Cofactor biosynthesis: an organic chemist's treasure trove†
Tadhg P.
Begley
*
Department of Chemistry and Chemical Biology, Cornell University, Ithaca, NY, 14853. E-mail: tpb2@cornell.edu
First published on 4th January 2006
Abstract
Covering: primarily 2000–2005
This Highlight focuses on reactions involved in cofactor biosynthesis that pose interesting mechanistic problems.
 Tadhg P. Begley Tadhg P. Begley | Tadhg P. Begley is a Professor of Chemistry and Chemical Biology at Cornell University, Ithaca, New York. He received his B.Sc. from University College Cork in 1977 and his Ph.D. from the California Institute of Technology in 1983. He carried out postdoctoral studies at the University of Geneva (W. Oppolzer) and at MIT (C. Walsh) before joining the Cornell Faculty in 1986. Dr. Begley’s research is focused on the mechanistic enzymology of complex organic transformations, particularly those found on the vitamin biosynthetic pathways. He is currently working on the biosynthesis of NAD, thiamin, and pyridoxal phosphate. He recently co-authored with John McMurry ‘The organic chemistry of biological pathways’. |
Introduction
Proteins contain a limited range of functional groups that can be used for enzymatic catalysis. These include acids, bases, simple nucleophiles and electrophiles. Redox chemistry is limited, with rare exceptions, to the thiol group of cysteine. These functional groups are inadequate for the efficient catalysis of many reactions found in living systems, and cells have evolved a set of small molecules, called cofactors, that bind at the active site and augment the catalytic potential of the enzyme. Many of these cofactors are biosynthesized from vitamins, so called because of the essential roles that they play in the human diet. This Highlight will focus on a subset of the reactions involved in the biosynthesis of thiamin, nicotinamide adenine dinucleotide, riboflavin, pyridoxal phosphate, biotin, vitamin B12, heme, coenzyme A, 5-methylene-3,5-dihydroimidazol-4-one, and topaquinone, reactions that pose particularly interesting mechanistic problems. The coverage is selective rather than comprehensive and reflects the research interests of the author. A more complete description of cofactor biosynthetic pathways can be found in a recent volume of the ‘Vitamins and Hormones’ series.1Thiamin biosynthesis: formation of the pyrimidine and thiazole heterocycles
The thiamin thiazole in Bacillus subtilis is biosynthesized from 1-deoxy-D-xylulose-5-phosphate (1, DXP), glycine imine 11 and a sulfur carrier protein thiocarboxylate (4, ThiS–COS−) as outlined in Fig. 1. In this mechanism, DXP forms an imine with lysine 96 of the thiazole synthase (ThiG), which then tautomerizes to 3. Addition of ThiS-thiocarboxylate 4, followed by an acyl shift and loss of water gives 7. Tautomerization of 7 followed by elimination of ThiS–COO−10 gives 9 which then adds to the glycine imine 11, formed by the oxidation of glycine,2 to give 12. Transimination followed by decarboxylation completes the thiazole formation. This mechanism is supported by isotope exchange experiments, intermediate trapping, the demonstration of oxygen transfer from DXP to ThiS–COO− and the structural characterization of thiazole synthase (ThiG) complexed to the sulfur carrier protein.2–6 This thiazole biosynthesis is different from any of the characterized chemical or biochemical routes to the thiazole heterocycle. A related oxygen-sensitive thiazole biosynthesis in Escherichia coli that uses tyrosine instead of glycine has been reconstituted but not yet mechanistically characterized.7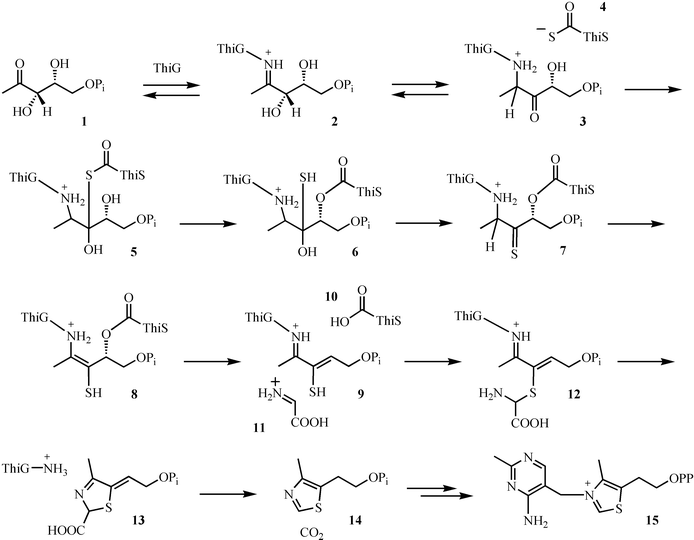 | ||
| Fig. 1 Mechanistic proposal for the formation of the thiazole moiety (14) of thiamin pyrophosphate (15). | ||
Studies on the mechanistic enzymology of thiazole biosynthesis led to the discovery of a novel sulfur transfer strategy (Fig. 2).8 In this strategy, the sulfide carrier protein (10, ThiS–COO−) is first activated by conversion to its acyl adenylate 16. This then reacts with the persulfide of the IscS protein to form 19, which is then reduced to the sulfide carrier protein thiocarboxylate (4, ThiS–COS−). This mechanism is supported by the reconstitution of the individual reactions, by the detection of the ThiS acyl adenylate intermediate and by structural characterization of ThiF complexed to ATP or to ThiS–COO−.3,9–15 The structural studies reveal that ThiS–COO− has a similar fold to ubiquitin. This, coupled with the functional similarity between ThiF and the ubiquitin-activating system suggests that ThiF/ThiS and related systems may have been the bacterial ancestors of the ubiquitinating system.11 The mechanism for the formation of the IscS persulfide, an intermediate also involved in the biosynthesis of iron sulfur clusters, is shown in Fig. 3. Imine formation between the active site PLP and cysteine followed by tautomerization gives 21. Sulfur transfer to the active site cysteine generates the IscS persulfide 18. Protonation of the resonance-stabilized carbanion 22 followed by tautomerization and transimination completes the reaction.16
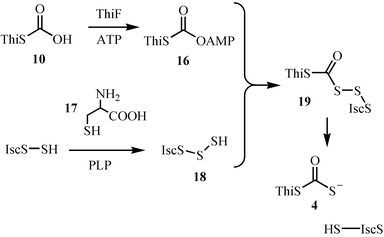
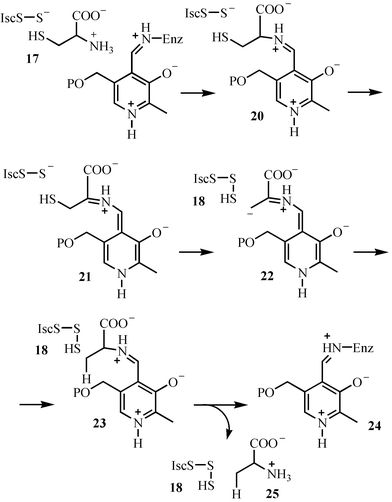 | ||
| Fig. 3 Mechanistic proposal for the formation of the IscS-persulfide 18. | ||
The use of sulfide carrier proteins is an emerging biosynthetic motif that has been experimentally confirmed for the biosynthesis of molybdopterin17–19 and in a new cysteine biosynthetic pathway.20 This motif is also likely to occur, based on sequence analysis, in the biosynthesis of the quinolobactin and pyridine bisthiocarboxylate siderophores.21,22
The biosynthesis of the pyrimidine moiety of thiamin (49, HMP) involves a remarkable rearrangement of aminoimidazole ribotide 30. To the best of our knowledge, this is the most complex unsolved rearrangement in primary metabolism. Only a single gene has been identified by mutagenesis studies (ThiC). The results of comprehensive carbon and hydrogen labeling studies, carried out in a cell-free system, are shown in Fig. 4.23
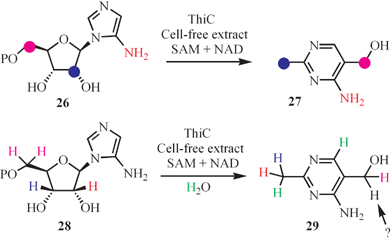 | ||
| Fig. 4 Labeling studies mapping out the conversion of aminoimidazole to the thiamin pyrimidine. | ||
A mechanism that is consistent with the results of this labeling study is outlined in Fig. 5. In this mechanism, dephosphorylation of AIR (30) followed by oxidation of the resulting alcohol gives aldehyde 31. N–glycosyl bond cleavage followed by a retroaldol reaction gives 33, 34, and 35. Loss of C1 as formate followed by tautomerization of ene-diols 35 and 37 generates the C5- and C2-containing carbon fragments required for the assembly of pyrimidine 49. Electrophilic substitution at C2 of the aminoimidazole by 40 gives 41. Loss of water to generate 42 followed by a hydride shift gives 43. Loss of formate from 43 completes the biogenesis of the methyl group. The next set of steps address the mechanism of the ring expansion reaction. Addition of aldehyde 39 to C4 of the aminoimidazole gives 45. Loss of water to form cation 46 followed by a 1,2-shift gives 47. Aromatization of 47 completes the formation of HMP (49). While this mechanism is consistent with the labeling pattern, it is not particularly elegant, and we believe that the underlying chemistry involved in the formation of the pyrimidine moiety of thiamin still eludes us.
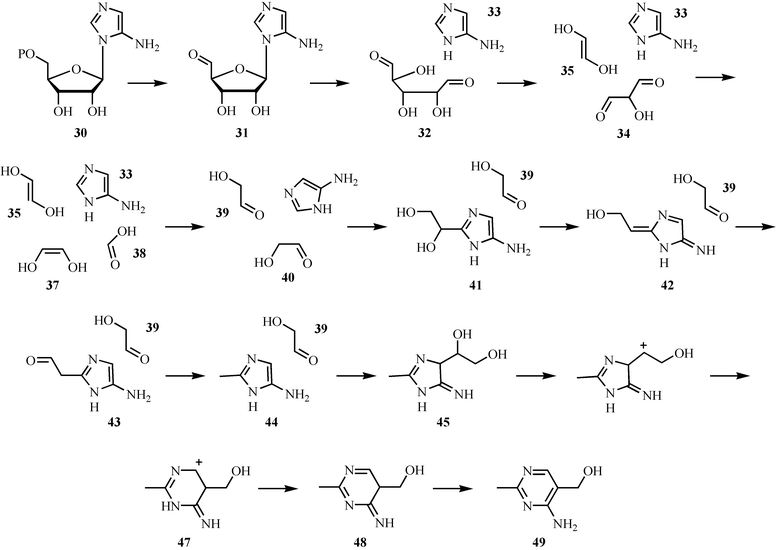 | ||
| Fig. 5 Mechanistic proposal for the formation of the pyrimidine moiety (49) of thiamin pyrophosphate. | ||
The biosynthesis of the thiamin thiazole and pyrimidine in yeast occurs by a different route, and there are no orthologs of the corresponding bacterial genes in this organism. Labeling studies demonstrate that the thiazole is derived from glycine, sulfide and an unidentified C5 sugar,24–29 and that the pyrimidine is formed from PLP and histidine (Fig. 6).30 The mechanistic enzymology of these remarkable transformations is still at a very early stage of investigation.
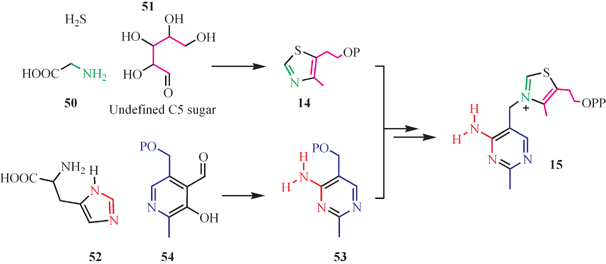 | ||
| Fig. 6 The labeling pattern for thiamin biosynthesis in yeast. | ||
NAD biosynthesis: formation of the pyrimidine ring
Two routes to quinolinic acid (58), the precursor to the pyridine ring of NAD, have been identified: one involving the oxidation of hydroxyanthranilate (55), the other involving the condensation of the imine of aspartic acid 56 with dihydroxyacetone phosphate 57 (Fig. 7).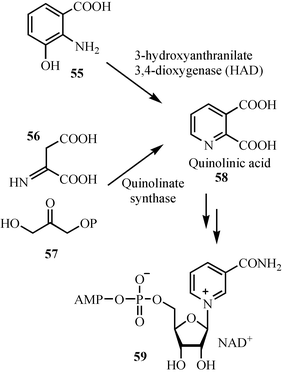 | ||
| Fig. 7 The two pathways for the biosynthesis of the pyridine ring of NAD. | ||
The mechanism for the oxidation of hydroxyanthranilate 55 to quinolinic acid 58 is now relatively well understood (Fig. 8). In this mechanism, the substrate binds to the active site iron as a bidentate ligand. Electron transfer to oxygen gives 61, which can also be written as the biradical resonance form 62. Radical coupling followed by Criegee rearrangement gives 64. Ester hydrolysis completes the reaction. This mechanism is supported by a structure of the enzyme with a bound substrate analog, by mutagenesis studies31,32 as well as extensive mechanistic characterization of closely related enzymes.33–37
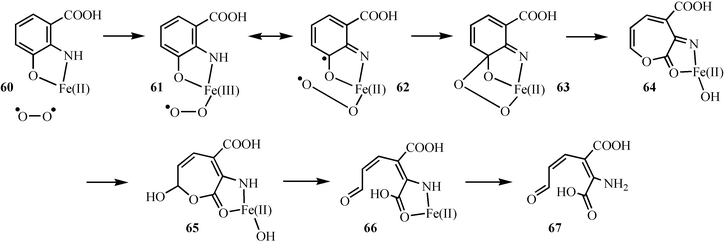 | ||
| Fig. 8 Mechanistic proposal for the oxidation of hydroxyanthranilic acid 55 to 2-amino-3-carboxymuconic acid semialdehyde 67. | ||
The conversion of 2-amino-3-carboxymuconic acid semialdehyde 67 to quinolinic acid 58 does not require enzymatic catalysis. A mechanistic proposal for this reaction, based primarily on the mechanistic characterization of the Zinke reaction, is shown in Fig. 9. In this mechanism, 67 undergoes a facile isomerization reaction to give 69, which undergoes an electrocyclic ring closure to 70. Dehydration completes the reaction.38
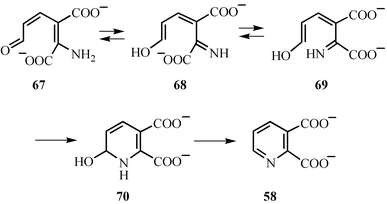 | ||
| Fig. 9 Mechanistic proposal for the formation of quinolinic acid from 2-amino-3-carboxymuconic acid semialdehyde 67. | ||
The second route to quinolinic acid is still poorly characterized, and the enzyme involved has only recently been reconstituted.39 It contains an iron sulfur cluster and a structure of the apoenzyme has recently been solved.40 No mechanistic studies have yet been reported.
Flavin adenine dinucleotide biosynthesis: formation of the isoalloxazine heterocycle
The formation of the isoalloxazine ring of riboflavin 79 by the dismutation of 6,7-dimethyl-8-ribityllumazine 71 is one of the most remarkable reactions in cofactor biosynthesis.41,42 The current mechanistic proposal for this reaction is outlined in Fig. 10. Deprotonation of one molecule of dimethyl ribityllumazine 71 and water addition to a second molecule gives 72 and 73 respectively. Nucleophilic addition of 72 to 73 gives 74, which tautomerizes to 75. Loss of water and a second tautomerization gives 76, which then cyclizes to 77. Two elimination reactions complete the formation of riboflavin 79. This proposal is consistent with the observed regiochemistry of riboflavin formation from labeled 6,7-dimethyl-8-ribityllumazine,43,44 and is also supported by the demonstrated acidity of the C7a methyl group (pKa = 8.5),45 the susceptibility of C7 to nucleophilic addition,46–49 and by the trapping of intermediate 77 using the S41A mutant of the E. coli riboflavin synthase.43,50 Remarkably, this complex reaction can also occur under mild conditions in the absence of the enzyme.51–53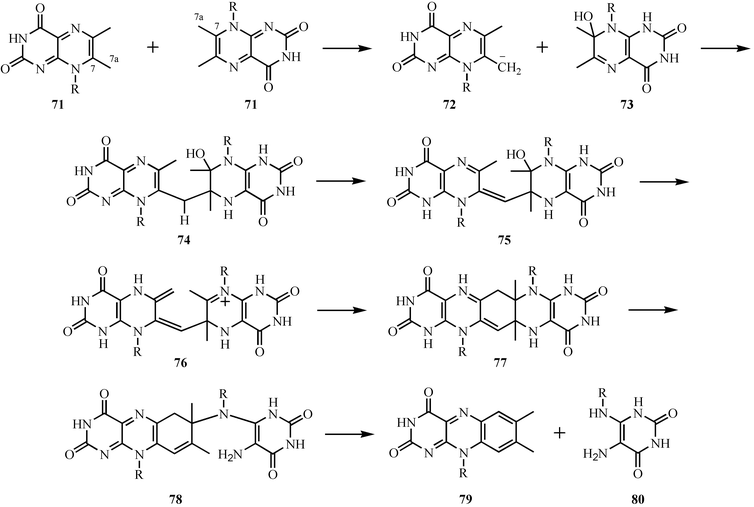 | ||
| Fig. 10 Mechanistic proposal for the formation of riboflavin. | ||
Pyridoxal phosphate biosynthesis: formation of the pyridine ring
The key reaction for the formation of the pyridine ring of pyridoxal phosphate (PLP) in E. coli involves the condensation of 1-deoxy-D-xylulose 5-phosphate 81 and 3-amino-1-hydroxyacetone-1-phosphate 82.54 The mechanism of this reaction is shown in Fig. 11. Imine formation followed by elimination of water gives 84. Phosphate hydrolysis by a surprising elimination–addition sequence gives 87. An aldol condensation to 88 followed by elimination of water and a final tautomerization gives pyridoxol phosphate 90. This mechanism is supported by structural studies and isotopic labeling experiments.55–57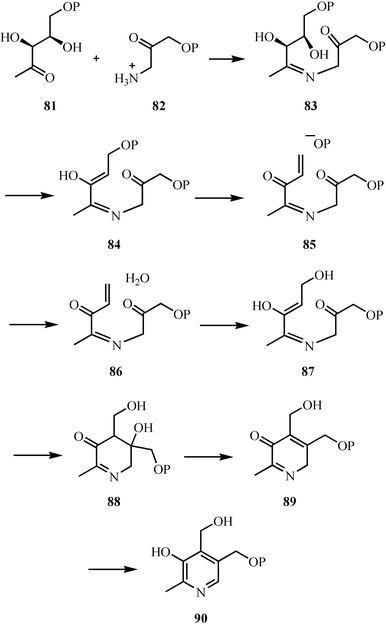 | ||
| Fig. 11 Mechanistic proposal for the formation of the pyridine ring of pyridoxal phosphate in E. coli. | ||
The complexity of PLP biosynthesis in E. coli is remarkable (7 enzymes). Much of this complexity arises during the biosynthesis of 3-amino-1-hydroxyacetone 1-phosphate 82, which requires four enzymes. This is surprising, because in principle this amine could be made in one step by the amination of glyceraldehyde-3-phosphate. A much more direct route to PLP that involves the condensation of ammonia, glyceraldehyde phosphate 92 and ribulose phosphate 91 has recently been identified (Fig. 12).58,59 This reaction is catalyzed by a single enzyme, which has now been structurally characterized.60,61 Comparative genome analysis indicates that this shorter route to PLP is the most commonly used pathway.62
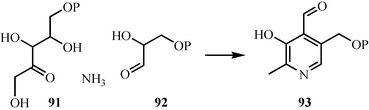 | ||
| Fig. 12 A new biosynthetic route to pyridoxal phosphate. | ||
Biotin biosynthesis: formation of the tetrahydrothiophene ring
The last step in biotin biosynthesis has fascinated enzymologists for more than four decades, and substantial progress with the mechanistic studies has now been achieved (Fig. 13).63,64 Biotin synthase generates the deoxyadenosyl radical 95 by the reductive cleavage of S-adenosylmethionine 94.65–67 This radical abstracts a hydrogen atom from the methyl group of dethiobiotin 97 to give 98, which then adds to one of the sulfurs of a [2Fe–2S] cluster to give 99. A second hydrogen atom abstraction generates 100, which again adds to the iron–sulfur cluster to give 101. Dissociation of biotin 102 disrupts the cluster, and so far all efforts to achieve multiple turnovers of this enzyme in vitro have failed.68 | ||
| Fig. 13 Mechanistic proposal for the C–S bond forming steps in biotin biosynthesis. A) Formation of the deoxyadenosyl radical. B) Mechanistic proposal for the insertion of sulfur into dethiobiotin. | ||
This proposal is supported by the identification of 5′-deoxyadenosine as a reaction product, by the demonstration that the sulfur of biotin is derived from the iron–sulfur cluster, by spectroscopic characterization of the iron sulfur clusters, and most recently by a structure of the enzyme complexed to SAM 94 and dethiobiotin 97.69,70–77
Vitamin B12 biosynthesis: ring contraction and axial ligand formation
The mechanism for the ring contraction/methylation reactions involved in the conversion of precorrin-3A 103 to precorrin-6A 111 is outlined in Fig. 14.78–80 Hydroxylation of 103 gives 104, which then cyclizes to 105.81 Methylation of the D-ring pyrrole followed by opening of the lactone gives carbocation 107. A 1→2 shift gives 108. Methylation of the C-ring pyrrole followed by tautomerization gives 110. Addition of water to the acetyl group, loss of acetic acid, methylation of the resulting resonance-stabilized carbanion and a final tautomerization gives precorrin-6A 111. Many mechanistic details of these reactions remain to be experimentally confirmed.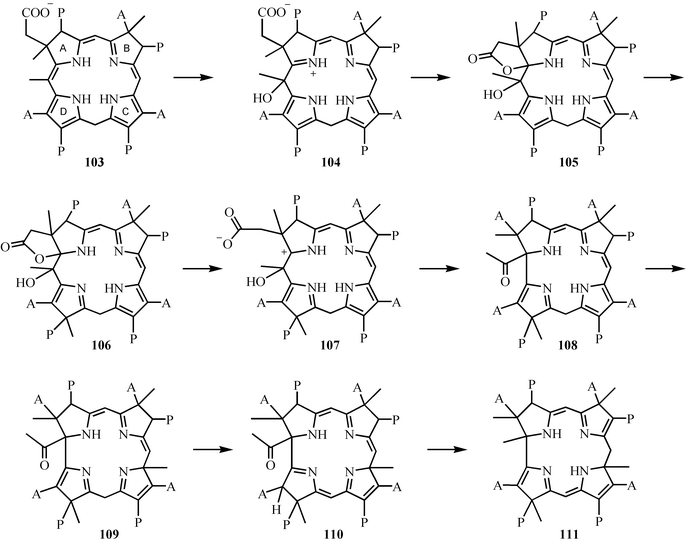 | ||
| Fig. 14 The conversion of precorrin-3A 103 to precorrin-6A 111. | ||
The formation of the dimethylbenzimidazole ligand of vitamin B12 from riboflavin is also of mechanistic interest.82–84 None of the enzymes involved have yet been characterized. A mechanistic proposal is outlined in Fig. 15. In this proposal, the pyrimidine moiety of riboflavin is removed by two consecutive hydrolysis reactions to give 116 which undergoes a facile nonenzymatic oxidative cyclization.85 This cyclization reaction involves the oxidation of the diaminobenzene 116 to the bisimine 118. Tautomerization followed by cyclization gives 120. A second two-electron oxidation followed by a retroaldol reaction gives dimethylbenzimidazole 122. Several analogous oxidative cascades have been identified.86
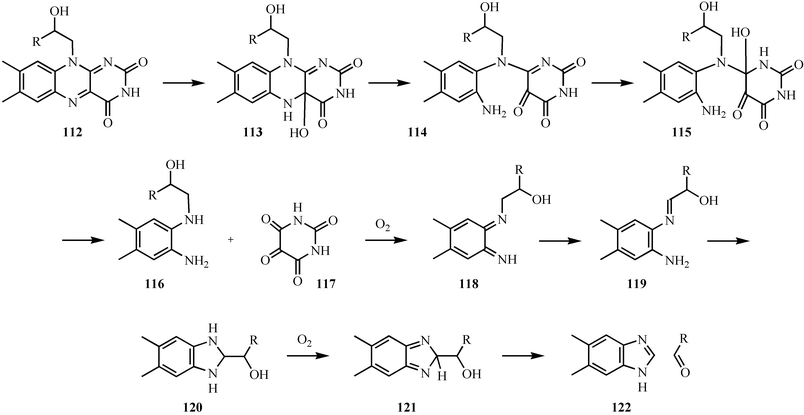 | ||
| Fig. 15 Mechanistic proposal for the formation of the dimethylbenzimidazole ligand of vitamin B12122. | ||
Heme biosynthesis: the formation of uroporphyrinogen III
The cyclization of hydroxymethylbilane 123 to uroporphyrinogen III 129 involves a remarkable isomerization of the D-ring pyrrole.87 This reaction is located at the branch point where heme, vitamin B12 and chlorophyll biosynthesis diverge. A possible mechanism is outlined in Fig. 16. Loss of water from 123 generates carbocation 124, which adds to the D-ring pyrrole as indicated to give 125. Reversal of the electrophilic addition with cleavage of the bond linking the C- and D-ring pyrroles gives 127, which then undergoes a normal electrophilic substitution reaction to give uroporphyrinogen III 129. This mechanism is supported by the inhibition of the enzyme with an analog of 125.88 While the structure of uroporphyrinogen III synthase has been solved, no structures of the enzyme complexed to a substrate analog have yet been reported.89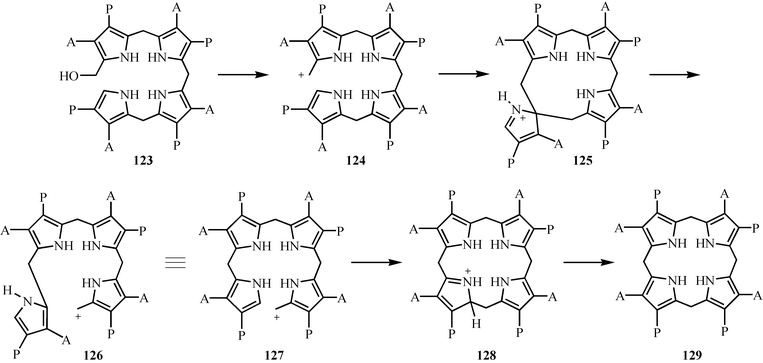 | ||
| Fig. 16 Mechanistic proposal for the formation of uroporphyrinogen III 129. | ||
Coenzyme A biosynthesis: the decarboxylation of phosphopantothenoylcysteine
The decarboxylation of phosphopantothenoylcysteine 130 was a puzzling reaction for a long time, because it was not clear how the carbanion intermediate could be stabilized.90 The identification of this enzyme as a flavoenzyme clarified this, and a mechanistic proposal is outlined in Fig. 17. Oxidation of phosphopantothenoylcysteine 130 to the thioaldehyde 131 generates the functionality capable of stabilizing the carbanion. Decarboxylation gives ene-thiolate 132, which tautomerizes to the thioaldehyde 133. Reduction of this by the reduced flavin completes the reaction. This proposal is supported by isotope-effect studies, a mechanism-based inactivation, and by an informative structure of the enzyme ene-thiolate complex.91–95 Phosphopantothenoylcysteine decarboxylase is one of the key gate-keepers in coenzyme A biosynthesis, preventing the incorporation of serine into CoA, and thus preventing the formation of toxic oxo-CoA. The selectivity for cysteine over serine is due to the greater difficulty of oxidizing an alcohol relative to a thiol.96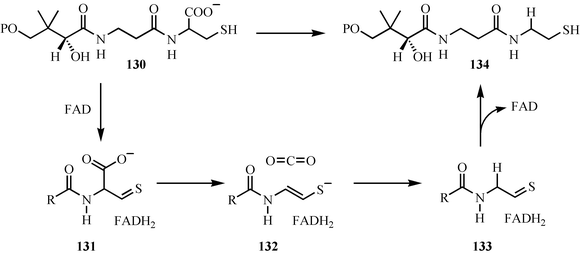 | ||
| Fig. 17 Mechanistic proposal for the decarboxylation of phosphopantothenoylcysteine. | ||
Cofactor biosynthesis by autocatalytic post-translational modification
Cofactor assembly by autocatalytic post-translational modification of proteins is an important emerging motif in cofactor biosynthesis, and a growing number of enzymes in which a cluster of active site residues undergo an autocatalytic reaction to generate a new cofactor have now been identified.97,98 Here we will look at two examples of this process.The 5-methylene-3,5-dihydroimidazol-4-one cofactor 138 functions as a finely tuned electrophile that catalyzes the elimination of ammonia from histidine (histidine ammonia lyase) and phenylalanine (phenylalanine ammonia lyase), potentially by carrying out an electrophilic addition to the aromatic ring of the aminoacid. The proposed biosynthesis of this novel cofactor is outlined in Fig. 18, and involves the addition of an amide nitrogen to an adjacent carbonyl to generate 136 which is converted to the cofactor by the elimination of two molecules of water.99
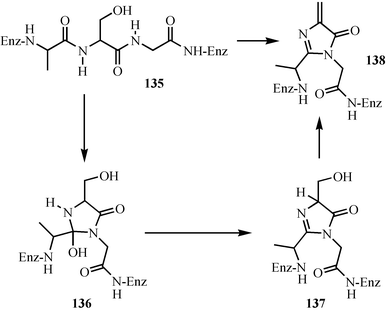 | ||
| Fig. 18 Mechanistic proposal for the biosynthesis of the 5-methylene-3,5-dihydroimidazol-4-one cofactor of phenylalanine and histidine ammonia lyase. | ||
Topaquinone is a cofactor used by some amine oxidases.98 A mechanistic proposal for its formation is outlined in Fig. 19. In this proposal, oxygen binds to the active site copper to give the copper(III) superoxide complex 140, which then oxidizes the active site tyrosine to give 141. Radical coupling followed by aromatization gives 143. Cleavage of the oxygen–oxygen bond followed by catechol oxidation gives quinone 145. Addition of water to this quinone, followed by aromatization, gives 147, which is then oxidized to topaquinone 150.
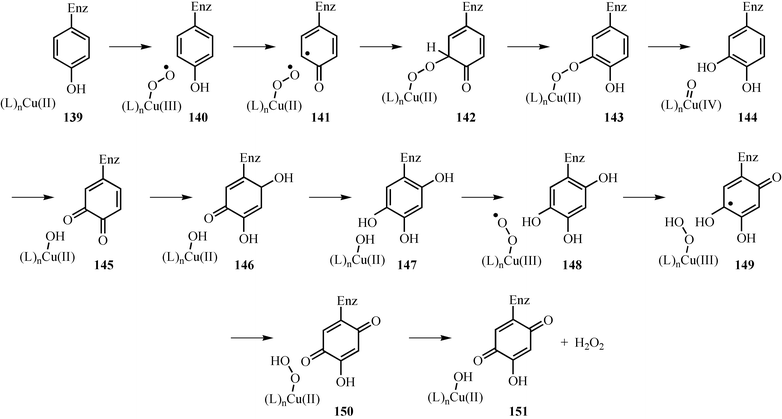 | ||
| Fig. 19 Mechanistic proposal for the formation of the topaquinone cofactor. | ||
Evolution of cofactor biosynthetic pathways
Most of the cofactors contain heterocyclic systems that can be formed in the absence of enzymes, and analogs of the pyridine, pyrimidine, thiazole, isoalloxazine and heme heterocyclic systems discussed here can all be readily assembled non-enzymatically. This suggests that cofactor formation in the early biosphere may have been non-enzymatic and that as life proliferated, these cofactors became growth-limiting substances, at which point cells evolved cofactor biosynthetic pathways. This hypothesis is consistent with the following three interesting features of cofactor biosynthesis.1) If uroporphyrinogen was initially synthesized non-enzymatically by the condensation of 3,4-heterodisubstituted pyrroles with aldehydes, the most abundant uroporphorinogen isomer in the resulting statistical mixture would have the XY–XY–XY–YX regiochemistry. As cells would preferentially use this isomer, the biosynthetic pathway had to evolve chemistry to flip the D-ring pyrrole, hence the surprising chemistry catalyzed by uroporphyrinogen III synthase.
2) Several of the cofactor biosynthetic pathways use an enzyme that requires the final cofactor. For example: DXP synthase, an enzyme on the thiamin biosynthetic pathway, uses thiamin pyrophosphate (1, Fig. 1) as a cofactor; acetylCoA carboxylase, an enzyme on the biotin biosynthetic pathway, uses biotin as a cofactor; and the formation of 3-amino-1-hydroxyacetone 1-phosphate 82 (Fig. 11), an intermediate on the PLP biosynthetic pathway, requires PLP. These observations are consistent with the proposal that some form of the cofactor existed before the evolution of the biosynthetic pathway.
Since cofactors are used catalytically, evolutionary pressure towards pathway optimization is probably weak. In support of this, many of the enzymes involved in cofactor biosynthesis are slow, and the chemistry used is much more complex than that found for the biosynthesis of other primary metabolites. This lack of evolutionary pressure might explain the very unusual chemistry used for the biosynthesis of the pyrimidine moiety of thiamin, given that closely related pyrimidines can be readily biosynthesized much more efficiently from cytosine. A similar explanation might explain the rather inefficient biosynthesis of PLP in E. coli.
Acknowledgements
I would like to thank my capable coworkers for their tireless efforts over the past decade in exploring the chemistry of cofactor biosynthesis. Their individual contributions are cited in the references. Our research was also greatly enhanced by collaborations with Steve Ealick and Fred McLafferty on the structure and mass spectrometry of the cofactor biosynthetic enzymes. The Cornell component of the described research was funded by NIH grants DK44083 and GM069618.References
- Cofactor biosynthesis: a mechanistic perspective (‘Vitamins and Hormones’, vol. 61), ed. T. P. Begley, Academic Press, San Diego, CA, 2001 Search PubMed.
- E. C. Settembre, P. C. Dorrestein, J.-H. Park, A. M. Augustine, T. P. Begley and S. E. Ealick, Biochemistry, 2003, 42, 2971–2981 CrossRef CAS.
- P. C. Dorrestein, H. Zhai, F. W. McLafferty and T. P. Begley, Chem. Biol., 2004, 11, 1373–1381 CrossRef CAS.
- E. C. Settembre, P. C. Dorrestein, H. Zhai, A. Chatterjee, F. W. McLafferty, T. P. Begley and S. E. Ealick, Biochemistry, 2004, 43, 11647–11657 CrossRef CAS.
- P. C. Dorrestein, H. Zhai, S. V. Taylor, F. W. McLafferty and T. P. Begley, J. Am. Chem. Soc., 2004, 126, 3091–3096 CrossRef CAS.
- J.-H. Park, P. C. Dorrestein, H. Zhai, C. Kinsland, F. W. McLafferty and T. P. Begley, Biochemistry, 2003, 42, 12430–12438 CrossRef CAS.
- R. Leonardi, S. A. Fairhurst, M. Kriek, D. J. Lowe and P. L. Roach, FEBS Lett., 2003, 539, 95–99 CrossRef CAS.
- T. P. Begley, J. Xi, C. Kinsland, S. Taylor and F. McLafferty, Curr. Opin. Chem. Biol., 1999, 3, 623–629 CrossRef CAS.
- C. Kinsland, S. V. Taylor, N. L. Kelleher, F. W. McLafferty and T. P. Begley, Protein Sci., 1998, 7, 1839–1842 CAS.
- S. V. Taylor, N. L. Kelleher, C. Kinsland, H.-J. Chiu, C. A. Costello, A. D. Backstrom, F. W. McLafferty and T. P. Begley, J. Biol. Chem., 1998, 273, 16555–16560 CrossRef CAS.
- C. Wang, J. Xi, T. P. Begley and L. K. Nicholson, Nat. Struct. Biol., 2001, 8, 47–51 CrossRef CAS.
- J. Xi, Y. Ge, C. Kinsland, F. W. McLafferty and T. P. Begley, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 8513–8518 CrossRef CAS.
- C. T. Lauhon and R. Kambampati, J. Biol. Chem., 2000, 275, 20096–20103 CrossRef CAS.
- D. M. Duda, H. Walden, J. Sfondouris and B. A. Schulman, J. Mol. Biol., 2005, 349, 774–786 CrossRef CAS.
- C. Lehmann, T. P. Begley and S. E. Ealick, Biochemistry, 2006, 45, 11–19 CrossRef CAS.
- L. Zheng, R. H. White, V. L. Cash and D. R. Dean, Biochemistry, 1994, 33, 4714–4720 CrossRef CAS.
- S. Leimkuehler, A. Freuer, J. A. S. Araujo, K. V. Rajagopalan and R. R. Mendel, J. Biol. Chem., 2003, 278, 26127–26134 CrossRef CAS.
- M. M. Wuebbens and K. V. Rajagopalan, J. Biol. Chem., 2003, 278, 14523–14532 CrossRef CAS.
- M. J. Rudolph, M. M. Wuebbens, O. Turque, K. V. Rajagopalan and H. Schindelin, J. Biol. Chem., 2003, 278, 14514–14522 CrossRef CAS.
- K. E. Burns, S. Baumgart, P. C. Dorrestein, H. Zhai, F. W. McLafferty and T. P. Begley, J. Am. Chem. Soc., 2005, 127, 11602–11603 CrossRef CAS.
- S. Matthijs, C. Baysse, N. Koedam, K. A. Tehrani, L. Verheyden, H. Budzikiewicz, M. Schaefer, B. Hoorelbeke, J.-M. Meyer, H. De Greve and P. Cornelis, Mol. Microbiol., 2004, 52, 371–384 CrossRef CAS.
- T. A. Lewis, M. S. Cortese, J. L. Sebat, T. L. Green, C.-H. Lee and R. L. Crawford, Environ. Microbiol., 2000, 2, 407–416 CrossRef CAS.
- B. G. Lawhorn, R. A. Mehl and T. P. Begley, Org. Biomol. Chem., 2004, 2, 2538–2546 RSC.
- K. Tazuya, M. Tanaka, M. Morisaki, K. Yamada and H. Kumaoka, Biochem. Int., 1987, 14, 769–777 Search PubMed.
- K. Tazuya, K. Yamada, K. Nakamura and H. Kumaoka, Biochim. Biophys. Acta, 1987, 924, 210–215 CrossRef CAS.
- K. Tazuya, M. Morisaki, K. Yamada, H. Kumaoka and K. Saiki, Biochem. Int., 1987, 14, 153–160 Search PubMed.
- R. L. White and I. D. Spenser, Biochem. J., 1979, 179, 315–325 CAS.
- R. L. White and I. D. Spenser, J. Am. Chem. Soc., 1979, 101, 5102–5104 CrossRef CAS.
- R. L. White and I. D. Spenser, J. Am. Chem. Soc., 1982, 104, 4934–4943 CrossRef CAS.
- J. Zeidler, B. G. Sayer and I. D. Spenser, J. Am. Chem. Soc., 2003, 125, 13094–13105 CrossRef CAS.
- Y. Zhang, K. L. Colabroy, T. P. Begley and S. E. Ealick, Biochemistry, 2005, 44, 7632–7643 CrossRef CAS.
- K. L. Colabroy, H. Zhai, T. Li, Y. Ge, Y. Zhang, A. Liu, S. E. Ealick, F. W. McLafferty and T. P. Begley, Biochemistry, 2005, 44, 7623–7631 CrossRef CAS.
- S. Mendel, A. Arndt and T. D. H. Bugg, Biochemistry, 2004, 43, 13390–13396 CrossRef CAS.
- T. D. H. Bugg and C. J. Winfield, Nat. Prod. Rep., 1998, 15, 513–530 RSC.
- T. D. H. Bugg and G. Lin, Chem. Commun., 2001, 941–952 RSC.
- S. L. Groce and J. D. Lipscomb, Biochemistry, 2005, 44, 7175–7188 CrossRef CAS.
- S. L. Groce, M. A. Miller-Rodeberg and J. D. Lipscomb, Biochemistry, 2004, 43, 15141–15153 CrossRef CAS.
- K. L. Colabroy and T. P. Begley, J. Am. Chem. Soc., 2005, 127, 840–841 CrossRef CAS.
- R. M. Cicchillo, L. Tu, J. A. Stromberg, L. M. Hoffart, C. Krebs and S. J. Booker, J. Am. Chem. Soc., 2005, 127, 7310–7311 CrossRef CAS.
- H. Sakuraba, H. Tsuge, K. Yoneda, N. Katunuma and T. Ohshima, J. Biol. Chem., 2005, 280, 26645–26648 CrossRef CAS.
- M. Fischer, W. Roemisch, B. Illarionov, W. Eisenreich and A. Bacher, Biochem. Soc. Trans., 2005, 33, 780–784 CrossRef CAS.
- M. Fischer and A. Bacher, Nat. Prod. Rep., 2005, 22, 324–350 RSC.
- B. Illarionov, W. Eisenreich and A. Bacher, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 7224–7229 CrossRef CAS.
- M. Fischer, A.-K. Schott, W. Roemisch, A. Ramsperger, M. Augustin, A. Fidler, A. Bacher, G. Richter, R. Huber and W. Eisenreich, J. Mol. Biol., 2004, 343, 267–278 CrossRef CAS.
- W. Pfleiderer and W. Hutzenlaub, Chem. Ber., 1973, 106, 3149–3174 CrossRef CAS.
- R. L. Beach and G. W. E. Plaut, J. Org. Chem., 1971, 36, 3937–3943 CrossRef CAS.
- R. Beach and G. W. E. Plaut, Biochemistry, 1970, 9, 760–770 CrossRef CAS.
- D. H. Bown, P. J. Keller, H. G. Floss, H. Sedlmaier and A. Bacher, J. Org. Chem., 1986, 51, 2461–2467 CrossRef CAS.
- W. Pfleiderer, R. Mengel and P. Hemmerich, Chem. Ber., 1971, 104, 2273–2292 CrossRef CAS.
- B. Illarionov, I. Haase, M. Fischer, A. Bacher and N. Schramek, Biol. Chem., 2005, 386, 127–136 CrossRef CAS.
- T. Rowan and H. C. S. Wood, Proc. Chem. Soc., 1963, 21–22 Search PubMed.
- T. Rowan and H. C. S. Wood, J. Chem. Soc. C, 1968, 452–458 RSC.
- R. Beach and G. W. Plaut, Tetrahedron Lett., 1969, 40, 3489–3492 CrossRef CAS.
- D. E. Cane, Chimia, 2003, 57, 75–76 CAS.
- D. E. Cane, S. Du and I. D. Spenser, J. Am. Chem. Soc., 2000, 122, 4213–4214 CrossRef CAS.
- J. I. Yeh, S. Du, E. Pohl and D. E. Cane, Biochemistry, 2002, 41, 11649–11657 CrossRef CAS.
- M. Garrido-Franco, B. Laber, R. Huber and T. Clausen, J. Mol. Biol., 2002, 321, 601–612 CrossRef CAS.
- K. E. Burns, Y. Xiang, C. L. Kinsland, F. W. McLafferty and T. P. Begley, J. Am. Chem. Soc., 2005, 127, 3682–3683 CrossRef CAS.
- T. Raschle, N. Amrhein and T. B. Fitzpatrick, J. Biol. Chem., 2005, 280, 32291–32300 CrossRef CAS.
- J. Zhu, J. W. Burgner, E. Harms, B. R. Belitsky and J. L. Smith, J. Biol. Chem., 2005, 280, 27914–27923 CrossRef CAS.
- J. A. Bauer, E. M. Bennett, T. P. Begley and S. E. Ealick, J. Biol. Chem., 2004, 279, 2704–2711 CAS.
- G. Mittenhuber, J. Mol. Microbiol. Biotechnol., 2001, 3, 1–20 CAS.
- J. T. Jarrett, Arch. Biochem. Biophys., 2005, 433, 312–321 CrossRef CAS.
- M. Lotierzo, B. T. S. Bui, D. Florentin, F. Escalettes and A. Marquet, Biochem. Soc. Trans., 2005, 33, 820–823 CrossRef CAS.
- G. Layer, D. W. Heinz, D. Jahn and W.-D. Schubert, Curr. Opin. Chem. Biol., 2004, 8, 468–476 CrossRef CAS.
- E. N. G. Marsh, A. Patwardhan and M. S. Huhta, Bioorg. Chem., 2004, 32, 326–340 CrossRef CAS.
- P. A. Frey and O. T. Magnusson, Chem. Rev., 2003, 103, 2129–2148 CrossRef CAS.
- E. Choi-Rhee and J. E. Cronan, Chem. Biol., 2005, 12, 461–468 CrossRef CAS.
- F. Berkovitch, Y. Nicolet, J. T. Wan, J. T. Jarrett and C. L. Drennan, Science, 2004, 303, 76–80 CrossRef CAS.
- G. N. L. Jameson, M. Mader Cosper, H. L. Hernandez, M. K. Johnson and B. H. Huynh, Biochemistry, 2004, 43, 2022–2031 CrossRef CAS.
- N. B. Ugulava, K. K. Surerus and J. T. Jarrett, J. Am. Chem. Soc., 2002, 124, 9050–9051 CrossRef CAS.
- N. B. Ugulava, C. J. Sacanell and J. T. Jarrett, Biochemistry, 2001, 40, 8352–8358 CrossRef CAS.
- N. B. Ugulava, B. R. Gibney and J. T. Jarrett, Biochemistry, 2001, 40, 8343–8351 CrossRef CAS.
- S. Ollagnier-de Choudens, Y. Sanakis, K. S. Hewitson, P. Roach, J. E. Baldwin, E. Muenck and M. Fontecave, Biochemistry, 2000, 39, 4165–4173 CrossRef CAS.
- N. B. Ugulava, C. J. Sacanell and J. T. Jarrett, Biochemistry, 2001, 40, 8352–8358 CrossRef CAS.
- N. B. Ugulava, B. R. Gibney and J. T. Jarrett, Biochemistry, 2001, 40, 8343–8351 CrossRef CAS.
- F. Escalettes, D. Florentin, B. T. S. Bui, D. Lesage and A. Marquet, J. Am. Chem. Soc., 1999, 121, 3571–3578 CrossRef CAS.
- A. I. Scott, J. Org. Chem., 2003, 68, 2529–2539 CrossRef CAS.
- Multiple biosynthetic pathways for vitamin B12: Variations on a central theme (‘Vitamins and Hormones’, vol. 61), ed. C. A. Roessner, P. J. Santander and A. I. Scott, Academic Press, San Diego, CA, 2001, pp. 267–297 Search PubMed.
- D. Heldt, A. D. Lawrence, M. Lindenmeyer, E. Deery, P. Heathcote, S. E. Rigby and M. J. Warren, Biochem. Soc. Trans., 2005, 33, 815–819 CrossRef CAS.
- H. M. McGoldrick, C. A. Roessner, E. Raux, A. D. Lawrence, K. J. McLean, A. W. Munro, S. Santabarbara, S. E. J. Rigby, P. Heathcote, A. I. Scott and M. J. Warren, J. Biol. Chem., 2005, 280, 1086–1094 CAS.
- P. Renz, in: Chemistry and Biochemistry of B12, J. Wiley & Sons, New York, 1999, pp. 557–575 Search PubMed.
- B. Keck, M. Munder and P. Renz, Arch. Microbiol., 1998, 171, 66–68 CrossRef CAS.
- J. A. Horig and P. Renz, Eur. J. Biochem., 1980, 105, 587–592 CrossRef CAS.
- L. A. Maggio-Hall, P. C. Dorrestein, J. C. Escalante-Semerena and T. P. Begley, Org. Lett., 2003, 5, 2211–2213 CrossRef CAS.
- P. Dorrestein and T. P. Begley, Bioorg. Chem., 2005, 33, 136–148 CrossRef CAS.
- A. R. Battersby and F. J. Leeper, Chem. Rev., 1990, 90, 1261–1274 CrossRef CAS.
- M. A. Cassidy, N. Crockett, F. J. Leeper and A. R. Battersby, J. Chem. Soc., Perkin Trans. 1, 1996, 2079–2090 RSC.
- M. A. A. Mathews, H. L. Schubert, F. G. Whitby, K. J. Alexander, K. Schadick, H. A. Bergonia, J. D. Phillips and C. P. Hill, EMBO J., 2001, 20, 5832–5839 CrossRef CAS.
- T. P. Begley and S. E. Ealick, Curr. Opin. Chem. Biol., 2004, 8, 508–515 CrossRef CAS.
- E. Strauss, H. Zhai, L. A. Brand, F. W. McLafferty and T. P. Begley, Biochemistry, 2004, 43, 15520–15533 CrossRef CAS.
- S. Steinbacher, P. Hernandez-Acosta, B. Bieseler, M. Blaesse, R. Huber, F. A. Culianez-Macia and T. Kupke, J. Mol. Biol., 2003, 327, 193–202 CrossRef CAS.
- E. Strauss and T. P. Begley, Bioorg. Med. Chem. Lett., 2003, 13, 339–342 CrossRef CAS.
- T. Kupke, P. Hernandez-Acosta, S. Steinbacher and F. A. Culianez-Macia, J. Biol. Chem., 2001, 276, 19190–19196 CrossRef CAS.
- E. Strauss, C. Kinsland, Y. Ge, F. W. McLafferty and T. P. Begley, J. Biol. Chem., 2001, 276, 13513–13516 CAS.
- E. Strauss and T. P. Begley, ChemBioChem, 2005, 6, 284–286 CrossRef CAS.
- N. M. Okeley and W. A. Van der Donk, Chem. Biol., 2000, 7, R159–R171 CrossRef CAS.
- Mechanisms of biosynthesis of protein-derived redox cofactors (‘Vitamins and Hormones’, vol. 61), ed. B. Schwartz and J. P. Klinman, Academic Press, San Diego, CA, 2001, pp. 219–239 Search PubMed.
- L. Poppe and J. Retey, Angew. Chem., Int. Ed., 2005, 44, 3668–3688 CrossRef CAS.
Footnote |
| † This paper is dedicated to Peter Dervan on the occasion of his 60th birthday. |
| This journal is © The Royal Society of Chemistry 2006 |