2, MeOH, THF, rt; (3) p-TsOH, EtOH, 80 °C, 79% (2 steps); (4) TMSOTf, 2,6-lutidine, CH2Cl2, −78 °C, 100%; (5) DIBAL-H, PhMe, −78 °C; (6) Ph3PC(Me)CO2Et, PhMe, 0 °C → rt, then Ph3PCHCOMe, 100 °C, 67% (2 steps); (7) TMSOTf, py, CH2Cl2, −78 °C, 98%; (8) (a) 41, nBuLi, HMPA, THF, −78 °C, then 35, −78 °C → rt; (b) PPTS, CH2Cl2,–MeOH, rt, 68% (2 steps); (c) AgClO4, NaHCO3, SiO2, 4 Å MS, MeNO2, rt; (9) Ph3SnH, AIBN, PhMe, 110 °C; (10) TBAF, THF, rt, 71% (3 steps); (11) PCC, benzene, 80 °C, 51%; (12) HF·py, CH2Cl2, 0 °C, 72%. | ||
Fujiwara and co-workers have reported a formal total synthesis27 of hemibrevetoxin B28 by a route that reaches 53, and bisects the Yamamoto synthesis (Scheme 8). The synthesis commences with the synthesis of 50 from butyrolactone by an 18-step sequence that employs a ring-closing metathesis for the synthesis of the 7-membered ring (46 → 48). Aldehyde 50 is coupled with the anion derived from 49 (synthesized from tri-O-acetyl-D-glucal 47 by a 14-step sequence) to give 51 in 86% yield. Acidic hydrolysis of the (methylthio)(methylsulfinyl)acetal and removal of the TBS ether gave hydroxyl ketone 52, from which compound 53 was prepared by a 7-step sequence.
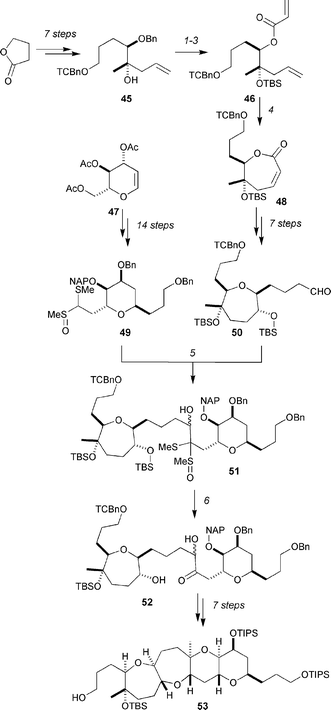 | ||
| Scheme 8 Fujiwara's formal synthesis of hemibrevetoxin B. Reagents and conditions: (1) TBSOTf, 2,6-lutidine, CH2Cl2, 99%; (2) DDQ, CH2Cl2–H2O (10 : 1), 93%; (3) acryloyl chloride, DIPEA, CH2Cl2, 94%; (4) Grubbs’ 2nd-generation catalyst, CH2Cl2, reflux, 89%; (5) 49, LDA then 50 (0.37 equiv.), −78 °C, 86%; (6) TFA–THF–H2O (1 : 5 : 5), 0 °C → rt, 71%. | ||
Inoue and Hirama have described the end-game of a 2nd-generation total synthesis of ciguatoxin CTX3C29 that involves milder conditions for the preparation of O,S-acetals (Scheme 9).30 Aldehyde 55 was converted to the α-chlorosulfide 58 by a sequence of three straightforward reactions (reduction, sulfide formation and chlorination). The authors note that ‘the reproducibility of the chlorination reaction … required the addition of 1 equiv. of NCS in CH2Cl2 to a solution of 57 in CCl4 at room temperature. The obtained solution of 58 in CH2Cl2/CCl4 (1:6) was directly used in the subsequent reaction because of the instability of 58 to any standard workup.’ Coupling of this α-chlorosulfide with alcohol 54 was effected by treating a CH2Cl2 solution of the alcohol, DTBMP, and AgOTf (2.0 equiv.) with the α-chlorosulfide. Removal of the C21 TIPS ether (TBAF, 85%) and conversion to the β-alkoxyacrylate (ethyl propiolate, NMM, 100%) set the stage for the radical cyclization to form the G ring. To this end, treatment of 60 with Bu3SnH and AIBN in PhMe led to 61 in 54% yield (along with 27% of the compound obtained from cyclization onto the C21 olefin). Two further steps (DIBAL-H reduction of the ester and olefination with Ph3PCH2, 92% over two steps) provided the substrate for a RCM analogous to that of the 1st-generation synthesis. Subjecting 62 to Grubbs’ I catalyst in CH2Cl2 at reflux produced 90% of protected CTX3C, and subsequent removal of the naphthylmethyl (NAP) ethers with DDQ gave 63% of CTX3C. This sequence is 4 steps shorter than the 1st-generation synthesis. Inoue and Hirama have also provided a detailed account of their 1st-generation synthesis31 and also a review of their recently developed methods for the synthesis of CTX3C,32 and Inoue has reviewed the use of acetal-linked intermediates for the synthesis of complex polyethers.33
 | ||
Scheme 9 Key steps from the Inoue–Hirama 2nd-generation synthesis of ciguatoxin CTX3C. Reagents and conditions: (1) NaBH4, MeOH–CH2Cl2, 81%; (2) PhSSPh, Bu3P, py, 93%; (3) NCS, CH2Cl2–CCl4 (1 : 6); (4) 54 (1.2 equiv.), AgOTf (2.0 equiv.), DTBMP, 4 Å MS, CH2Cl2–CCl4, 70% from 58; (5) (a) TBAF, THF, 85%; (b) methyl propiolate, NMM, CH2Cl2, 100%; (6) nBu3SnH, AIBN, PhMe, 54%; (7) (a) DIBAL-H, CH2Cl2; (b) Ph3PCH3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Br, tBuOK, THF, 90% (2 steps); (8) (Cy3P)2Cl2RuCHPh (30 mol%), CH2Cl2, 40 °C, 90%; (b) DDQ (6.0 equiv.), CH2Cl2–H2O (20 : 1), 63%. Br, tBuOK, THF, 90% (2 steps); (8) (Cy3P)2Cl2RuCHPh (30 mol%), CH2Cl2, 40 °C, 90%; (b) DDQ (6.0 equiv.), CH2Cl2–H2O (20 : 1), 63%. | ||
In other studies on polyethers, Clark and co-workers34 have described a 12-step synthesis of the A ring fragment of the gambieric acids.35 The key step is a [2,3]-sigmatropic rearrangement of an allylic oxonium ylide generated from the intramolecular reaction of a crotyl ether with a copper carbenoid. Related studies have shown that sequential ring-closing enyne and cross metathesis36 of carbohydrate systems is a useful method for the synthesis of polyether building blocks. Isobe and co-workers have developed an approach to the HIJ ring system of ciguatoxin that features an intromolecular Nicholas reaction for the synthesis of the 8-membered I ring (Scheme 10).37 Full details of the Sasaki total synthesis of gambierol have been reported, along with initial SAR studies that delineate some of the features required for potent toxicity.38
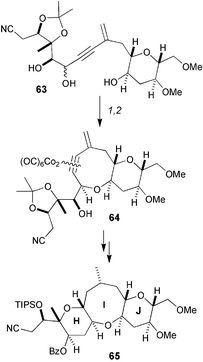 | ||
| Scheme 10 Isobe's intramolecular Nicholas approach to the I ring of ciguatoxin. Reagents and conditions: (1) Co2(CO)8, CH2Cl2, 87%; (2) BF3·OEt2, CH2Cl2, 60%. | ||
An improved synthesis of key intermediate 66 in the Hirama ciguatoxin CTX3C has been reported. The sequence to this compound is now 38 steps, and displays improved stereochemical control over the earlier route.39
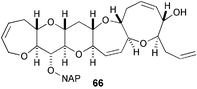
The synthesis of models of the CDE–FG ring systems of the prymnesins has suggested that a reassignment of relative stereochemistry should be considered in this region.40 A series of gambierol analogs have been synthesized from advanced intermediates in the Sasaki total synthesis and evaluated for toxicity against mice. These SAR studies indicate that the C28–C29 double bond (in the H ring) and the unsaturated side chain are crucial and that the C1 and C6 hydroxy groups, the C30 Me group, and the C37–C38 olefin have minimal influence on toxicity.41 Trost has described the Ru-catalyzed cycloisomerization and oxidative cyclization of bis-homopropargylic alcohols as a rapid iterative approach to the pyranopyran rings found in ladder toxins such as prymnesin and yessotoxin.42
5 Batzelladines
Several syntheses of members of the batzelladine family of the guanidinium alkaloids have been described in 2004. Overman's continuing work in this area has produced a synthesis of (−)-dehydrobatzelladine C43 that is based around the use of Biginelli condensations to produce the tricyclic core (Scheme 11). β-Ketoester 67 was transesterified with N-Boc-protected 4-aminobutanol under catalysis by DMAP to give 68 in 78% yield. Biginelli condensation of 68 with guanidinium aminal 69 (synthesized in 10 steps from commercial materials) in trifluoroethanol at elevated temperature produced 70 with >10 : 1 diastereoselectivity. Oxidation of the crude product with CAN and reverse-phase preparative HPLC gave compounds 71 and 72 in 45% combined yield for the two steps (this material consisted of 33% of the N-Boc compound, and 12% of the amine hydrochloride). Removal of the Boc group with TFA quantitatively led to 72, and the guanidinium group was then introduced by reaction with N,N′-di(tert-butoxycarbonyl)thiourea in the presence of HgCl2 and Et3N. Removal of the Boc groups then yielded (−)-dehydrobatzelladine C in 90% yield over the final two steps. Overman has also reviewed the applications of the tethered Biginelli reaction in natural products synthesis.44 | ||
| Scheme 11 Overman's synthesis of (−)-dehydrobatzelladine C. Reagents and conditions: (1) HO(CH2)4NHBoc, DMAP, PhMe, 100 °C, 78%; (2) morpholinium acetate, Na2SO4, CF3CH2OH, 65 → 70 °C; (3) CAN, MeCN, rt (45%, 2 steps); (4) 1 : 1 TFA–CH2Cl2, rt, ∼100%; (5) SC(NHBoc)2, HgCl2, Et3N, CH2Cl2, rt; (6) 1 : 1 TFA–CH2Cl2, rt, 90%. | ||
Nagasawa has described a synthesis of batzelladine A45 that is based around the use of diastereoselective 1,3-dipolar cycloadditions of nitrones with olefins to provide precursors to the bicyclic and tricyclic guanidinium units (Scheme 12).
 | ||
| Scheme 12 An overview of the retrosynthetic analysis of batzelladine A by Nagasawa and co-workers. | ||
The bicyclic guanidinium domain synthesis commenced with the 1,3-dipolar cycloaddition reaction between nitrone 74 and ester 80 to give isoxazolidine 81 as a single diastereomer (Scheme 13).46 Reduction of the ester with LiAlH4 and removal of the TIPS group with CsF gave 82 in 59% overall yield from 81. After selective protection of the primary alcohol with TBSCl and pyridine, removal of the secondary alcohol at C9 was achieved by a Barton–McCombie reaction to produce 83 (44% over 3 steps). A sequence of 9 steps then led to alcohol 84.
 | ||
| Scheme 13 Nagasawa's synthesis of batzelladine A. Reagents and conditions: (1) 1-undecene, PhMe, 90 °C, 75%; (2) methyl crotonate, PhMe, 100 °C, >60%; (3) (a) TBAF, THF, 97%; (b) Jones’ reagent, acetone; (4) PhMe, 90 °C; (5) LiAlH4, Et2O, 0 °C, then CsF, EtOH, 90 °C (59%, 3 steps); (6) (a) TBSCl, py, 81%; (b) ClC(S)OPh, py, DMAP, 58%; (c) nBu3SnH, AIBN, 94%; (7) EDCI, DMAP, CH2Cl2 (60%, 2 steps from acid); (8) HF·py, THF, 80%; (9) (a) Pd/C, H2; (b) PPh3, DEAD, PhMe; (c) TFA–CH2Cl2 (24%, 3 steps). | ||
The synthesis of acid 79, which ultimately serves as a progenitor of the tricyclic guanidine, commences with the diastereoselective 1,3-dipolar cycloaddition of 1-undecene and nitrone 74 to give 75 in 75% yield (see also Scheme 13). After a 4-step sequence of deprotection, acylation with ClC(S)OPh, Barton–McCombie deoxygenation, and oxidation with MCPBA, nitrone 76 was then subjected to cycloaddition with methyl crotonate to give 77. This material was advanced to compound 78, which was deprotected (TBAF, 97%) and oxidized with Jones' reagent to give acid 79. Subunit coupling of this acid with bicyclic guanidine 84 was achieved by esterification with EDCI in the presence of DMAP under carefully controlled conditions (epimerization at C24 occurred at room temperature, but could be supressed at 0 °C) to furnish 85 in 60% yield. Deprotection of the secondary alcohol with HF·pyridine yielded 86 in 80% yield. After removal of the Cbz protecting groups under standard hydrogenolysis conditions the resulting bicyclic guanidine was converted to the tricyclic guanidine by an intramolecular Mitsunobu reaction. The remaining four Boc groups were cleaved with TFA, and after purification by reverse-phase HPLC, batzelladine A was obtained as the trifluoroacetate salt in 24% yield for the 3 steps from 86.
In other studies on the batzelladines, Elliott has described the application of diastereoselective three-component coupling of an alkenylpyrrolidine, an aldehyde and Si(NCS)4 as a method to prepare the bicyclic core of the batzelladine alkaloids.47 In a collaboration between the Bewley and Overman groups, a synthetic library of 28 batzelladine analogs was tested for the ability to inhibit HIV-1 envelope-mediated cell–cell fusion.48 SAR relationships indicated that the best inhibitors of fusion were most similar in structure to natural batzelladine F, with IC50 values ranging from 0.8 to 3.0 µM.
6 Amphidinolides
The amphidinolides continue to provide a fertile environment for the development of new methods and strategies, and the review period has seen new syntheses of several members of this class. As noted in the previous review in this series, synthesis also continues to play an important role in structural studies for this class of compounds. The story of (+)-amphidinolide A149 provides a notable example of the value of synthesis to the structure elucidation of complex marine natural products. Kobayashi's initially proposed structure was synthesized in 2002 in independent efforts by the groups of Trost,50 Pattenden51 and Maleczka.52 However, none of these efforts produced material that matched the reported data, which suggested that there were questions regarding the stereochemistry of the molecule remaining to be answered. This puzzle was solved in 2004 by Trost and Harrington, when they described the structure elucidation of (+)-amphidinolide A1 by a combination of total synthesis and NMR analysis.53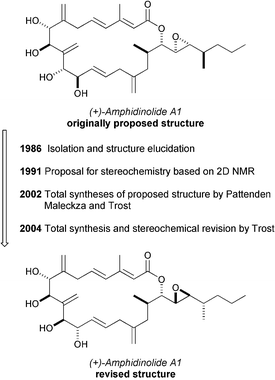
The synthesis highlights Trost's new methodology for the construction of 1,4-dienes by the Ru-catalyzed coupling of alkenes and alkynes (Scheme 14).54 The first subunit coupling was achieved by reaction of 89 with 90 in the presence of Cp*Ru(MeCN)3PF6 as catalyst (Scheme 14). This catalyst provided the branched product 91 in 23% yield (39% yield based on recovered starting material). A straightforward sequence of 3 steps provided acid 92 which was coupled to the potentially sensitive epoxy alcohol 93 under Kita's conditions55 to give ester 94 in 51% yield. After removal of the triethylsilyl ethers with TBAF–AcOH, [Cp*Ru(MeCN)3]PF6-catalyzed macrocyclization of 95 provided amphidinolide A1, 96. Although the yield may seem modest (33% or 38% based on recovered starting material) this an impressive example of the remarkable selectivity of the Ru-catalyzed alkene–alkyne addition. The spectral data for synthetic material matched very well to the natural product, with only two protons deviating by more than 0.01 ppm from the reported values (the two deviations were by 0.03 ppm and by 0.02 ppm). The 13C NMR spectrum deviated by 0.1 ppm or less in CDCl3, J values in three solvents were also in agreement, and the optical rotation was also consistent with reported data (synthetic [α]24D +56 (c 0.05, CHCl3) cf. reported [α]24D +46 (c 1.0, CHCl3)). Even with these excellent comparisons, in the absence of authentic material for comparison, Trost and Harrington conclude their paper with guarded comments: “In conclusion, we have employed a combination of synthesis and NMR spectroscopy as tools to determine the correct structure of amphidinolide A1. Although the lack of a sample of the natural product prevents a definitive comparison, the excellent correlation [of our synthetic compound] strongly suggests it is (+)-amphidinolide A1.”
![Trost's synthesis of amphidinolide A1. Reagents and conditions: (1) 89 (5 equiv.), [Cp*Ru(MeCN)3]PF6, 23% (39% brsm); (2) (i) [RuCl2(p-cymene)]2, ethoxyacetylene; (ii) 93, CSA, 51%; (3) TBAF, AcOH, 79%; (4) [Cp*Ru(MeCN)3]PF6, 33% (38% brsm).](/image/article/2006/NP/b501014b/b501014b-s14.gif) | ||
| Scheme 14 Trost's synthesis of amphidinolide A1. Reagents and conditions: (1) 89 (5 equiv.), [Cp*Ru(MeCN)3]PF6, 23% (39% brsm); (2) (i) [RuCl2(p-cymene)]2, ethoxyacetylene; (ii) 93, CSA, 51%; (3) TBAF, AcOH, 79%; (4) [Cp*Ru(MeCN)3]PF6, 33% (38% brsm). | ||
Trost has also completed a synthesis of amphidinolide P56 that employs the Ru-catalyzed alkene–alkyne coupling for subunit assembly (Scheme 15). Coupling of β-lactone 98 (3.5 equivalents) and enyne 97 in the presence of 10 mol% [CpRu(CH3CN)3]PF6 in acetone at room temperature gave 99 in 75% yield, and with no trace of linear product detectable. Four further steps led to amphidinolide P.
![The key Ru-catalyzed alkene–alkyne coupling en route to amphidinolide P. Reagents and conditions: (1) 10 mol% [CpRu(CH3CN)3]PF6, acetone, 75%.](/image/article/2006/NP/b501014b/b501014b-s15.gif) | ||
| Scheme 15 The key Ru-catalyzed alkene–alkyne coupling en route to amphidinolide P. Reagents and conditions: (1) 10 mol% [CpRu(CH3CN)3]PF6, acetone, 75%. | ||
Ghosh has synthesized amphidinolide W57 and corrected the stereochemistry at C6.58 The synthesis of the originally proposed structure employs a Ru-catalyzed cross-metathesis for subunit coupling (Scheme 16). Among various protecting groups and catalysts surveyed, an acetate protecting group on the secondary allylic alcohol of 102 and second generation Grubbs’ catalyst gave the best result, affording the desired compound 103 along with the Z-isomer (E/Z ratio 11 : 1) in 85% yield. A short sequence of protecting group and oxidation state manipulations provided seco-acid 104. Macrolactonization under Yamaguchi conditions provided the desired macrolactone 105 along with an epimer as a 1 : 1 mixture in 50% combined yield. Removal of dioxolane and MOM protecting groups produced the target compound, which was not identical to the natural material. After determining the stereochemistry at C6 to be the likely culprit, this sequence was repeated (details not discussed in the paper) to provide synthetic amphidinolide W whose spectral data (1H and 13C NMR) were identical to those of natural amphidinolide W.2

 | ||
| Scheme 16 Ghosh's synthesis of the proposed structure for (+)-amphidinolide W. Reagents and conditions: (1) (H2IMes)(PCy3)Cl2RuCHPh (5 mol%), CH2Cl2, 45 °C, 85%; (2) iPr2NEt, 2,4,6-trichlorobenzoyl chloride, then DMAP, PhH, 80 °C, 50%. | ||
Jamison has described a concise, modular synthesis of amphidinolide T159 that employs a Ni-catalyzed reductive macrocyclization of an alkyne with an aldehyde (Scheme 17).60 Esterification of alcohol 106 and carboxylic acid 107 under standard DCC-based conditions in the presence of 4-pyrrolidinopyridine gave ester 108. Removal of the TBS ether and oxidation with Dess–Martin periodinane gave the precursor to the macrocyclization, 109, in 59% yield for the three steps. Exposure of 109 as a 0.05 M solution to catalytic Ni(cod)2, Bu3P, and Et3B in toluene at 60 °C produced the desired allylic alcohol 110 with excellent stereocontrol (>10 : 1 dr) and in 44% yield. Protection of the secondary alcohol as the TBS ether, ozonolysis, selective methylenation using a modification of Takai's method,61 and finally desilylation with HF·pyridine led to amphidinolide T1, 111.
 | ||
| Scheme 17 Key steps of Jamison's synthesis of amphidinolide T1. Reagents and conditions: (1) DCC, 4-pyrrolidinopyridine; (2) TBAF, THF; (3) Dess–Martin periodinane, 59% (3 steps); (4) 20 mol% Ni(cod)2, 40 mol% Bu3P, Et3B, PhMe, 60 °C, 44% (>10 : 1 dr); (5) (a) TBSOTf, 2,6-lutidine; (b) O3, then Me2S; (c) CH2I2, Zn, ZrCl4, PbCl2; (d) HF·py, 25% (4 steps). | ||
Amphidinolide X62 has been synthesized by Fürstner.63 The synthesis features subunit coupling by esterification and B-alkyl Suzuki reactions (Scheme 18). Acid 112 and alcohol 113 were coupled using Yamaguchi's conditions to give ester 114 in 96% yield. Reaction of this compound with boronate 119 (which was derived from the corresponding iodide that was in turn synthesized by a 12-step sequence involving the application of a newly developed iron-catalyzed ring-opening reaction of a propargyl epoxide with a Grignard reagent as a method for the synthesis of an allenol en route to a dihydrofuran) in the presence of Pd(0) produced 115 in an excellent 74% yield. Removal of the methyl ester with LiI followed by unmasking of the ketone with aqueous AcOH led to 116 in 53% yield over the two steps, and subsequent removal of the PMB group with DDQ in the presence of pH 7 buffer produced seco-acid 117 in 84% yield. The synthesis was then completed by Yamaguchi macrolactonization to yield amphidinolide X, 118, in 62% yield.64
 | ||
| Scheme 18 Subunit couplings and the closing stages of Furstner's synthesis of amphidinolide X. Regents and conditions: (1) 112, 2,4,6-trichlorobenzoyl chloride, Et3N, PhMe, then 113, DMAP, 96%; (2) (dppf)PdCl2, Ph3As, K3PO4, aq. DMF, 74%; (3) LiI, py, 125 °C; (4) aq. AcOH, 53% (over 2 steps); (5) DDQ, CH2Cl2, pH 7 buffer, 84%; (6) 2,4,6-trichlorobenzoyl chloride, Et3N, THF, then DMAP, PhMe, 62%. | ||
In other studies on the amphidinolides, a synthesis of the C11–C29 fragment of amphidinolide F65 has been accomplished using a diastereoselective [3 + 2]-annulation reaction of an allylsilane and ethylglyxolate to produce the key tetrahydrofuran.66 A synthesis of the C19–C26 subunit of amphidinolides B1 and B2 has been completed using a boron-mediated aldol reaction, and a synthesis of the C19–C26 subunit of amphidinolide B3 has also been completed.67 Ghosh has also provided a detailed account of their synthesis of amphidinolide T1.68
7 Dictyostatin-1 and discodermolide
Dictyostatin-1, a macrolide that is closely related to discodermolide and also displays impressive growth inhibition of the P388 murine leukemia cell line (ED50 0.38 ng mL−1), along with differential cytotoxicity in the NCI 60-cell line screen, has received substantial attention in 2004. Dictyostatin's planar structure was first described by Pettit and co-workers who isolated the natural product from a Spongia sp. marine sponge collected in the Republic of Maldives.69 Re-isolation of the compound by Wright and co-workers at the Harbor Branch Oceanographic Institute from a Corallistidae sp. lithistid sponge provided material that was used to assign the relative stereochemistry.70 Shortly after the stereochemistry was available, simultaneous publications from the Curran71 and Paterson72 groups described the first syntheses of (−)-dictyostatin. Both syntheses employ similar strategies for the coupling of subunits, and conclude with Yamaguchi macrolactonization (Scheme 19).

 | ||
| Scheme 19 A comparison of key reactions in the Paterson and Curran dictyostatin-1 syntheses. | ||
The Paterson synthesis ultimately commences with methyl (3S)-hydroxy-2-methylpropionate (the ‘Roche ester’, 119, Scheme 20). A sequence of 14 relatively routine steps from the Roche ester led to β-ketophosphonate 120, and an 11-step synthesis produced aldehyde 121. These two fragments were coupled by a Horner–Wadsworth–Emmons reaction employing Ba(OH)2 in wet THF73 to give enone 122 in 92% yield. Conjugate reduction of the enone with the Stryker's reagent,74 followed by removal of both PMB groups with DDQ provided an intermediate β-hydroxyketone, which underwent reductione in a 1,3-syn-selective fashion when exposed to Zn(BH4)2 in Et2O to produce triol 123 (>20 : 1 dr).
![Paterson's intial fragment coupling en route to dictyostatin. Reagents and conditions: (1) 120, Ba(OH)2·8H2O, THF, 1 h, then 121, 40 : 1 THF–H2O, 92%; (2) [Ph3P·CuH]6, PhH, H2O, rt; (3) DDQ, pH 7 buffer, CH2Cl2, 0 °C; (4) Zn(BH4)2, Et2O, −30 °C, 66% (3 steps, >20 : 1 dr).](/image/article/2006/NP/b501014b/b501014b-s20.gif) | ||
| Scheme 20 Paterson's intial fragment coupling en route to dictyostatin. Reagents and conditions: (1) 120, Ba(OH)2·8H2O, THF, 1 h, then 121, 40 : 1 THF–H2O, 92%; (2) [Ph3P·CuH]6, PhH, H2O, rt; (3) DDQ, pH 7 buffer, CH2Cl2, 0 °C; (4) Zn(BH4)2, Et2O, −30 °C, 66% (3 steps, >20 : 1 dr). | ||
Key fragments 124 and 125 were coupled by a Still–Gennari-modified Horner–Wasdworth–Emmons olefination to give α,β-unsaturated ketone 126 with good levels of selectivity (Z/E = 5 : 1, 77%, Scheme 21). Stille coupling of the vinyl iodide with stannane 127 under Liebeskind conditions75 followed by cleavage of the TIPS ester with KF in THF–MeOH gave the seco-acid 128 in 83% overall yield for these two steps. Yamaguchi macrolactonization furnished the macrocycle in 77% yield, and reduction of the C9 ketone under Luche conditions provided the final stereocenter under macrocyclic stereocontrol (70%). Global deprotection of the TBS groups with HCl in methanol gave dictyostatin-1 in 87% yield.
![Final steps of the Paterson synthesis of dictyostatin-1. Reagents and conditions: (1) K2CO3, [18]-crown-6, PhMe, rt, Z/E = 5 : 1, 77% (from the primary alcohol preceding 124); (2) CuTC, NMP, rt, then KF, THF–MeOH, rt, 83% (2 steps); (3) 2,4,6-trichlorobenzoyl chloride, Et3N, DMAP, PhMe, 60 °C, 77%; (4) NaBH4, CeCl3·7H2O, EtOH, −30 °C, 70%; (5) 3 N HCl, MeOH, rt, 87%.](/image/article/2006/NP/b501014b/b501014b-s21.gif) | ||
| Scheme 21 Final steps of the Paterson synthesis of dictyostatin-1. Reagents and conditions: (1) K2CO3, [18]-crown-6, PhMe, rt, Z/E = 5 : 1, 77% (from the primary alcohol preceding 124); (2) CuTC, NMP, rt, then KF, THF–MeOH, rt, 83% (2 steps); (3) 2,4,6-trichlorobenzoyl chloride, Et3N, DMAP, PhMe, 60 °C, 77%; (4) NaBH4, CeCl3·7H2O, EtOH, −30 °C, 70%; (5) 3 N HCl, MeOH, rt, 87%. | ||
Curran's synthesis of dictyostatin is also predicated on the observation that the Roche ester can serve as the starting material for the β-ketophosphonate 129 and alkyne 130 (Scheme 22). The intial subunit coupling was achieved by the reaction of the lithiated acetylide derived from 130 with Weinreb amide 131 to give ynone 132 in 93% yield. The C9 stereocenter was set by reduction with Noyori's Ru(II)-(S,S)-N-p-toluenesulfonyl-1,2-diphenylethylenediamine catalyst with iPrOH as hydrogen donor, and subsequent Lindlar reduction of the alkyne and silylation with TBSOTf gave 133 (91% and 99% for the last two steps).
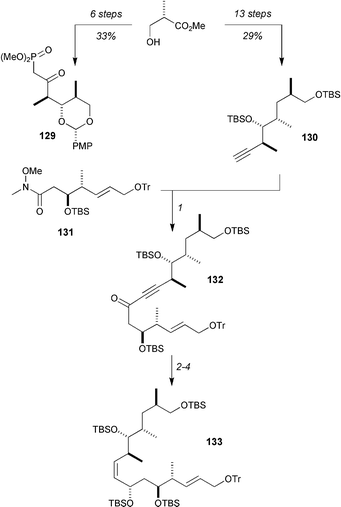 | ||
| Scheme 22 Fragment coupling of the C10–C18 and C3–C9 domains. Reagents and conditions: (1) 130, nBuLi, THF, then 131, 93%; (2) S,S Noyori catalyst (20 mol%), iPrOH, 79%; (3) Lindlar catalyst, H2, PhMe, 91%; (4) TBSOTf, 2,6-lutidine, CH2Cl2, 99%. | ||
Introduction of the final fragment was achieved by a Horner–Wadsworth–Emmons reaction between aldehyde 134 (obtained from 133 by desilylation with HF·pyridine in 67% yield and Dess–Martin oxidation) employing the Ba(OH)2 system (Scheme 23). Reduction of the enone was achieved by treatment with NiCl2–NaBH4, and the ketone was reduced with NaBH4 to give the desired alcohol 136 in 70% yield (along with 29% of the α-diastereoisomer). This compound was advanced to dictyostatin by a 12-step sequence similar to the approach employed by Paterson that featured Yamaguchi macrolactonization after the installation of the diene.
 | ||
| Scheme 23 The second key fragment coupling of the Curran synthesis of dictyostatin-1. Reagents and conditions: (1) Ba(OH)2, 129 (see Scheme 21), then 134, THF–H2O, 80% (2 steps from the alcohol); (2) NiCl2, NaBH4, MeOH–THF, 76%; (3) NaBH4, MeOH–THF, 70% (β), 29% (α). | ||
In other work on dictyostatin, an approach to mixed polyacetate-polypropionates based on the cyclization of (silyloxy)enynes has been reported in the context of a synthesis of the C9–C19 subunit.
Discodermolide also continues to draw attention, and one of the highlights of 2004 in this area is the description of work at Novartis that resulted in the preparation of 60 g of discodermolide. The route is a hybrid of the Smith and Paterson synthetic route and involves 39 steps in totala (26 steps in the longest linear sequence).76 This work has also been reviewed by Mickel.77,78 Paterson has reported a 3rd-generation synthesis that takes advantage of a Still–Gennari-modified HWE reaction that is similar to the one employed en route to dictyostatin-1.79 Several subunit syntheses have been described including: (i) an approach to the C15–C24 subunit employing desymmetrization of a meso dialdehyde with the Haffner–Duthaler crotyltitanium reagent,80 (ii) syntheses of the C1–C8 and C15–C21 subunits by diastereoselective dihydroxylation of dihydropyranones and subsequent α-deoxygenation,81 (iii) a highly diastereoselective and practical route to a lactone that serves as a key building block for the Novartis synthesis82 and (iv) an approach that exploits the pseudosymmetry of the C1–C13 region.83 A series of 7-deoxydiscodermolide analogs in which the lactone was replaced by aromatics were designed, synthesized, and evaluated for cytotoxicity.84 The majority of the compounds in this study displayed low micromolar IC50 values against a variety of cancer cell lines. In other studies a series of analogs (which are minor side products generated during the final stages in the synthesis of (+)-discodermolide) were evaluated for in vitro cytotoxicity against 6 cell lines.85 These compounds showed significant variation of cytotoxicity, and suggest the relevance of the C11 hydroxyl group, the C13 double bond, and the 16S stereochemistry are important for potent cytotoxocity.
8 Total syntheses of other compounds
The polycyclic alkaloid dragmacidin F86 has been synthesized by Stoltz (Scheme 24).87 Weinreb amide 138, which was prepared from the corresponding acid (synthesized in 6 steps from quinic acid), was reacted with lithiated pyrrole 139 to give ketone 140 in 66% yield from the acid. Exposure of this compound to Pd(OAc)2 produced bicylic pyrrole 141 in 74% yield, which was advanced to tosyl oxime 142 by a 9-step sequence. Sequential treatment of this compound with KOH in EtOH–H2O, followed by 6 N HCl, and finally K2CO3 produced amino ketone 143 (isolated as the trifluoroacetate) in an excellent 86% yield for the Neber rearrangement (as well as removal of the SEM and Ts protecting groups). Treatment with trimethylsilyl iodide to remove the methyl ethers, followed by reaction with cyanamide, gave dragmacidin F in 82% yield for these two operations. | ||
| Scheme 24 Key steps of the Stoltz synthesis of dragmacidin F. Reagents and conditions: (1) lithiopyrrole 139, THF, 66% from the acid; (2) Pd(OAc)2, DMSO, tBuOH–AcOH, 74%; (3) KOH, H2O, EtOH, then 6 N HCl, then K2CO3, THF–H2O, 96% (3 steps); (4) (a) TMSI, CH3CN; (b) H2NCN, NaOH, H2O, 82% (2 steps). | ||
Birman et al.88 and Baran et al.89 have independently described sequences that lead to members of the sceptrin class. The Baran synthesis of sceptrin proceeds in an excellent 24% overall yield from dimethylacetylene dicarbocylate and employs the acid-catalyzed rearrangement of 3-oxaquadricyclane 144 to cyclobutane 145 as the key step (Scheme 25).
 | ||
| Scheme 25 The key step from the Baran synthesis of sceptrin. Reagents and conditions: (1) H2SO4, MeOH, 24 h, 50%. | ||
Based on their synthetic studies, Baran has synthesized ageliferin90 in a single step from sceptrin by heating sceptrin in H2O in a microwave (Scheme 26).91 Based on this result, Baran and co-workers have proposed an alternative hypothesis to the long-held [4 + 2]-cycloaddition biogenesis of these compounds and related structures such as axinellamine and palau'amine (Scheme 27).
 | ||
| Scheme 26 Conversion of sceptrin to ageliferin. Reagents and conditions: (1) H2O, 195 °C, microwave, 1 min, 40% (plus 52% recovered sceptrin) | ||
 | ||
| Scheme 27 Baran's proposed sequence for the conversion of dibromosceptrin to axinellamine A in nature. A similar sequence can be drawn for palau'amine. | ||
Spongidepsin92 has been synthesized by both the Forsyth and Ghosh groups, and the relative and stereochemistry has been established to be as shown.93,94

A synthesis of bistramide A95 that confirms Wipf's stereochemical assignment for bistramide C has been completed by Kozmin.96 The synthesis involves 15 steps in the longest linear sequence and features a nice application of the ring-opening cross-metathesis of a cyclopropene acetal (Scheme 28) as a novel method for the rapid assembly of precursors to spiroacetals. Readily accesible alkene 150 was subjected to 10 mol% Grubbs’ 2nd-generation catalyst in the presence of 1.5 equivalents of cyclopropenone acetal 152, and then aqueous acid to give dienone 151 in 63% over the two steps. Cross-metathesis with alkene 153 then provided 156 in 68% yield. Hydrogenolysis of the benzyl groups and the dienone resulted in acetalization, and subsequent Dess–Martin oxidation gave aldehyde 157 in 53% yield over the two steps. A sequence of three steps led to amino alcohol 158, and completed a very concise approach to this fragment. The synthesis was then completed by PyBOP-mediated acylation of the amine with 154 to produce 159. Removal of the Fmoc protecting group and acylation with activated ester 155 produced bistramide A in 65% over these final two steps.
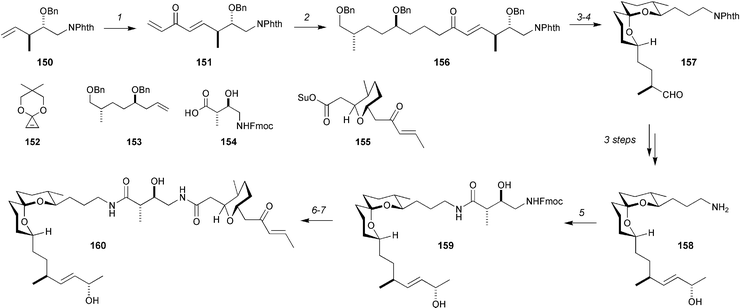 | ||
| Scheme 28 The Kozmin synthesis of bistramide A. Reagents and conditions: (1) 10 mol% Grubbs’ 2nd-generation catalyst, 1.5 equiv. 152, PhH, 60 °C then 1 M H2SO4, MeCN, 63%; (2) 153, 10 mol% Grubbs’ 2nd-generation catalyst, 68%; (3) H2 (80 psi), Pd(OH)2/C; (4) Dess–Martin periodinane, 53% (2 steps); (5) 154, PyBOP, DMF, 85%; (6) Et2NH, DMF; (7) 155, DMF, 20 °C, 65%. | ||
A large number of other total synthesis of marine natural products were reported in the review period, and papers describing first total syntheses are presented in Table 1. New total syntheses of compounds previously prepared are summarized in Table 2.
| Compound | Reference | Notes |
|---|---|---|
(+)-Xyloketal D |
Krohn et al.98 | • 5 Steps |
| • Non-racemic | ||
| • Bioactivity: potent acetylcholine esterase inhibition | ||
Cyercene A  |
Baldwin et al.99 | • 4 Steps from known compound |
| • Bioactivity: may act as mediators in tissue regeneration and chemical defence in the mollusc | ||
Plakortone G  |
Nishiyama et al.100 | • 19 Steps |
| • Non-racemic | ||
| • Bioactivity: cytotoxic | ||
Austrodoric acid  |
Gavagnin et al.101 | • 7 Steps from known compound |
| • Non-racemic | ||
| • Bioactivity: unknown | ||
(+)-Agelasine D  |
Gundersen et al.102 | • 4 Steps from (+)-manool |
| • Non-racemic | ||
| • Bioactivity: related compounds are cytotoxic and antimicrobial | ||
Hachijodine B  |
Lee et al.103 | • 10 Steps |
| • Non-racemic | ||
| • Bioactivity: cytotoxic against the P388 cell line | ||
(18S)-Variabilin  |
Takabe et al.104 | • 11 Steps |
| • Non-racemic | ||
| • Bioactivity: compounds of this class have shown antiviral and cytotoxic activity | ||
(−)-Presphaerene  |
Lee et al.105 | • 18 Steps |
| • Non-racemic | ||
| • Bioactivity: none described | ||
Dolastatin 18  |
Pettit et al.106 | • 5 steps from N-(benzyloxycarbonyl)-N-methylphenylalanine (Z-N-Me PheOH) |
| • Non-racemic | ||
| • Bioactivity: human cancer cell-growth inhibitor | ||
Barettin  |
Bergman et al.107 | • 3 Steps from known compound |
| • Non-racemic | ||
| • Bioactivity: not stated | ||
No name  |
Kende et al.108 | • 5 Steps from known compound (Kende) |
| Bewley and Fetterolf109 | • 7 Steps from 3,5-dibromotyrosine (Bewley and Fetterolf) | |
| • Bioactivity: inhibits the mycobacterial enzyme mycothiol S-conjugate amidase | ||
(+)-Phomopsidin  |
Nakada et al.110 | • 37 Steps |
| • Non-racemic | ||
| • Bioactivity: inhibitor of microtubule assembly | ||
Phakellistatins 3 and 13  |
Van Vranken et al.111 | • 3 Steps from linear precursor synthesized on solid phase |
| • Non-racemic | ||
| • Bioactivity: cytotoxic against the P388 cell line | ||
(−)-Aurilide  |
Suenaga and Kigoshi et al.112 | • 16 Steps |
| • Non-racemic | ||
| • Bioactivity: cytotoxic | ||
Eurypamide A  |
Nishiyama et al.113 | • 19 Steps from known compound |
| • Non-racemic | ||
| • Bioactivity: none described, although related compounds are known to exhibit antimicrobial, cytotoxic and enzyme inhibition activity | ||
(±)-Maculalactones A and B  |
Brown and Wong114 | • 6 Steps each |
| • Racemic | ||
| • Bioactivity: inhibitor of marine herbivore foraging and barnacle settling | ||
(+)-Milnamide A  |
Molinski et al.115 | • 8 Steps from indole |
| • Non-racemic | ||
| • Bioactivity: disrupts microtubule assembly during cell division and inhibits growth of cultured tumor cells | ||
Subarine  |
Delfourne et al.116 | • 5 Steps from phenanthroline |
| • Bioactivity: no significant antitumor activity | ||
(+)-SCH 351448  |
Lee et al.117 | • 27 Steps |
| • Non-racemic | ||
| • Bioactivity: activator of low-density lipoprotein receptor promoter with IC50 25 µM | ||
Xestodecalactone A  |
Bringmann et al.118 | • 5 Steps |
| • Non-racemic | ||
| • Bioactivity: antifungal activity against Candida albicans | ||
Lepadins B, D, E and H  |
Pu and Ma119 | • 21 Steps to lepadins B, E, and H |
| • 20 Steps to lepadin D | ||
| • Non-racemic | ||
| • Bioactivity: significant in vitro cytotoxicity against several human cancer cell lines | ||
(+)-Tricycloclavulone  |
Iguchi et al.120 | • 21 Steps |
| • Non-racemic | ||
| • Bioactivity: related compounds have antiproliferative activity against human cancer cell lines | ||
Cribostatin 6  |
Nakahara and Kubo121 | • 10 Steps from known compound |
| • Bioactivity: growth inhibitor of cancer cell lines and antibacterial activity | ||
Oscillarin 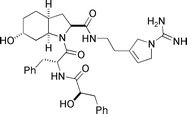 |
Hanessian et al.122 | • 13 Steps from dimethyl-N-BocGlyOH |
| • Non-racemic | ||
| • Bioactivity: potent inhibition of thrombin | ||
(+)-Eupenoxide and (+)-Phomoxide  |
Mehta and Roy123 | • 9 Steps from known compound to eupenoxide |
| • 8 Steps from known compound to phomoxide | ||
| • Non-racemic | ||
| • Bioactivity: antibiotic | ||
Basiliskamides A and B  |
Panek et al.124 | • 11 Steps to each compound |
| • Non-racemic | ||
| • Bioactivity: potent in vivo activity against Candida albicans and Aspergillus fumigatus | ||
Myriaporones 1, 3, and 4  |
Taylor and Fleming125 and Echavarren126 | • 20 Steps from known compound (Taylor) |
| • 14 Steps from known compound (Echavarren) | ||
| • Non-racemic | ||
| • Bioactivity: inhibits growth of a range of cancer cell lines | ||
Norrisolide 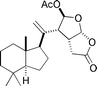 |
Theodorakis et al.127 | • 24 Steps from known compound |
| • Non-racemic | ||
| • Bioactivity: not described | ||
6-Hydroxy-7-methoxyisoquinolinemethanol  |
Saito et al.128 | • 5 Steps from known compound |
| • Bioactivity: not described | ||
Plakortone E 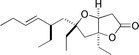 |
Hayes and Kitching129 | • 10 Steps |
| • Racemic | ||
| • Bioactivity: no significant antitumor activity | ||
Caulibugulones A–E 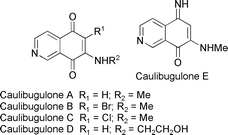 |
Wipf et al.130 (A–E) and Tamagnan et al.131 (A–D) | • 2–3 Steps each from 5-hydroxyisoquinoline (Wipf) |
| • 4–5 Steps (Tamangan) | ||
| • Bioactivity: cytotoxic; potent inhibitors of dual specificity phosphatase Cdc25B | ||
Cyclomarin C 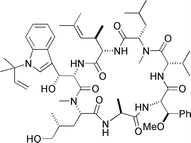 |
Wen and Yao132 | • 21 Steps from known compound |
| • Non-racemic | ||
| • Bioactivity: not described | ||
Aigialomycin D 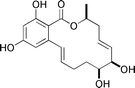 |
Geng and Danishefsky133 | • 18 Steps from 2-deoxyribose |
| • Non-racemic | ||
| • Bioactivity: potent antimalarial and antitumor activity | ||
Kalihinol C 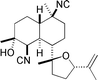 |
Wood et al.134 | • 24 Steps |
| • Non-racemic | ||
| • Bioactivity: related compounds show potent antimalarial activity | ||
(6R,7S)-7-Amino-7,8-dihydro-α-bisabolene  |
Kochi and Ellman135 | • 9 Steps |
| • Non-racemic | ||
| • Bioactivity: antimicrobial |
| Compound | Reference | Compound | Reference |
|---|---|---|---|
| Helianane | Venkateswaran and Sabui136 | (±)-Pseudopterosins A–F and K–L aglycones | Harrowven and Tyte137 |
| (−)-Salicylihalamides A and B | Yadav and Srihari138 | (R)-Strongylodiols A and B | Lee and Baldwin et al.139 |
| Cryptophycin-24 | Tripathy and Georg140 | (+)-Cylindricine C | Kibayashi et al.141 |
| (−)-Agelastatin A | Hale et al.142 | Siphonarienolone | Negishi et al.143 |
| (+)-Anatoxin-a | Brenneman and Martin144 | Haliclorensin and isohaliclorensin | Huang et al.145 |
| Lamellarin G trimethyl ether | Handy et al.146 | (−)-Callystatin A | Langille and Panek147 |
| Bacillariolide III | Suh et al.148 | Macrosphelides A, B, and E | Nemoto et al.149 |
| Caulersin | Bergman et al.150 | (−)-Myclamaide A | Trost et al.151 |
| Octalactins A and B | Burton and Holmes et al.152 | (+)-Tanikolide | Borhan and Schomaker153 |
| (+)-Pinnatoxin A | Inoue and Hirama et al.154 | Lamellarins K and L | Ruchirawat et al.155 |
| (+)-Tanikolide | Masaki et al.156 | 6-Chlorohyellazole | Knölker et al.157 |
| Lyngbic Acid | Braddock and Matsuno158 | Hapalosin | Mandai et al.159 and Palomo et al.160 |
| Pulchellalactam | Takabe et al.161 | (−)-Dysibetaine | Wardrop and Burge162 |
| Bistratamide G | Shin et al.163 | Pulchellalactam | Mangaleswaran and Argade164 |
| (−)-Pateamine A | Pattenden et al.165 | (+)-Kalkitoxin | White et al.166 |
| (±)-Pinnaic acid and (±)-tauropinnaic acid | Christie and Heathcock167 | (−)-Doliculide | Hanessian et al.168 |
| (−)-Flustramine B | MacMillan et al.169 | Peridinin | Katsumura et al.170 |
| Petrobactin | Phanstiel et al.171 | (−)-Cycloepoxydon | Mehta and Islam172 |
| (−)-α-Kainic Acid | Hoppe and Martinez173 | (−)-Apicularen A | Rizzacasa et al.174 |
| (±)-Halichlorine and (±)-pinnaic Acid | Kibayashi et al.175 | (+)-Brasilenyne | Denmark and Yang176 |
| Callipeltoside A | Panek and Huang177 | (+)-Acanthodoral | Zhang and Koreeda178 |
| (±)-Dibromophakellstatin | Austin et al.179 | Hamigerans A, B and E | Nicolaou et al.180 |
| 7-Epicyclindrospermopsin | Looper and Williams181 |
9 Acknowledgements
We would like to thank Professor John Blunt and Professor Murray Munro (University of Canterbury, Christchurch, New Zealand) for a copy of the 2005 version MarinLit database97 which facilitated data collection for this review. We thank Jim Henderson for assistance in drawing structures.10 References
- J. W. Blunt, B. R. Copp, M. H. G. Munro, P. T. Northcote and M. R. Prinsep, Nat. Prod. Rep., 2006, 23 10.1039/b502792f (the preceding paper in this issue).
- P. Wipf, J. T. Reeves and B. W. Day, Curr. Pharm. Des., 2004, 10, 1417 Search PubMed.
- C.-G. Yang, H. Huang and B. Jiang, Curr. Org. Chem., 2004, 8, 1691 CrossRef CAS.
- M. Sasaki and H. Fuwa, Synlett, 2004, 1851 CrossRef CAS.
- H. H. Wasserman and J. Parr, Acc. Chem. Res., 2004, 37, 687 CrossRef CAS.
- D. Fernandez, A. Ahaidar, G. Danelon, P. Cironi, M. Marfil, O. Perez, C. Cuevas, F. Albericio, J. A. Joule and M. Alvarez, Monatsh. Chem., 2004, 135, 615 CrossRef CAS.
- K. Suenaga, Bull. Chem. Soc. Jpn., 2004, 77, 443 CrossRef CAS.
- K. Fujiwara, Kenshu, A. Murai and Akio, Bull. Chem. Soc. Jpn., 2004, 77, 2129 CrossRef CAS.
- G. Zanoni and M. Franzini, Angew. Chem., Int. Ed., 2004, 43, 4837 CrossRef CAS.
- M. C. Nora de Souza, TheScientificWorld, 2004, 4, 415 Search PubMed.
- C. Bailly, Curr. Med. Chem.: Anti-Cancer Agents, 2004, 4, 363 Search PubMed.
- J. Kobayashi and M. Tsuda, Nat. Prod. Rep., 2004, 21, 77 RSC.
- J. W. Blunt, B. R. Copp, M. H. G. Munro, P. T. Northcote and M. R. Prinsep, Nat. Prod. Rep., 2004, 21, 1 RSC.
- V. S. C. Yeh, Tetrahedron, 2004, 60, 11995 CrossRef CAS.
- A. B. Smith, III and C. M. Adams, Acc. Chem. Res., 2004, 37, 365 CrossRef.
- (a) K. C. Nicolaou, S. Vyskocil, T. V. Koftis, Y. M. A. Yamada, T. Ling, D. Y.-K. Chen, W. Tang, G. Petrovic, M. O. Frederick, Y. Li and M. Satake, Angew. Chem., Int. Ed., 2004, 43, 4312 CrossRef CAS; (b) K. C. Nicolaou, T. V. Koftis, S. Vyskocil, G. Petrovic, T. Ling, Y. M. A. Yamada, W. Tang and M. O. Frederick, Angew. Chem., Int. Ed., 2004, 43, 4318 CrossRef CAS.
- K. C. Nicolaou, Y. Li, N. Uesaka, T. V. Koftis, S. Vyskocil, T. Ling, M. Govindasamy, W. Qian, F. Bernal and D. Y. K. Chen, Angew. Chem., Int. Ed., 2003, 42, 3643 CrossRef CAS.
- K. C. Nicolaou, D. Y. K. Chen, Y. Li, W. Qian, T. Ling, S. Vyskocil, T. V. Koftis, M. Govindasamy and N. Uesaka, Angew. Chem., Int. Ed., 2003, 42, 3649 CrossRef CAS.
- M. Satake, K. Ofuji, H. Naoki, K. J. James, A. Furey, T. McMahon, J. Silke and T. Yasumoto, J. Am. Chem. Soc., 1998, 120, 9967 CrossRef CAS.
- C. Hopmann and D. J. Faulkner, Tetrahedron Lett., 1997, 38, 169–170 CrossRef CAS.
- H. Nakamura, M. Ono, Y. Shida and H. Akita, Tetrahedron: Asymmetry, 2002, 13, 705 CrossRef CAS.
- L. K. Geisler, S. Nguyen and C. J. Forsyth, Org. Lett., 2004, 6, 4159 CrossRef CAS.
- X. T. Zhou and R. G. Carter, Chem. Commun., 2004, 2138 RSC.
- Y. Ishikawa and S. Nishiyama, Heterocycles, 2004, 63, 885 CrossRef CAS.
- (a) Y. Ishikawa and S. Nishiyama, Tetrahedron Lett., 2004, 45, 351 CrossRef CAS; (b) Y. Ishikawa and S. Nishiyama, Heterocycles, 2004, 63, 539 CrossRef CAS.
- Y.-Y. Lin, M. Risk, S. M. Ray, D. Van Engen, J. Clardy, J. Golik, J. C. James and K. Nakanishi, J. Am. Chem. Soc., 1981, 103, 6773 CrossRef CAS.
- K. Fujiwara, D. Sato, M. Watanabe, H. Morishita, A. Murai, H. Kawi and T. Suzuki, Tetrahedron Lett., 2004, 45, 5243 CrossRef CAS.
- A. V. Krishna and Y. J. Shimizu, J. Am. Chem. Soc., 1989, 111, 6476 CrossRef CAS.
- M. Satake, M. Murata and T. Yasumoto, Tetrahedron Lett., 1993, 34, 1975 CrossRef CAS.
- M. Inoue, K. Miyazaki, H. Uehara, M. Maruyama and M. Hirama, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 12013 CrossRef CAS.
- M. Inoue and M. Hirama, Synlett, 2004, 577 CrossRef CAS.
- M. Inoue and M. Hirama, Acc. Chem. Res., 2004, 37, 961 CrossRef CAS.
- M. Inoue, Org. Biomol. Chem., 2004, 2, 1811 RSC.
- J. S. Clark, T. C. Fessard and C. Wilson, Org. Lett., 2004, 6, 1773 CrossRef CAS.
- H. Nagai, M. Murata, K. Torigoe, M. Satake and T. Yasumoto, J. Org. Chem., 1992, 57, 5448 CrossRef CAS; H. Nagai, K. Torigoe, M. Satake, M. Murata, T. Yasumoto and H. Hirota, J. Am. Chem. Soc., 1992, 114, 1102 CrossRef CAS.
- J. S. Clark, F. Elustondo and M. C. Kimber, Chem. Commun., 2004, 2470 RSC.
- T. Baba, S. Takai, N. Sawada and M. Isobe, Synlett, 2004, 603 CAS.
- H. Fuwa, N. Kainuma, K. Tachibana, C. Tsukano, M. Satake and M. Sasaki, Chem. Eur. J., 2004, 10, 4894 CrossRef CAS.
- S. Kobayashi, Y. Takahashi, K. Komano, B. H. Alizadeh, Y. Kawada, T. Oishi, S. Tanaka, Y. Ogasawara, S. Sasaki and M. Hirama, Tetrahedron, 2004, 60, 8375 CrossRef CAS.
- M. Sasaki, M. Ebine, H. Takagi, H. Takakura, T. Shida, M. Satake, Y. Oshima, T. Igarashi and T. Yasumoto, Org. Lett., 2004, 6, 1501 CrossRef CAS.
- H. Fuwa, N. Kainuma, K. Tachibana, C. Tsukano, M. Satake and M. Sasaki, Chem. Eur. J., 2004, 10, 4894 CrossRef CAS.
- B. M. Trost and Y. H. Rhee, Org. Lett., 2004, 6, 4311 CAS.
- J. C. Braekman, D. Daloze, R. Tavares, E. Hajdu and R. W. M. Van Soest, J. Nat. Prod., 2000, 63, 193 CrossRef CAS.
- Z. D. Aron and L. E. Overman, Chem. Commun., 2004, 253 RSC.
- (a) S. H. Mai, V. K. Nagulapalli, A. D. Patil, A. Truneh and J. W. Westley, PCT Int. Appl. W09301, 193 (Chem. Abstr., 1993, 118, 225662) Search PubMed; (b) A. D. Patil, N. V. Kumar, W. C. Kokke, M. F. Bean, A. J. Freyer, C. De Brosse, S. H. Mai, A. Truneh, D. J. Faukner, B. Carte, A. L. Breen, R. P. Hertzberg, R. K. Johnson, J. W. Westley and B. C. M. Potts, J. Org. Chem., 1995, 60, 1182 CrossRef CAS.
- A. Goti, M. Cacciarini, F. Cardona and A. Brandi, Tetrahedron Lett., 1999, 40, 2853 CrossRef CAS.
- M. C. Elliott and M. S. Long, Org. Biomol. Chem., 2004, 2, 2003 RSC.
- C. A. Bewley, S. Ray, F. Cohen, S. K. Collins and L. E. Overman, J. Nat. Prod., 2004, 67, 1319 CrossRef CAS.
- (a) J. Kobayashi, M. Ishibashi, H. Nakamura, Y. Ohizumi, T. Yamasu, T. Sasaki and Y. Hirata, Tetrahedron Lett., 1986, 27, 5755 CrossRef CAS; (b) J. Kobayashi, M. Ishibashi and H. Hirota, J. Nat. Prod., 1991, 54, 1435 CrossRef CAS.
- B. M. Trost, J. D. Chisholm, S. A. Wrobleski and M. Jung, J. Am. Chem. Soc., 2002, 124, 12420 CrossRef CAS.
- H. W. Lam and G. Pattenden, Angew. Chem., Int. Ed., 2002, 41, 508 CrossRef CAS.
- R. E. Maleczka, Jr, L. R. Terrell, F. Geng and J. S. Ward, III, Org. Lett., 2002, 4, 2841 CrossRef CAS.
- B. M. Trost and P. E. Harrington, J. Am. Chem. Soc., 2004, 126, 5028 CrossRef CAS.
- (a) B. M. Trost and A. Indolese, J. Am. Chem. Soc., 1993, 115, 4361 CrossRef CAS; (b) B. M. Trost, A. F. Indolese, T. J. J. Mueller and B. Treptow, J. Am. Chem. Soc., 1995, 117, 615 CrossRef CAS; (c) B. M. Trost and F. D. Toste, Tetrahedron Lett., 1999, 40, 7739 CrossRef CAS; (d) B. M. Trost and F. D. Toste, J. Am. Chem. Soc., 2000, 122, 714 CrossRef CAS; (e) M. Schnaderbeck, Ph.D. Thesis, Stanford University, 1998 Search PubMed; (f) M. L. Sundermann, Ph.D. Thesis, Stanford University, 2000 Search PubMed.
- Y. Kita, H. Maeda, K. Omori, T. Okuno and Y. Tamura, Synlett, 1993, 273 CrossRef CAS.
- M. Ishibashi, M. Takahashi and J. Kobayashi, J. Org. Chem., 1995, 60, 6062 CrossRef CAS.
- K. Shimbo, M. Tsuda, N. Izui and J. Kobayashi, J. Org. Chem., 2002, 67, 1020 CrossRef CAS.
- A. K. Ghosh and G. Gong, J. Am. Chem. Soc., 2004, 126, 3704 CrossRef CAS.
- (a) M. Tsuda, T. Endo and J. Kobayashi, J. Org. Chem., 2001, 66, 134 CrossRef CAS; J. Kobayashi, T. Kubota, M. Tsuda and T. Endo, J. Org. Chem., 2000, 65, 1349 CrossRef CAS; (b) T. Kubota, T. Endo, M. Tsuda, M. Shiro and J. Kobayshi, Tetrahedron, 2001, 57, 6175 CrossRef CAS.
- E. A. Colby, K. C. O'Brien and T. F. Jamison, J. Am. Chem. Soc., 2004, 126, 998 CrossRef CAS.
- K. Takai, T. Kakiuchi, Y. Kataoka and K. Utimoto, J. Org. Chem., 1994, 59, 2668 CrossRef CAS.
- M. Tsuda, N. Izui, K. Shimbo, M. Sato, E. Fukushi, J. Kawabata, K. Katsumata, T. Horiguchi and J. Kobayashi, J. Org. Chem., 2003, 68, 5339 CrossRef CAS.
- O. Lepage, E. Kattnig and A. Fürstner, J. Am. Chem. Soc., 2004, 126, 15970 CrossRef CAS.
- O. Lepage, E. Kattnig and A. Fürstner, J. Am. Chem. Soc., 2004, 126, 15970 CrossRef CAS.
- J. Kobayashi, M. Tsuda, M. Ishibashi, H. Shigemori, T. Yamasu, H. Hirota and T. Sasaki, J. Antibiot., 1991, 44, 1259 CAS.
- J. B. Shotwell and W. R. Roush, Org. Lett., 2004, 6, 3865 CrossRef CAS.
- W. Zhang, R. G. Carter and A. F. T. Yokochi, J. Org. Chem., 2004, 69, 2569 CrossRef CAS.
- A. K. Ghosh and C. Liu, Strategies Tactics Org. Synth., 2004, 5, 255 Search PubMed.
- (a) G. R. Pettit, Z. A. Cichacz, F. Gao, M. R. Boyd and J. M. R. Schmidt, J. Chem. Soc., Chem. Commun., 1994, 1111 RSC; (b) G. R. Pettit and Z. A. Cichacz, US Pat., 1995, 5,430,052 Search PubMed.
- I. Paterson, R. Britton, O. Delgado and A. E. Wright, Chem. Commun., 2004, 632 RSC.
- Y. Shin, J.-H. Fournier, Y. Fukui, A. M. Brückner and D. P. Curran, Angew. Chem., Int. Ed., 2004, 43, 4634 CrossRef CAS.
- I. Paterson, R. Britton, O. Delgado and A. Meyer, K. G., Angew. Chem., Int. Ed., 2004, 43, 4629 CrossRef CAS.
- I. Paterson, K.-S. Yeung and J. B. Smaill, Synlett, 1993, 774 CrossRef CAS.
- W. S. Mahoney, D. M. Brestensky and J. M. Stryker, J. Am. Chem. Soc., 1988, 110, 291 CrossRef CAS.
- G. D. Allred and L. S. Liebeskind, J. Am. Chem. Soc., 1996, 118, 2748 CrossRef CAS.
- (a) S. J. Mickel, G. H. Sedelmeier, D. Niederer, R. Daeffler, A. Osmani, K. Schreiner, M. Seeger-Weibel, B. Berod, K. Schaer, R. Gamboni, S. Chen, W. Chen, Chen, C. T. Jagoe, F. R. Kinder, M. Loo, K. Prasad, O. Repic, W.-C. Shieh, R.-M. Wang, L. Waykole, D. D. Xu and S. Xue, Org. Process Res. Dev., 2004, 8, 92 Search PubMed; (b) S. J. Mickel, G. H. Sedelmeier, D. Niederer, F. Schuerch, D. Grimler, G. Koch, R. Daeffler, A. Osmani, A. Hirni, K. Schaer, R. Gamboni, A. Bach, A. Chaudhary, S. Chen, W. Chen, B. Chen, B. Hu, C. T. Jagoe, H.-Y. Kim, F. R. Kinder, Y. Liu, Y. Lu, J. McKenna, M. Prasad, T. M. Ramsey, O. Repic, L. Rogers, W.-C. Shieh, R.-M. Wang and L. Waykole, Org. Process Res. Dev., 2004, 8, 101 Search PubMed; (c) S. J. Mickel, G. H. Sedelmeier, D. Niederer, F. Schuerch, G. Koch, E. Kuesters, R. Daeffler, A. Osmani, M. Seeger-Weibel, E. Schmid, A. Hirni, K. Schaer, R. Gamboni, A. Bach, S. Chen, W. Chen, P. Geng, C. T. Jagoe, F. R. Kinder, G. T. Lee, J. McKenna, T. M. Ramsey, O. Repic, L. Rogers, W.-C. Shieh, R.-M. Wang and L. Waykole, Org. Process Res. Dev., 2004, 8, 107 Search PubMed; (d) S. J. Mickel, G. H. Sedelmeier, D. Niederer, F. Schuerch, M. Seger, K. Schreiner, R. Daeffler, A. Osmani, D. Bixel, O. Loiseleur, J. Cercus, H. Stettler, K. Schaer and R. Gamboni, Org. Process Res. Dev., 2004, 8, 113 Search PubMed; (e) S. J. Mickel, D. Niederer, R. Daeffler, A. Osmani, E. Kuesters, E. Schmid, K. Schaer, R. Gamboni, W. Chen, E. Loeser, F. R. Kinder, K. Konigsberger, K. Prasad, T. M. Ramsey, O. Repic, R.-M. Wang, G. Florence, I. Lyothier and I. Paterson, Org. Process Res. Dev., 2004, 8, 122 Search PubMed.
- S. J. Mickel, Curr. Opin. Drug Dev., 2004, 7, 869 Search PubMed.
- S. J. Mickel, R. Fischer and W. Marterer, Chimia, 2004, 58, 640 CrossRef CAS.
- I. Paterson and I. Lyothier, Org. Lett., 2004, 6, 4933 CrossRef CAS.
- (a) S. BouzBouz and J. Cossy, Synlett, 2004, 2034 CrossRef CAS; (b) A. Hafner, R. O. Duthaler, R. Marti, G. Rihs, P. Rothe-Streit and F. Schwarzenbach, J. Am. Chem. Soc., 1992, 114, 2321 CrossRef CAS.
- P. V. Ramachandran, B. Prabhudas, J. S. Chandra and M. V. R. Reddy, J. Org. Chem., 2004, 69, 6294 CrossRef CAS.
- O. Loiseleur, G. Koch and T. Wagner, Org. Process Res. Dev., 2004, 8, 597 Search PubMed.
- K. A. Parker and I. A. Katsoulis, Org. Lett., 2004, 6, 1413 CrossRef CAS.
- M. A. Burlingame, S. J. Shaw, K. F. Sundermann, D. Zhang, J. Petryka, E. Mendoza, F. Liu, D. C. Myles, M. J. LaMarche, T. Hirose, B. S. Freeze and A. B. Smith, Bioorg. Med. Chem. Lett., 2004, 14, 2335 CrossRef CAS.
- S. P. Gunasekera, S. J. Mickel, R. Daeffler, D. Niederer, A. E. Wright, P. Linley and T. Pitts, J. Nat. Prod., 2004, 67, 749 CrossRef CAS.
- A. Cutignano, G. Bifulco, I. Bruno, A. Casapullo, L. Gomez-Paloma and R. Riccio, Tetrahedron, 2000, 56, 3743 CrossRef CAS.
- N. K. Garg, D. D. Caspi and B. M. Stoltz, J. Am. Chem. Soc., 2004, 126, 9552 CrossRef CAS.
- V. B. Birman and X.-T. Jiang, Org. Lett., 2004, 6, 2369 CrossRef CAS.
- P. S. Baran, A. L. Zografos and D. P. O'Malley, J. Am. Chem. Soc., 2004, 126, 3726 CrossRef.
- (a) K. L. Rinehart, Jr., US Pat., 4737510, 1988 Search PubMed; (b) J. Kobayashi, M. Tsuda, T. Murayama, J. Nakamura, Y. Ohizumi, M. Ishibashi, M. Iwamura, T. Ohta and S. Nozoe, Tetrahedron, 1990, 46, 5579 CrossRef CAS; (c) P. A. Keifer, R. E. Schwartz, M. E. S. Koker, R. G. Hughes, Jr., D. Rittschof and K. L. Rinehart, Jr., J. Org. Chem., 1991, 56, 2965 CrossRef CAS.
- P. S. Baran, D. P. O'Malley and A. L. Zografos, Angew. Chem., Int. Ed., 2004, 43, 2674 CrossRef CAS.
- A. Grassia, I. Bruno, C. Debitus, S. Marzocco, A. Pinto, L. Gomez-Paloma and R. Riccio, Tetrahedron, 2001, 57, 6257 CrossRef CAS.
- J. Chen and C. J. Forsyth;, Angew. Chem., Int. Ed., 2004, 43, 2148 CrossRef CAS.
- A. K. Ghosh and X. Xu, Org. Lett., 2004, 6, 2055 CrossRef CAS.
- D. Gouiffes, M. Juge, N. Grimaud, L. Welin, M. P. Sauviat, Y. Barbin, D. Laurent, C. Roussakis, J. P. Henichart and J. F. Verbist, Toxicon, 1988, 26, 1129 CrossRef CAS.
- A. V. Statsuk, D. Liu and S. A. Kozmin, J. Am. Chem. Soc., 2004, 126, 9546 CrossRef CAS.
- MarinLit database, Department of Chemistry, University of Canterbury: http://www.chem.canterbury.ac.nz/marinlit/marinlit.shtml Search PubMed.
- K. Krohn and M. Riaz, Tetrahedron Lett., 2004, 45, 293 CrossRef CAS.
- J. E. Moses, J. E. Baldwin and R. M. Adlington, Tetrahedron Lett., 2004, 45, 6447 CrossRef CAS.
- S. Kowashi, T. Ogamino, J. Kamei, Y. Ishikawa and S. Nishiyama, Tetrahedron Lett., 2004, 45, 4393 CrossRef CAS.
- V. Kulcitki, N. Ungur, M. Gavagnin, M. Carbone and G. Cimino, Tetrahedron: Asymmetry, 2004, 15, 423 CrossRef CAS.
- B. T. Utenova and L.-L. Gundersen, Tetrahedron Lett., 2004, 45, 4233 CrossRef CAS.
- S. P. Romeril, V. Lee and J. E. Baldwin, Tetrahedron Lett., 2004, 45, 3273 CrossRef CAS.
- K. Takabe, H. Hashimoto, H. Sugimoto, M. Nomoto and H. Yoda, Tetrahedron: Asymmetry, 2004, 15, 909 CrossRef CAS.
- J. Lee and J. Hong, J. Org. Chem., 2004, 69, 6433 CrossRef CAS.
- G. R. Pettit, F. Hogan and D. L. Herald, J. Org. Chem., 2004, 69, 4019 CrossRef CAS.
- A.-L. Johnson, J. Bergman, M. Sjögren and L. Bohlin, Tetrahedron, 2004, 60, 961 CrossRef CAS.
- A. S. Kende, J. Lan and J. Fan, Tetrahedron Lett., 2004, 45, 133 CrossRef CAS.
- B. Fetterolf and C. A. Bewley, Bioorg. Med. Chem. Lett., 2004, 14, 3785 CrossRef CAS.
- T. Suzuki, K. Usui, Y. Miyake, M. Namikoshi and M. Nakada, Org. Lett., 2004, 6, 553 CrossRef CAS.
- K. L. Greenman, D. M. Hach and D. L. Van Vranken, Org. Lett., 2004, 6, 1713 CrossRef CAS.
- K. Suenaga, T. Mutou, T. Shibata, T. Itoh, T. Fujita, N. Takada, K. Hayamizu, M. Takagi, T. Irifune, H. Kigoshi and K. Yamada, Tetrahedron, 2004, 60, 8509 CrossRef CAS.
- M. Ito, M. Yamanaka, N. Kutsumura and S. Nishiyama, Tetrahedron, 2004, 60, 5623 CrossRef CAS.
- G. D. Brown and H.-F. Wong, Tetrahedron, 2004, 60, 5439 CrossRef CAS.
- C. Liu, M. N. Masuno, J. B. MacMillan and T. F. Molinski, Angew. Chem., Int. Ed., 2004, 43, 5951 CrossRef CAS.
- L. Bijeire, L. Legentil, J. Bastide, F. Darro, C. Rochart and E. Delfourne, Eur. J. Org. Chem., 2004, 1891 CrossRef CAS.
- E. J. Kang, E. J. Cho, Y. E. Lee, M. K. Ji, D. M. Shin, Y. K. Chung and E. Lee, J. Am. Chem. Soc., 2004, 126, 2680 CrossRef CAS.
- G. Bringmann, G. Lang, M. Michel and M. Heubes, Tetrahedron Lett., 2004, 45, 2829 CrossRef CAS.
- X. Pu and D. Ma, Angew. Chem., Int. Ed., 2004, 43, 4222 CrossRef CAS.
- H. Ito, M. Hasegawa, Y. Takenaka, T. Kobayashi and K. Iguchi, J. Am. Chem. Soc., 2004, 126, 4520 CrossRef CAS.
- S. Nakahara and A. Kubo, Heterocycles, 2004, 63, 2355 CrossRef CAS.
- S. Hanessian, M. Tremblay and J. F. W. Petersen, J. Am. Chem. Soc., 2004, 126, 6064 CrossRef CAS.
- G. Mehta and S. Roy, Org. Lett., 2004, 6, 2389 CrossRef CAS.
- D. J. Lipomi, N. F. Langille and J. S. Panek, Org. Lett., 2004, 6, 3533 CrossRef CAS.
- K. N. Fleming and R. E. Taylor, Angew. Chem., Int. Ed., 2004, 43, 1728 CrossRef CAS.
- M. Pérez, C. del Pozo, F. Reyes, A. Rodriguez, A. Francesch, A. M. Echavarren and C. Cuevas, Angew. Chem., Int. Ed., 2004, 43, 1724 CrossRef CAS.
- T. P. Brady, S. H. Kim, K. Wen and E. A. Theodorakis, Angew. Chem., Int. Ed., 2004, 43, 739 CrossRef CAS.
- N. Saiato, C. Tanaka, T. Satomi, C. Oyama and A. Kubo, Chem. Pharm. Bull., 2004, 52, 282 CrossRef.
- P. Y. Hayes and W. Kitching, Heterocycles, 2004, 62, 173 CrossRef CAS.
- P. Wipf, B. Joo, T. Nguyen and J. S. Lazo, Org. Biomol. Chem., 2004, 2, 2173 RSC.
- D. Alagille, R. M. Baldwin and G. D. Tamagnan, Tetrahedron Lett., 2004, 45, 6179 CrossRef CAS.
- S.-J. Wen and Z.-J. Yao, Org. Lett., 2004, 6, 2721 CrossRef CAS.
- X. Geng and S. J. Danishefsky, Org. Lett., 2004, 6, 413 CrossRef CAS.
- R. D. White, G. F. Keaney, C. D. Slown and J. L. Wood, Org. Lett., 2004, 6, 1123 CrossRef CAS.
- T. Kochi and J. A. Ellman, J. Am. Chem. Soc., 2004, 126, 15652 CAS.
- S. K. Sabui and R. V. Venkateswaran, Tetrahedron Lett., 2004, 45, 9653 CrossRef.
- D. C. Harrowven and M. J. Tyte, Tetrahedron Lett., 2004, 45, 2089 CrossRef CAS.
- J. S. Yadav and P. Srihari, Tetrahedron: Asymmetry, 2004, 15, 81 CrossRef CAS.
- J. E. D. Kirkham, T. D. L. Courtney, V. Lee and J. E. Baldwin, Tetrahedron Lett., 2004, 45, 5645 CrossRef CAS.
- N. K. Tripathy and G. I. Georg, Tetrahedron Lett., 2004, 45, 5309 CrossRef CAS.
- T. Arai, H. Abe, S. Aoyagi and C. Kibayashi, Tetrahedron Lett., 2004, 45, 5921 CrossRef CAS.
- M. M. Domostoj, E. Irving, F. Scheinmann and K. J. Hale, Org. Lett., 2004, 6, 2615 CrossRef CAS.
- M. Magnin-Lachaux, Z. Tan, B. Liang and E.-I. Negishi, Org. Lett., 2004, 6, 1425 CrossRef CAS.
- J. B. Brenneman and S. F. Martin, Org. Lett., 2004, 6, 1329 CrossRef CAS.
- J.-F. Zheng, L.-R. Jin and P.-Q. Huang, Org. Lett., 2004, 6, 1139 CrossRef CAS.
- S. T. Handy, Y. Zhang and H. Bergman, J. Org. Chem., 2004, 69, 2362 CrossRef CAS.
- N. F. Langille and J. S. Panek, Org. Lett., 2004, 6, 3203 CrossRef CAS.
- S.-Y. Seo, J.-K. Jung, S.-M. Paek, Y.-S. Lee, S.-H. Kim, K.-O. Lee and Y.-G. Suh, Org. Lett., 2004, 6, 429 CrossRef CAS.
- T. Kawaguchi, N. Funamori, Y. Matsuya and H. Nemoto, J. Org. Chem., 2004, 69, 505 CrossRef CAS.
- N. Wahlström, B. Stensland and J. Bergman, Tetrahedron, 2004, 60, 2147 CrossRef CAS.
- B. M. Trost, H. Yang and G. D. Probst, J. Am. Chem. Soc., 2004, 126, 48 CrossRef CAS.
- P. T. O'Sullivan, W. Buhr, M. A. M. Fuhry, J. R. Harrison, J. E. Davies, N. Feeder, D. R. Marshall, J. W. Burton and A. B. Holmes, J. Am. Chem. Soc., 2004, 126, 2194 CrossRef CAS.
- J. M. Schomaker and B. Borhan, Org. Biomol. Chem., 2004, 2, 621 RSC.
- S. Sakamoto, H. Sakazaki, K. Hagiwara, K. Kamada, K. Ishii, T. Noda, M. Inoue and M. Hirama;, Angew. Chem., Int. Ed., 2004, 43, 6505 CrossRef CAS.
- P. Ploypradith, C. Mahidol, P. Sahakitpichan, S. Wongbundit and S. Ruchirawat, Angew. Chem., Int. Ed., 2004, 43, 866 CrossRef CAS.
- H. Arasaki, M. Iwata, M. Makida and Y. Masaki, Chem. Pharm. Bull., 2004, 52, 848 CrossRef CAS.
- H.-J. Knölker, W. Fröhner and R. Heinrich, Synlett, 2004, 15, 2705 CrossRef.
- D. C. Braddock and A. Matsuno, Synlett, 2004, 14, 2521 CrossRef.
- T. Oshitari, Saiyinbilige and T. Mandai, Heterocycles, 2004, 62, 185 CrossRef CAS.
- C. Palomo, M. Oiarbide, J. M. García, A. González, R. Pazos, J. M. Odriozola, P. Bañuelos, M. Tello and A. Linden, J. Org. Chem., 2004, 69, 4126 CrossRef CAS.
- J.-I. Bessho, Y. Shimotsu, S. Mizumoto, N. Mase, H. Yoda and K. Takabe;, Heterocycles, 2004, 63, 1013 CrossRef CAS.
- D. J. Wardrop and M. S. Burge, Chem. Commun., 2004, 1230 RSC.
- C.-G. Shin, C. Abe and Y. Yonezawa, Chem. Lett., 2004, 33, 664 CrossRef CAS.
- S. Mangaleswaran and N. P. Argade, Synthesis, 2004, 10, 1560.
- G. Pattenden, D. J. Critcher and M. Remuiñán, Can. J. Chem., 2004, 82, 353 CrossRef CAS.
- J. D. White, Q. Xu, C.-S. Lee and F. A. Valerlote, Org. Biomol. Chem., 2004, 2, 2092 RSC.
- H. S. Christie and C. H. Heathcock, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 12079 CrossRef CAS.
- S. Hanessian, V. Mascitti and S. Giroux, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 11996 CrossRef CAS.
- J. F. Austin, S.-G. Kim, C. J. Sinz, W.-J. Xiao and D. W. C. MacMillan, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 5482 CrossRef CAS.
- N. Furuichi, H. Hara, T. Osaki, M. Nakano, H. Mori and S. Katsumura, J. Org. Chem., 2004, 69, 7949 CrossRef CAS.
- R. A. Gardner, R. Kinkade, C. Wang and O. Phanstiel, IV, J. Org. Chem., 2004, 69, 3530 CrossRef CAS.
- G. Mehta and K. Islam, Org. Lett., 2004, 6, 807 CrossRef CAS.
- M. M. Martinez and D. Hoppe, Org. Lett., 2004, 6, 3743 CrossRef CAS.
- F. Hilli, J. M. White and M. A. Rizzacasa, Org. Lett., 2004, 6, 1289 CrossRef CAS.
- Y. Matsumura, S. Aoyagi and C. Kibayashi, Org. Lett., 2004, 6, 965 CrossRef CAS.
- S. E. Denmark and S. Yang, J. Am. Chem. Soc., 2004, 126, 12432 CrossRef CAS.
- H. Huang and J. S. Panek, Org. Lett., 2004, 6, 4383 CrossRef CAS.
- L. Zhang and M. Koreeda, Org. Lett., 2004, 6, 4–537.
- R. Chung, E. Yu, C. D. Incarvito and D. J. Austin, Org. Lett., 2004, 6, 3881 CrossRef CAS.
- K. C. Nicolaou, D. L. Gray and J. Tae, J. Am. Chem. Soc., 2004, 126, 613 CrossRef CAS.
- R. E. Looper and R. M. Williams, Angew. Chem., Int. Ed., 2004, 43, 2930 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2006 |