Surface modification of nylon 6,6 using a carbene insertion approach
Anton
Blencowe
a,
Kevin
Cosstick
b and
Wayne
Hayes
*a
aThe School of Chemistry, The University of Reading, Whiteknights, Reading, UK RG6 6AD. E-mail: w.c.hayes@reading.ac.uk
bDuPont, P.O. Box 401, Wilton Site, Middlesbrough, Cleveland, UK TS6 8JJ
First published on 17th November 2005
Abstract
The diazirine functionalised fluorenone, 3-[3-(trifluoromethyl)diazirin-3-yl]phenyl-9-oxo-9H-fluorene-2-carboxylate was synthesised to act as a model compound capable of modifying a wide variety of polymeric substrates. Photochemical activation of the diazirine moiety of the fluorenone derivative was utilised to afford highly reactive carbenes capable of insertion into or addition to a wide variety of functionalities. In this paper the photoinduced attachment of a fluorenone derivative to nylon 6,6 has been studied using UV-visible spectroscopic analysis. Incorporation of the fluorenone chromophore onto the backbone of nylon at different loading levels and after different coating cycles has been investigated and is detailed in this paper.
1. Introduction
The development of compounds capable of functionalising multiple substrates has been the subject of intense research. An area that has attached considerable interest is the modification of polymeric substrates with multifunctional dyes and reactants. Included amongst these types of reagents are ‘universal dyes’ which possess high affinities for multiple substrate types. A notable example is the vinyl sulfone azo dye developed1 by Lee et al., which possesses the ability to act as a disperse dye and a reactive dye. When applied to polyester–cotton blends the vinyl sulfone moiety reacts with the nucleophilic groups present in the cotton whilst the dye is also dispersed efficiently into the hydrophobic polyester fibres. Freeman and Sokolowska-Gajda have developed naphtholazo dyes that can act as disperse or ionic dyes if they are converted into their cationic form.2 More recently, Freeman and co-workers have developed a pyrazoline based reactive-disperse dye that has high affinities for polyester, nylon and wool fibres.3 Although such universal dyes have shown significant promise these reagents are still limited to use with particular polymeric substrates as a result of their chemical reactivity and selectivity.An alternative class of reactants that is capable of reacting with almost all organic functionalities are carbenes. The ability of carbenes to react rapidly with any groups within their vicinity, including C–H, O–H, N–H and S–H bonds and C–C double bonds, render these reagents ideal for functionalising organic substrates. As a result of the highly reactive and indiscriminate nature of carbenes they have been applied widely for the modification of substrates to afford biofunctional materials.4 In addition, carbene insertion approaches have also been adopted for the general modification of polymeric materials to afford functionalised substrates with enhanced physical and chemical characteristics.5 Braybrook et al. have developed tosylhydrazone and diazirine functionalised azobenzene dyes,6 which, under photolytic conditions decompose with extrusion of nitrogen to afford carbenes. In preliminary experiments the tosylhydrazone-dye was converted to its sodium salt and then photolysed in methanol and 2,2,2-trifluoroethanol. The reaction in methanol afforded the desired O–H insertion product only in low yield, whereas exclusive formation of the desired ether occurred with 2,2,2-trifluoroethanol. Photolysis of the diazirine-dye in cyclohexane and methanol resulted in low yields of the desired C–H and O–H insertion products, respectively. More recently, Moloney and co-workers have employed carbenes derived from the thermal treatment of diaryldiazomethanes to introduce electron-rich aromatic systems onto the surface of various polymeric substrates, including polystyrene, polythene, nylon, silica, controlled pore glass and poly(tetrafluoroethylene).7 Reaction of the electron-rich aromatic systems introduced onto the substrate surface with diazonium salts resulted in intense coloration which was found to be consistent with initial modification of the polymeric substrate via the carbene insertion approach employed.
In the light of these developments our studies have focused on the development of a functionalised carbene that is capable of reacting in a single step with any substrate type upon exposure to suitable stimuli (for example light). The carbene derivative has been designed so that it can be mixed with the substrate in an inert form as a diazirine. The carbene is then liberated from the diazirine8 upon irradiation. In the present study, the ability of a photoinduced carbene containing fluorenone to react with nylon 6,6 was investigated. The diazirine functionalised dye was mixed initially with nylon 6,6 in different weight to weight ratios and irradiated using a 125 W high pressure mercury vapour lamp. After exhaustive washing, the amount of chromophore incorporated on to the nylon substrate was assessed using UV-visible spectroscopic analysis.
2. Results and discussion
2.1 Synthesis of diazirine functionalised fluorenone 1
The diazirine functionalised fluorenone 1 was synthesised efficiently (87%) from 3-(3-hydroxylphenyl)-3-(trifluoromethyl)diazirine 2 and fluorenone-2-carboxylic acid (Scheme 1) using the coupling reagent N,N′-dicyclohexylcarbodiimide (DCC) and a catalytic amount of 4-(N,N-dimethylamino)pyridine (DMAP).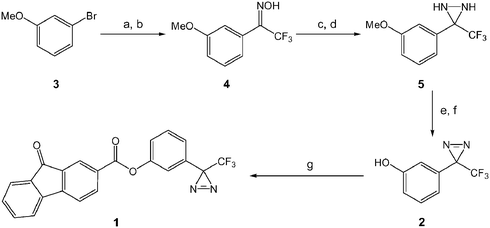 | ||
| Scheme 1 Synthesis of diazirine functionalised fluorenone derivative 1, (a) n-BuLi, Et2NCOCF3 (b) NH2OH·HCl (c) TsCl, NEt3, DMAP (d) NH3, −78 °C (e) Ag2O (f) BBr3 (g) fluorenone-2-carboxylic acid, DCC, DMAP. | ||
The hydroxyl diazirine 2 was constructed in 6-steps (overall yield of 78%) from commercially available 3-bromophenol (Scheme 1) using a method based upon the synthesis of trifluoromethyl aryl diazirines first reported by Brunner et al.9 3-Bromophenol was protected initially as the methyl ether 3 using sodium hydroxide in the presence of iodomethane. Reaction of the methyl ether 3 with n-butyllithium and N,N-diethyltrifluoroacetamide10 afforded the respective trifluoromethyl ketone, 2,2,2-trifluoro-1-(3-methoxyphenyl)ethanone, which was then converted to the corresponding oxime 4via reaction with hydroxylamine hydrochloride. Formation of the diaziridine 5 was achieved by converting the oxime 4 to its O-tosyl derivative, O-tosyl-2,2,2-trifluoro-1-(3-methoxyphenyl)ethanone oxime, which was then reacted with liquid ammonia to yield the diaziridine 5. Oxidation of 5 using Ag2O afforded 3-(trifluoromethyl)-3-(3-methoxyphenyl)diazirine, which was then deprotected with boron tribromide to afford the hydroxyl diazirine 2.
The fluorenone derivative 1 was a bright yellow crystalline solid similar in appearance to that of fluorenone-2-carboxylic acid. UV-visible spectroscopic analysis of 1 in 2,2,2-trichloroethanol revealed a strong absorption band (λmax = 277 nm) with a pronounced shoulder at λ = 304 nm resulting from the conjugated aromatic system of the fluorenone moiety. The adsorption resulting from the π–π* transition of the diazo moiety of the diazirine could be observed as a weak adsorption band centred at λ = 369 nm. The molar absorptivity coefficient of 1 in 2,2,2-trichloroethanol was determined to be 62![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 407 m2 mol−1 at λ = 277 nm.
407 m2 mol−1 at λ = 277 nm.
2.2 Photochemisty of diazirine functionalised fluorenone 1
In order to simulate the reaction between the carbene generated from 1 and nylon 6,6, model reactions were conducted involving representative substrates. Irradiation (using a 125 W high pressure mercury lamp) of the fluorenone 1 in acetic acid, n-butylamine and N-butylbutyramide11 (0.03 M) for a period of 12 hours resulted in high yields (>95%) of the desired insertion products. Photochemical reaction of 1 with acetic acid resulted in exclusive formation of the ester 6 (Scheme 2) as shown by 1H NMR spectroscopic analysis.12 Photolysis of 1 in n-butylamine afforded secondary amines (such as 7, Scheme 2) arising from insertion of carbenes into the amine N–H bonds and C–H insertion products (such as 8, Scheme 2) as revealed by 1H and 19F NMR spectroscopic analysis.13–1519F NMR spectroscopic analysis of the product obtained upon photolysis of 1 in N-butylbutyramide revealed an extensive number of resonances corresponding to the trifluoromethyl group of a complex mixture of products arising from insertion of the carbene into N–H and C–H bonds. In addition, the carbonyl oxygen could have acted as an election donor, reacting with a carbene to initially form an ylide which could react further to afford secondary products.15,16 UV-visible spectroscopic analysis of the crude product mixtures obtained from photochemical reaction of the fluorenone 1 with acetic acid, n-butylamine and N-butylbutyramide exhibited strong adsorption maxima at 277 nm (in 2,2,2-trichloroethanol) corresponding to the fluorenone moiety.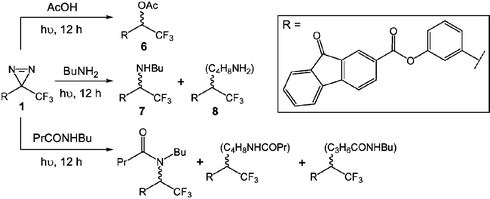 | ||
| Scheme 2 Insertion products generated via photochemical reaction of 1 with acetic acid, n-butylamine and N-butylbutyramide. | ||
The ability of the carbene generated from 1 to insert into the X–H (X = C, O or N) bonds of acetic acid, n-butylamine and N-butylbutyramide demonstrated that, upon photochemical activation, the carbene would be capable of inserting into the chain ends and polymer backbone of nylon 6,6. In such circumstances it would be possible to attach several fluorenone molecules to a single polymer chain.
Photolysis of benzophenone derivatives such as 9-fluorenone can also generate diradical species that are capable of reacting with nylon. To determine if the fluorenone moiety of 1 would react with nylon in this manner, 9-fluorenone was dissolved in cyclohexane and acetic acid (0.15 M) and irradiated for a period of 12 hours. Subsequent removal of the solvent and analysis of the residue via1H NMR spectroscopic analysis revealed that ca. 10% of 9-fluorenone reacted with cyclohexane via hydrogen abstraction (Scheme 3),17 whereas no reaction occurred when acetic acid was used. These results imply that, upon irradiation, the fluorenone carbonyl of 1 could become photo-excited leading to hydrogen abstraction from the alkyl backbone of nylon and the formation of a 9-hydroxyfluorenyl radical (see 9, Scheme 3).18 The radicals produced in this process could then react further to afford a variety of products including alcohols, alkanes, ketones and carboxylic acids or undergo radical–radical coupling.17
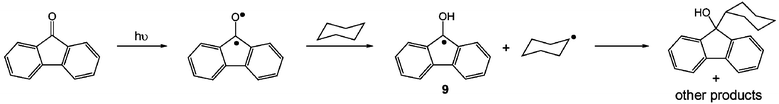 | ||
| Scheme 3 Photochemical reaction of 9-fluorenone with cyclohexane. | ||
2.3 Dyeing of nylon 6,6
As a result of the poor solubility of nylon 6,6 in common organic solvents, attachment of the model fluorenone derivative via photochemical means was conducted in the solid phase. Anhydrous diethyl ether solutions of 1 were added to nylon 6,6 powder (with dimensions varying from 44 to 440 μm) in a 5 mL vial, vortexed and then sonicated under argon. To produce thin layers of the nylon powder suitable for irradiation the solvent was removed from the mixture under reduced pressure (0.1 mbar) at 50 °C, whilst being spun at ca. 400 rpm. The vials were flushed with argon, sealed and placed 3 cm away from a 125 watt high pressure mercury vapour lamp and irradiated for 12 hours. The resultant nylon powder was then washed with chloroform, tetrahydrofuran (THF) and methanol, dried (0.1 mbar) at 40 °C and analysed using UV-visible spectroscopic analysis. To eliminate chemisorption as a possible mechanism of dyeing, blanks were run in parallel using the same procedure without irradiation. In order to eliminate nylon photolytic degradation as a possible source of contaminates that could absorb within the UV-visible region under investigation, nylon powder was irradiated for 72 hours under argon. Samples of nylon powder were removed every 12 hours and analysed via UV-visible spectroscopic analysis. Changes were not observed in the UV-visible spectrum of the nylon powder after each 12 hour period of irradiation and thus it was concluded that the nylon used was stable under the photolytic conditions employed.UV-visible spectroscopic analysis of the modified nylon obtained using different ratios of diazirine to nylon indicated that the fluorenone derivative 1 was incorporated successfully at each of the mixing ratios employed (diazirine∶nylon, 1∶1, 1∶2, 1∶5, 1∶10, 1∶20, 1∶100, 1∶1000). A graph of absorbance at 277 nm versus the mixing ratio (diazirine/nylon) employed revealed an exponential relationship in which the absorbance was found to increase as the mixing ratio increased (Fig. 1, ▲). At mixing ratios of <0.1 (diazirine∶nylon 1∶10) the absorbance of the modified nylon was found to increase steadily with an increase in the mixing ratio. However, at higher mixing ratios (>0.1) the absorbance of the modified nylon only increased slightly with an increase in the mixing ratio, reaching a plateau at a diazirine∶nylon mixing ratio of 1∶2. These results indicate that there is a limit to the loading of the diazirine derivative at which additional chromophore is not incorporated onto the substrate regardless of the amount of diazirine employed. At higher loadings (diazirine∶nylon 1∶1) it is proposed that the carbenes produced from the fluorenone derivative 1 are in close enough proximity to react with other molecules of 1, thus limiting the reaction of the carbene with the nylon substrate.
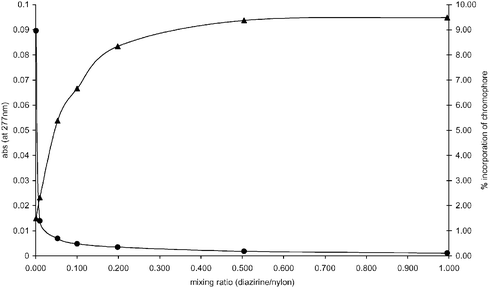 | ||
| Fig. 1 ▲ = Absorbance of functionalised nylon at 277 nm vs. mixing ratio (diazirine/nylon). ● = Percentage incorporation of chromophore vs. mixing ratio of diazirine and nylon (diazirine/nylon). | ||
The percentage of chromophore incorporated into the nylon compared to the amount of fluorenone derivative 1 employed initially is shown in Fig. 1 (●). At high mixing ratios the amount of chromophore incorporated is <1% and was found to increase exponentially as the mixing ratio decreased resulting in ca. 9% incorporation of the chromophore at a diazirine∶nylon mixing ratio of 1∶1000. The results confirm that at high mixing ratios more of the fluorenone derivative 1 is in contact with itself as opposed to the substrate. Thus, at high loadings only a small proportion of the applied fluorenone derivative 1 is in close enough proximity to react with the nylon polymer chains upon photoactivation of the diazirine moiety. In addition, the adsorption of radiation by the fluorenone derivative 1 at the surface of the solid mixture may hinder the activation of the diazirine moiety of other molecules of 1 just below the surface. UV-visible and NMR spectroscopic analyses of the washings obtained when high mixing ratios (e.g. diazirine∶nylon, 1∶1) were employed provided evidence for unreacted diazirine. In addition, a singlet resonance at δF −68.5 ppm in the 19F NMR spectrum19 of the washings revealed the presence of the azine derivative20 (see 10, Fig. 2) of the fluorenone 1. The formation of azine derivatives in the solution phase chemistry of diazirine functionalised azobenzene dyes has also been reported by Braybrook et al.6 At lower diazirine∶nylon mixing ratios (<1∶10) the diazirine 1 was not detected via either UV-visible or 19F NMR spectroscopic analysis of the washings, implying complete decomposition of the diazirine moiety of 1.
 | ||
| Fig. 2 Structure of azine 10. | ||
To determine if the amount of chromophore incorporated after additional dyeing cycles followed a linear relationship the nylon was mixed with the fluorenone derivative 1, irradiated, washed and then the procedure was repeated a total of six times. The modified nylon was analysed via UV-visible spectroscopic analysis after each coating cycle. A graph of absorbance at 277 nm against the coating cycle at four different diazirine∶nylon mixing ratios (1∶1, 1∶10, 1∶100 and 1∶1000) revealed a linear relationship for each mixing ratio employed, in which the absorbance increased with each coating cycle (Fig. 3). In addition to the UV-visible spectroscopic results obtained it was possible to observe a difference in the depth of colour after each coating cycle. For example, after six coating cycles (at a 1∶1 diazirine∶nylon mixing ratio) the colour of the nylon powder had changed from a bright white to a light yellow.
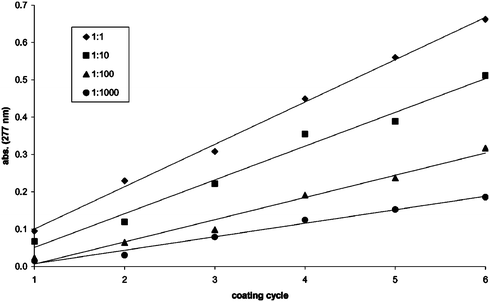 | ||
| Fig. 3 Absorbance of functionalised nylon at 277 nm vs. the coating cycle for the diazirine∶nylon mixing ratios of 1∶1, 1∶10, 1∶100 and 1∶1000, respectively. | ||
3. Conclusions
A diazirine functionalised fluorenone has been synthesised efficiently and employed as a model compound to demonstrate the applicability of carbene insertion processes for the modification of polymeric substrates such as nylon 6,6. Photochemical reaction of the fluorenone derivative with substrates representative of the functional groups present in nylon afforded high yields of carbene insertion products. Solid state photochemical reactions of the fluorenone derivative with nylon were conducted at different diazirine∶nylon mixing ratios and UV-visible spectroscopic analysis of the modified nylon revealed that the fluorenone moiety was incorporated successfully into the nylon and enabled quantification of the amount of chromophore present. In addition, repeating the photo-induced modification cycle resulted in a linear increase in the amount of chromophore incorporated.4. Experimental
Materials
Reagents were purchased from either Lancaster, Acros Chimica or the Aldrich Chemical Company and were used without further purification, unless stated. Diethyl ether and tetrahydrofuran (THF) were distilled from benzophenone and sodium. Dichloromethane was distilled from CaH2. Samples of nylon 6,6 (stabilised with copper halide) were supplied by DuPont. GC grade 2,2,2-trichloroethanol (99.9+%) was used for UV-visible spectroscopic analysis.Instrumentation
Thin-layer chromatography (TLC) was performed on aluminium sheets coated with Merck silica gel 60 F254. Developed TLC plates were stained with potassium permanganate solution or scrutinised under 254 nm UV light. Column chromatography was performed using SI60 Sorbent silica (40–63 μm) supplied from VWR international. Melting points were determined using a Mettler FP61 melting point apparatus and are uncorrected. 1H Nuclear magnetic resonance (NMR) spectroscopy was performed on either a Bruker DPX250 (250 MHz) or a Bruker AMX400 (400 MHz) spectrometer (using TMS and the deuterated solvent as lock and residual solvent). 13C NMR spectroscopy was performed on Bruker AC250 (62.8 MHz) or Bruker AMX400 (100 MHz) spectrometers. 19F NMR spectroscopy was performed on a Bruker DPX250 (235 MHz) spectrometer (using CFCl3 as internal reference). Infrared spectroscopy was performed using a Perkin Elmer 1720-X spectrometer with the samples analysed as thin films. UV-visible analysis was performed on a Perkin Elmer Lambda 25 UV/Vis spectrometer using methanol or 2,2,2-trichloroethanol as the solvent. Mass spectra (MS) were obtained either using a Fisons VG Autospec instrument or Finnigan MAT 95 instrument operating in chemical ionisation mode, using ammonia as the impact gas.Preparation of fluorenone derivative 1
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N); IR νmax/cm−1 (thin film) 739, 1056, 1151, 1188, 1241, 1605, 1713, 1719 and 3070; 1H NMR (250 MHz, CDCl3, TMS) δH 7.07–7.08 (m, 1H, ArH), 7.12 (dquin, J = 1.8, 7.9 Hz, 1H, ArH), 7.33 (ddd, J = 1.0, 2.3, 8.2 Hz, 1H, ArH), 7.42 (td, J = 1.1, 7.4 Hz, 1H, ArH), 7.49 (t, J = 7.9 Hz, 1H, ArH), 7.58 (td, J = 1.2, 7.5 Hz, 1H, ArH), 7.65 (dt, J = 1.0, 7.5 Hz, 1H, ArH), 7.68 (dd, J = 0.6, 7.8 Hz, 1H, ArH), 7.75 (dt, J = 1.0, 7.4 Hz, 1H, ArH), 8.35 (dd, J = 1.6, 7.8 Hz, 1H, ArH), 8.45 (dd, J = 0.6, 1.6 Hz, 1H, ArH) ppm; 13C NMR (62.8 MHz, CDCl3) δC 28.7 (q, J = 40.3 Hz, CN2), 119.9 (ArCH), 120.4 (ArCH), 121.4 (ArCH), 121.5 (q, J = 271.5 Hz, CF3), 123.2 (ArCH), 124.1 (ArCH), 124.8 (ArCH), 125.8 (ArCH), 130.1 (ArCH), 130.2 (ArCC), 130.5 (ArCH), 131.3 (ArCC), 134.8 (ArCC), 135.1 (ArCC), 135.1 (ArCH), 137.0 (ArCH), 143.5 (ArCC), 149.7 (ArCC), 151.4 (CO2), 164.2 (ArCO), 192.8 (CO) ppm; 19F NMR (235 MHz, CDCl3, CFCl3) δF
−65.5 (s, CF3) ppm; CI-MS ([M]+
− N2) calculated for C22H11F3O3: 380.0660, found: 380.0651.
N); IR νmax/cm−1 (thin film) 1154, 1204, 1272, 1355, 1454, 1500, 1588, 1612 and 3334; 1H NMR (250 MHz, CDCl3, TMS) δH 5.00 (s, 1H, OH), 6.67 (m, 1H, ArH), 6.71–6.75 (m, 1H, ArH), 6.85–6.90 (m, 1H, ArH), 7.27 (t, J = 7.9 Hz, 1H, ArH) ppm; 13C NMR (62.8 MHz, CDCl3) δC 28.7 (q, J = 40.5 Hz, CN2), 113.9 (ArCH), 117.2 (ArCH), 119.2 (ArCH), 122.4 (q, J = 274.6 Hz, CF3), 130.6 (ArCH), 131.2 (ArCC), 156.2 (ArCO) ppm; 19F NMR (235 MHz, CDCl3, CFCl3) δF
−65.6 (s, CF3) ppm; CI-MS ([MH]+
− N2) calculated for C8H5F3O: 175.0371, found: 175.0365.
N); IR νmax/cm−1 (thin film) 1154, 1214, 1267, 1355, 1467, 1496, 1584, 1607 and 2962; 1H NMR (250 MHz, CDCl3, TMS) δH 3.73 (s, 3H, OCH3), 6.61 (s, 1H, ArH), 6.68–6.72 (m, 1H, ArH), 6.84–6.89 (m, 1H, ArH), 7.23 (t, J = 8.1 Hz, 1H, ArH) ppm; 13C NMR (62.8 MHz, CDCl3) δC 28.8 (q, J = 40.2 Hz, CN2), 55.7 (OCH3), 112.6 (ArCH), 115.6 (ArCH), 119.1 (ArCH), 122.5 (q, J = 274.7 Hz, CF3), 130.0 (ArCC), 130.4 (ArCH), 160.2 (ArCO) ppm; 19F NMR (235 MHz, CDCl3, CFCl3) δF
−65.6 (s, CF3) ppm; CI-MS ([M]+
− N2) calculated for C9H7F3O: 188.0449, found: 188.0442.
N); IR νmax/cm−1 (thin film) 739, 1056, 1151, 1188, 1241, 1605, 1713, 1719 and 3070; 1H NMR (250 MHz, CDCl3, TMS) δH 7.07–7.08 (m, 1H, ArH), 7.12 (dquin, J = 1.8, 7.9 Hz, 1H, ArH), 7.33 (ddd, J = 1.0, 2.3, 8.2 Hz, 1H, ArH), 7.42 (td, J = 1.1, 7.4 Hz, 1H, ArH), 7.49 (t, J = 7.9 Hz, 1H, ArH), 7.58 (td, J = 1.2, 7.5 Hz, 1H, ArH), 7.65 (dt, J = 1.0, 7.5 Hz, 1H, ArH), 7.68 (dd, J = 0.6, 7.8 Hz, 1H, ArH), 7.75 (dt, J = 1.0, 7.4 Hz, 1H, ArH), 8.35 (dd, J = 1.6, 7.8 Hz, 1H, ArH), 8.45 (dd, J = 0.6, 1.6 Hz, 1H, ArH) ppm; 13C NMR (62.8 MHz, CDCl3) δC 28.7 (q, J = 40.3 Hz, CN2), 119.9 (ArCH), 120.4 (ArCH), 121.4 (ArCH), 121.5 (q, J = 271.5 Hz, CF3), 123.2 (ArCH), 124.1 (ArCH), 124.8 (ArCH), 125.8 (ArCH), 130.1 (ArCH), 130.2 (ArCC), 130.5 (ArCH), 131.3 (ArCC), 134.8 (ArCC), 135.1 (ArCC), 135.1 (ArCH), 137.0 (ArCH), 143.5 (ArCC), 149.7 (ArCC), 151.4 (CO2), 164.2 (ArCO), 192.8 (CO) ppm; 19F NMR (235 MHz, CDCl3, CFCl3) δF
−65.5 (s, CF3) ppm; CI-MS ([M]+
− N2) calculated for C22H11F3O3: 380.0660, found: 380.0651.
N); IR νmax/cm−1 (thin film) 1154, 1204, 1272, 1355, 1454, 1500, 1588, 1612 and 3334; 1H NMR (250 MHz, CDCl3, TMS) δH 5.00 (s, 1H, OH), 6.67 (m, 1H, ArH), 6.71–6.75 (m, 1H, ArH), 6.85–6.90 (m, 1H, ArH), 7.27 (t, J = 7.9 Hz, 1H, ArH) ppm; 13C NMR (62.8 MHz, CDCl3) δC 28.7 (q, J = 40.5 Hz, CN2), 113.9 (ArCH), 117.2 (ArCH), 119.2 (ArCH), 122.4 (q, J = 274.6 Hz, CF3), 130.6 (ArCH), 131.2 (ArCC), 156.2 (ArCO) ppm; 19F NMR (235 MHz, CDCl3, CFCl3) δF
−65.6 (s, CF3) ppm; CI-MS ([MH]+
− N2) calculated for C8H5F3O: 175.0371, found: 175.0365.
N); IR νmax/cm−1 (thin film) 1154, 1214, 1267, 1355, 1467, 1496, 1584, 1607 and 2962; 1H NMR (250 MHz, CDCl3, TMS) δH 3.73 (s, 3H, OCH3), 6.61 (s, 1H, ArH), 6.68–6.72 (m, 1H, ArH), 6.84–6.89 (m, 1H, ArH), 7.23 (t, J = 8.1 Hz, 1H, ArH) ppm; 13C NMR (62.8 MHz, CDCl3) δC 28.8 (q, J = 40.2 Hz, CN2), 55.7 (OCH3), 112.6 (ArCH), 115.6 (ArCH), 119.1 (ArCH), 122.5 (q, J = 274.7 Hz, CF3), 130.0 (ArCC), 130.4 (ArCH), 160.2 (ArCO) ppm; 19F NMR (235 MHz, CDCl3, CFCl3) δF
−65.6 (s, CF3) ppm; CI-MS ([M]+
− N2) calculated for C9H7F3O: 188.0449, found: 188.0442.
General procedure for dyeing of nylon 6,6 using the fluorenone dye 1
Nylon 6,6 powder was washed prior to modification with hexane, chloroform, THF and methanol (10 mL of each) and dried under vacuum (0.1 mbar). Nylon powder was weighed into a 5 mL vial and a solution of 1 in anhydrous diethyl ether (1 mL) was added to afford weight to weight diazirine∶nylon mixing ratios of 1∶1, 1∶2, 1∶5, 1∶10, 1∶20, 1∶100 and 1∶1000, respectively. The vial was spun at 400 rpm and the solvent removed under reduced pressure (0.1 mbar) at 50 °C for 2 hours. The vial was flushed with argon, sealed and irradiated using a 125 W high pressure mercury lamp for 12 hours. The modified nylon was washed three times with chloroform, THF and methanol (3 × 10 mL of each) and dried under vacuum (0.1 mbar) at 40 °C. Blanks were prepared in an identical manner and washed in an identical manner, however, they were stored in the dark for 12 hours as opposed to being irradiated.Acknowledgements
AB would like to thank DuPont Fellows Forum for funding.References
- J. J. Lee, N. K. Han, W. J. Lee, J. H. Choi and J. P. Kim, Color. Technol., 2003, 119, 134 CAS.
- J. Sokolowska-Gajda and H. S. Freeman, Dyes Pigm., 1990, 14, 35 CrossRef CAS.
- P. Suwanruji, H. S. Freeman and D. Zhao, Color. Technol., 2004, 120, 220 CAS.
- A. Blencowe and W. Hayes, Soft Matter, 2005, 1, 178 RSC.
- (a) U. G. Kang, J. W. Raksis, C. L. Kehr and C. A. Ferrin, in Polymer Surface Modification using Aqueous Stable Diazo Solution, US Patent 5,075,427, 1991 Search PubMed; (b) R. Reiner and H. Kaiser, in Process and Compounds for Surface Modification of Macromolecular Substances, US Patent 4,309,453, 1982 Search PubMed; (c) D. A. Olsen and A. J. Osterras, in Carbene Modified Organic Surfaces, Canadian Patent 812,736, 1969 Search PubMed; (d) A. W. Morgan and J. S. Swenson, in Process for the Chemical Modification of the Solid Surface of a Polymer, US Patent 3,376,278, 1968 Search PubMed; (e) P. J. Pullan, in Dyeing Textile Fibres, Br. Patent 1,395,885, 1975 Search PubMed; (f) P. J. Pullan, in A Method of Colouring a Fabric or Film, Br. Patent 1,412,963, 1975 Search PubMed; (g) C. Holstead, R. A. Jeffreys, L. Shuttleworth and R. S. Wickham, in Photographic Materials, Br. Patent 1,344,993, 1974 Search PubMed; (h) C. Holstead, L. Shuttleworth, J. Griffiths and P. J. Pullan, in Dyeing or Treating Films and Fibres, Br. Patent 1,344,991, 1974 Search PubMed; (i) M. G. Moloney, W. Ebenezer and K. Awenat, in Process for the Surface Functionalisation of Polymeric Substrates using Diarylcarbenes as Reactive Intermediates, US Patent 6,699,527, 2004 Search PubMed; (j) K. Awenat, W. Ebenezer and M. G. Moloney, in A Method for the Functionalisation of Polymeric Substrates, Br. Patent PCT/GB99/03629, 1999 Search PubMed.
- D. R. Braybrook, M. G. Moloney, H. M. I. Osborn and W. J. Norris, J. Photochem. Photobiol., A, 1993, 70, 171 CrossRef CAS.
- K. M. Awenat, P. J. Davis, M. G. Moloney and W. Ebenezer, Chem. Commun., 2005, 990 RSC.
- Chemistry of Diazirines, ed. M. T. H. Liu, CRC Press, Boca Raton, 1987, vol. 1 Search PubMed.
- (a) J. Brunner, H. Senn and F. M. Richards, J. Biol. Chem., 1980, 255, 3313 CAS; (b) J. Brunner and G. Semenza, Biochemistry, 1981, 20, 7174 CrossRef CAS.
- J. H. Robson and J. Reinhart, J. Am. Chem. Soc., 1955, 77, 498 CrossRef CAS.
- C. Aubert, C. Huard-Perrio and M.-C. Lasne, J. Chem. Soc., Perkin Trans. 1, 1997, 2837 RSC.
- (a) A. D. Allen, I. C. Ambridge, C. Che, H. Micheal, R. J. Muir and T. T. Tidwell, J. Am. Chem. Soc., 1983, 105, 2343 CrossRef CAS; (b) J. Gaspar and A. Guerrero, Tetrahedron: Asymmetry, 1995, 6, 231 CrossRef CAS.
- (a) J. P. Richard, J. Am. Chem. Soc., 1989, 111, 6735 CrossRef CAS; (b) F. Levrat, H. Stoeckli-Evans and N. Engel, Tetrahedron: Asymmetry, 2002, 13, 2335 CrossRef CAS; (c) Y. Gong, K. Kato and H. Kimoto, Bull. Chem. Soc. Jpn., 2002, 75, 2637 CrossRef CAS.
- T. Okano, H. Sugiura, M. Fumoto, H. Matsubara, T. Kusukawa and M. Fujita, J. Fluorine Chem., 2002, 114, 91 CrossRef CAS.
- A. Admasu, A. D. Gudmundsdóttir, M. S. Platz, D. S. Watt, S. Kwiatkowski and P. J. Crocker, J. Chem. Soc., Perkin Trans. 2, 1998, 1093 RSC.
- M. S. Platz, W. R. White and D. A. Modarelli, Res. Chem. Intermed., 1994, 20, 175 CrossRef CAS.
- J.-F. Rontani, Trends Photochem. Photobiol., 1997, 4, 125 Search PubMed.
- (a) R. S. Davidson, P. F. Lambeth, F. A. Younis and R. Wilson, J. Chem. Soc. C, 1969, 2203 RSC; (b) R. S. Davidson and R. Wilson, J. Chem. Soc. B, 1970, 71 RSC.
- S. Benomar, B. Patel and A. E. Tipping, J. Fluorine Chem., 1990, 50, 207 CrossRef CAS.
- (a) M. T. H. Liu and K. Ramakrishnan, J. Org. Chem., 1977, 42, 3450 CrossRef CAS; (b) M. P. Doyle, A. H. Devia, K. E. Bassett, J. W. Terpstra and S. N. Mahapatro, J. Org. Chem., 1987, 52, 1619 CrossRef CAS.
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2006 |