Creation of monomeric La complexes on apatite surfaces and their application as heterogeneous catalysts for Michael reactions
Kohsuke
Mori
,
Michitaka
Oshiba
,
Takayoshi
Hara
,
Tomoo
Mizugaki
,
Kohki
Ebitani
and
Kiyotomi
Kaneda
*
Department of Materials Engineering Science, Graduate School of Engineering Science, Osaka University, 1-3 Machikaneyama, Toyonaka, Osaka 560-8531, Japan. E-mail: kaneda@cheng.es.osaka-u.ac.jp; Fax: +81-6-6850-6260; Tel: +81-6-6850-6260
First published on 9th November 2005
Abstract
Using a cation-exchange method, an equimolar substitution of La3+ for Ca2+ occurred by the treatment of stoichiometric hydroxyapatite (HAP: Ca10(PO4)6(OH)2) with an aqueous solution of La(OTf)3, affording a monomeric hydroxyapatite-bound La complex (LaHAP). Physicochemical characterization by means of XRD, XPS, IR, and La K-edge XAFS analyses proved that a monomeric La3+ phosphate complex was generated on its surface. Such monomeric La3+ species function as an efficient heterogeneous catalyst for the Michael reaction of 1,3-dicarbonyls with enones under aqueous or solvent-free conditions. The work-up procedure is straightforward and the spent catalyst could be recycled without any loss of the catalytic activity. Further application to an asymmetric version was also investigated using various apatite catalysts modified with chiral organic ligands. Enantioselectivity was found to depend on the chiral ligand, solvent, and rare earth metal triflate precursor (RE(OTf)3) for the reaction of methyl 1-oxoindan-2-carboxylate with methyl vinyl ketone. Under optimized reaction conditions, a monomeric fluoroapatite-bound La complex catalyst modified with (R,R)-tartaric acid (TA-LaFAP) provided the Michael adduct quantitatively in up to 60% ee.
Introduction
In the drive toward environmentally friendly organic synthesis routes, the development of nanostructured heterogeneous catalysts is one of the most exciting challenges in modern chemistry.1 A general principle for the design of these heterogeneous catalysts requires precise control of the coordination sphere at which the elementary catalytic steps proceed while maintaining robust stabilization of the catalytically active species on the solid surface. To date, most attempts have focused on the encapsulation of efficient soluble systems within a zeolite cavity or anchoring on an insoluble matrix via covalent bond formation.2 It is also possible to immobilize cationic or anionic complexes by ion paring with anionic or cationic solids, respectively. These hybrid catalysts, however, often have encountered serious disadvantages such as tedious multi-step preparations and limited stability against metal leaching. Another approach involves the doping of reactive species into sol–gel materials.3 Unfortunately, this method usually causes a decrease in the structural ordering and inferior activity compared with homogeneous analogues because a large proportion of metal centers is buried within the pore walls.Apatites and related compounds, most notably hydroxyapatite (HAP: Ca10(PO4)6(OH)2) and fluoroapatite (FAP: Ca10(PO4)6F2), are of considerable interest in many areas owing to their multiple functionalities.4 Our strategy for the design of nanostructured heterogeneous catalysts focuses on the utilization of promising ability of HAP, which can be considered as a rigid macroligand for catalytically active centers.5 The cation-exchange ability of the HAP enables an equimolar substitution of transition metal cations, e.g. Ru3+ or Zn2+, for Ca2+ on its surface, which gave monomeric metal species stabilized by phosphate ligands.5a,k Such transition metal-substituted HAPs exhibit remarkably high catalytic activities for various aerobic oxidation reactions and carbon–carbon bond-forming reactions with high reusability. The use of apatite surfaces is a powerful method of creating stable active sites, and also provides unexpected novel functions including site isolation of well-defined active species, steric control of the reaction pathway, and specific performances in aqueous media.
We envisaged that the accommodation of rare earth metals, which prefer high coordination numbers from 6–12 due to their large ionic radii,6 into the apatite framework could generate a heterogeneous catalyst that combines potential vacant coordination sites as well as robust stability. These characteristics could allow further binding of organic modifiers, e.g., chiral ligands, to form an enantioselective sphere on a solid surface. Herein we describe our detailed studies concerning the synthesis and characterization of a hydroxyapatite-bound La complex (LaHAP) and its evaluation as a recyclable heterogeneous catalyst for the Michael reaction of 1,3-dicarbonyls with enones under aqueous or solvent-free conditions. Conducting the reactions by heterogeneous catalysts under aqueous7 or solvent-free8 conditions instead of using harmful organic solvents could make a contribution to improve environmental outcomes and simplify organic syntheses. We also describe the extension to an asymmetric version by a fluoroapatite-bound La complex catalyst modified with (R,R)-tartaric acid (TA-LaFAP).
Results and discussion
Catalyst preparation and characterization
The hexagonal apatite structure comprises Ca2+ sites surrounded by PO43– tetrahedral; OH− or F− ions occupy columns parallel to the hexagonal axis, as illustrated in Fig. 1.4 The crystal structure presents two nonequivalent Ca2+ sites: one set of Ca2+ ions is aligned in columns (site I: CaI), while the other set forms equilateral triangles centered on the screw axes (site II, CaII). As mentioned above, various kinds of transition metal cations can readily be accommodated into the apatite framework by taking advantage of their cation-exchange ability.5a,k Using this approach, various rare earth metal-exchanged HAPs were synthesized by treatment of stoichiometric HAP, Ca10(PO4)6(OH)2, with an aqueous solution of the corresponding rare earth metal triflates (RE(OTf)3) at room temperature for 9 h. In this way, the LaHAP was obtained from La(OTf)3 as a white powder (La content: 0.16 mmol g−1).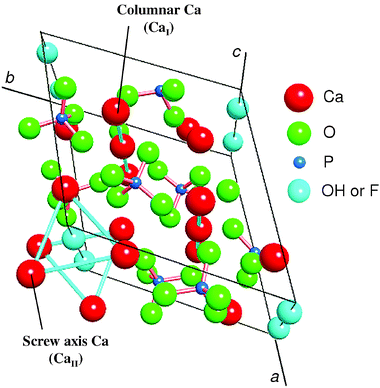 | ||
| Fig. 1 Structure of apatite. | ||
To gain insight into the structure around La species of the LaHAP, characterization by means of physicochemical methods was carried out. Retention of the crystal structure of the LaHAP was confirmed by X-ray diffraction (XRD). Elemental analysis determined that both the Ca/P ratio of the parent HAP and the (La + Ca)/P ratio of the LaHAP were 1.67, demonstrating that the substitution of La3+ for Ca2+ was isomorphic in the above preparation sequence. It has been well documented that larger cations compared to Ca2+ (ionic radius: r = 1.14 Å) replace Ca2+ at site II, whereas smaller ones occupy site I.4 Due to its larger ionic radius, the La3+ cations (1.22 Å) is thought to be accommodated into the CaII site surrounded by both OH− and PO43– tetrahedra.9 No signals assignable to ν(C–F) band could be observed in the infrared spectra (IR), suggesting that OTf ligands originating from the La precursor were not present. The absence of OTf groups was also supported by X-ray photoelectron spectroscopy (XPS) and elemental analyses. The XPS spectra of the LaHAP and La2O3 showed identical binding energy values of 836.2 and 835.8 eV for La 3d5/2, respectively. Formation of a trivalent La species was also indicated by the edge position of the La K-edge X-ray absorption near-edge structure (XANES) spectrum of the LaHAP, which was identical to that of La2O3 (A and D in Fig. 2).10 The Fourier transforms (FT) of k3-weighted extended X-ray absorption fine structure (EXAFS) data are depicted in Fig. 3. There were no peaks suggestive of contiguous La–O–La bonds in the LaHAP (A), detectable in that of La2O3 (D) at around 3.7 Å. The inverse FT of the main peaks of the LaHAP was well fitted using five short (2.52 Å) and three longer (2.59 Å) La–O bonds. The La–O distances were slightly longer than the value of 2.43 Å in the La–Na–BINOL complex.11 We therefore propose a monomeric LaIII complex surrounded by hydroxyl, phosphate, and weakly coordinated aqua ligands on the HAP surface, as illustrated in Fig. 4. In the case of the LaFAP prepared by the same method with FAP in place of HAP, the shape and edge position of the XANES spectra resembled those of the LaHAP (B and A in Fig. 2). Additionally, comparing analogous FT-EXAFS spectra of the LaFAP with the LaHAP (B and A in Fig. 3) shows that the La species are supported on FAP in a monomeric form having +3 oxidation state and that the local structure around the La species is quiet similar to that of LaHAP.
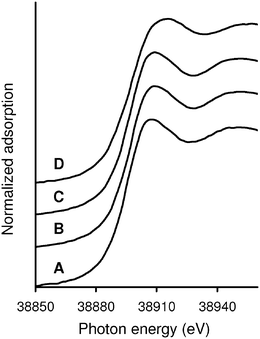 | ||
| Fig. 2 La K-edge XANES spectra of (A) LaHAP, (B) LaFAP, (C) TA-LaFAP, and (D) La2O3. | ||
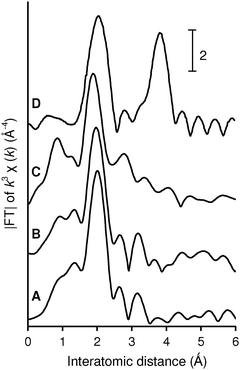 | ||
| Fig. 3 Fourier-transforms of k3-weighted La K-edge EXAFS experimental data for (A) LaHAP, (B) LaFAP, (C) TA-LaFAP, and (D) La2O3. | ||
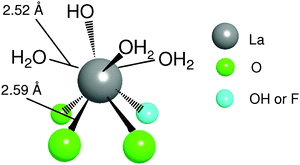 | ||
| Fig. 4 A proposed surface structure of apatite-bound La complexes. | ||
Michael reaction by LaHAP catalyst
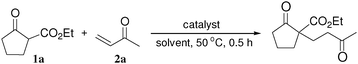
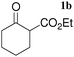
Michael reaction is widely recognized as one of the most important carbon–carbon bond-forming reactions, leading to functionalized adducts of high synthetic value.12 In particular, 1,5-dioxo synthons, which are available from the reaction of 1,3-dicarbonyl compounds with enones, can be transformed into cyclohexenone derivatives for use as paramount building blocks in steroid and terpenoid synthesis.13 In the quest to improve the reaction, various transition metal complexes, including chiral versions have been developed instead of traditional strong bases.14 Among them, RE(OTf)3 compounds are considered to be effective for aqueous Michael reactions.15In an effort to evaluate the potential catalytic activity of the LaHAP, the Michael reaction of 2-oxocyclopentanecarboxylate (1a) with methyl vinyl ketone (2a) was carried out under several sets of reaction conditions. As can be seen from Table 1, the LaHAP gave the highest yield of 2-oxo-1-(3-oxobutyl)cyclopentanecarboxylate under solvent-free conditions (entry 1), and also showed higher catalytic activity than the homogeneous La(OTf)3 precursor (entry 5). The activity of rare earth metal-exchanged HAPs decreased in the order LaHAP (>99%) > ScHAP (90%) >YHAP (67%) > YbHAP (39%) (entries 2, 3 and 6), which is not the order of decreasing Lewis acidity of the metal center: the Sc3+ ion is the smallest rare earth metal and is hence the most Lewis acidic cation, while the largest La3+ ion has the strongest Brønsted basicity among the corresponding rare earth metal hydroxides (RE(OH)3).6a The use of the LaFAP instead of the LaHAP gave a moderate yield of the Michael product (entry 4). The parent HAP16 and FAP17 are reported to act as heterogeneous catalysts for the Michael reaction of mercaptans with chalcone derivatives, but they were found to be less effective under the present reaction conditions (entries 7 and 8). Among the solvents examined, water was also effective for the present reaction, affording the Michael adduct in quantitative yield (entry 9). The use of toluene gave a moderate yield, whereas THF and DMF were poor solvents (entries 10, 14 and 15).
| Entry | Catalyst | Solvent | Yield (%)b |
|---|---|---|---|
| a Reaction Conditions: catalyst (0.5 mol%), 1a (2.0 mmol), 2a (3.0 mmol), 50 °C , 30 min, Ar atmosphere. b Yields of products were determined by GC. c Reaction Conditions: catalyst (0.5 mol%), 1a (0.5 mmol), 2a (0.75 mmol), solvent (3 ml), 50 °C, 30 min, Ar atmosphere. | |||
| 1 | LaHAP | Neat | >99 |
| 2 | ScHAP | Neat | 90 |
| 3 | YHAP | Neat | 67 |
| 4 | LaFAP | Neat | 41 |
| 5 | La(OTf)3 | Neat | 40 |
| 6 | YbHAP | Neat | 39 |
| 7 | HAP | Neat | 30 |
| 8 | FAP | Neat | 11 |
| 9c | LaHAP | Water | >99 |
| 10c | LaHAP | Toluene | 58 |
| 11c | LaHAP | Acetonitrile | 35 |
| 12c | LaHAP | Dichloromethane | 30 |
| 13c | LaHAP | n-hexane | 21 |
| 14c | LaHAP | THF | 9 |
| 15c | LaHAP | DMF | Trace |
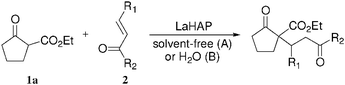
The LaHAP catalyzed-Michael reactions could be extended to other 1,3-dicarbonyl compounds and enones under solvent-free (method A) or aqueous conditions (method B), as demonstrated in Tables 2 and 3. In all cases, the desired 1,4-addition products were obtained exclusively without the formation of aldol-type compounds. Acyclic and cyclic enones were found to be good acceptors for the reaction with 1a (Table 2). It is said that the solvent-free Michael reactions with cyclic enones using FeCl3·6H2O18 and Cu(OAc)2·6H2O19 hardly occur. Moreover, the LaHAP catalyst was applicable to a less reactive acceptor methyl acrylate (2e) under solvent-free conditions (entry 9). 2a also successfully reacted with a variety of β-keto esters and 1,3-diketones (Table 3). The reaction rate in an aqueous medium was largely dependent on the solubility of the substrate, but a 25 mmol-scale experiment for the reaction of 1a with 2a under biphasic conditions was performed to give 90% isolated yield of Michael adduct after 12 h, in which the corresponding turnover number (TON) and turnover frequency (TOF) approached up to 4500 and 375 h−1, respectively.
| Entry | Acceptor | Methoda | La(mol%) | Time (h) | Yield (%)b |
|---|---|---|---|---|---|
| a Method A: 1a (2 mmol), acceptor (3 mmol), solvent-free, Ar atmosphere, 50 °C: method B: 1a (0.5 mmol), acceptor (0.75 mmol), H2O (3 ml), Ar atmosphere, 50 °C. b Determined by GC analysis. c The third recycling experiment. d 80 °C. e 100 °C. | |||||
| 1 |
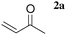
|
A | 0.5 | 0.5 | >99 |
| 2c | A | 0.5 | 0.5 | >99 | |
| 3 | B | 0.5 | 0.5 | >99 | |
| 4 |
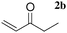
|
A | 0.5 | 1 | 97 |
| 5 | B | 0.5 | 4 | 85 | |
| 6 |
|
A | 1.0 | 6 | 86 |
| 7 | B | 1.0 | 24 | 89 | |
| 8d |
|
A | 1.5 | 13 | 95 |
| 9d | B | 1.5 | 27 | 64 | |
| 10d |

|
A | 2.0 | 24 | >99 |
| 11e |

|
A | 2.0 | 30 | 40 |
| Entry | Donor | Methoda | La (mol%) | Time (h) | Yield (%)b |
|---|---|---|---|---|---|
| a Method A: donor (2 mmol), 2a (3 mmol), solvent-free, Ar atmosphere, 50 °C; method B: donor (0.5 mmol), 2a (0.75 mmol), H2O (3 ml), Ar atmosphere, 50 °C. b Determined by GC analysis. c 80 °C. d 130 °C. | |||||
| 1c |
|
A | 1.0 | 1 | 98 |
| 2c |

|
A | 1.0 | 3 | 97 |
| 3c | B | 1.0 | 3 | 99 | |
| 4c |

|
A | 1.25 | 6 | 83 |
| 5c | B | 1.25 | 24 | 90 | |
| 6 |

|
A | 0.5 | 4 | 99 |
| 7 | B | 0.5 | 9 | 99 | |
| 8 |
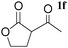
|
A | 0.5 | 2 | 95 |
| 9 | B | 0.5 | 3 | 99 | |
| 10d |
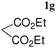
|
A | 2.0 | 24 | 65 |
The LaHAP catalyst was easily separated from the reaction mixture and retained its inherent catalytic activity; over 99% yields were attained for at least three recycling experiments for the reaction of 1a with 2a under solvent-free conditions (entry 2 in Table 2). Moreover, the catalyst was filtered off after ca. 60% conversion at the reaction temperature. Further treatment of the filtrate under identical reaction conditions did not afford any products. ICP analysis of the filtrate confirmed that the La content was determined to be negligible (less than 0.1 ppb). It can be said that the present reaction undoubtedly proceeds on the hydroxyapatite surface, which effectively serves as a promising macroligand for the catalytically active La species.
The present catalyst showed specific activity only toward 1,3-dicarbonyls as Michael donors. Other active methylene compounds such as ethylcyanoacetate (pKa = 9) having a similar pKa value to acetylacetone (1e) did not yield the Michael product under the same reaction conditions.20 Unfortunately, benzalacetone proved to be a poor acceptor, affording the corresponding Michael adduct in 40% yield, even when 2.0 mol% La catalyst was employed at 100 °C (entry 11 in Table 2). The reaction using diethyl malonate (1g) as a donor, which has a relatively high pKa value of 13.3, was also retarded using the present catalytic system (entry 10 in Table 3).
Mechanistic considerations
Generally, the Michael reaction proceeds in the presence of Brønsted base catalysts. Transition metal and rare-earth metal catalysts have also been extensively employed for the Michael reaction of 1,3-dicarbonyl compounds with enones, in which the reaction mechanism is considered to involve a ternary complex with both the 1,3-dicarbonyl compound and the enone coordinated to a metal center.14,21In order to elucidate the pathway of the present reaction, preliminary experiments were carried out. Treatment of 1a with D2O in the presence of the LaHAP (1.0 mol%) at 40 °C for 3.5 h led to H–D exchange reaction, giving the α-deuterated compounds in 15% yield as determined by 1H NMR spectroscopy:22 the exchange reaction did not proceed at all in the absence of the LaHAP. The LaHAP catalyst also successfully promoted typical base-catalyzed carbon–carbon bond-forming reactions, e.g., (i) aldol and (ii) knoevenagel reactions, to afford the corresponding condensation products in excellent yields (Scheme 1).23Vide supra, the LaHAP catalyst, which possesses the strongest Brønsted basicity among the corresponding RE(OH)3, showed the highest yield. These results apparently demonstrate that the LaHAP has a Brønsted base site, i.e., La–OH species.24 Additionally, the IR spectrum of the LaHAP upon treatment with 1e showed a shift of the ν(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) band toward 1560 cm−1 in comparison to the free carbonyl group at around 1700 cm−1, suggesting an 1,3-diketonato La species is formed as a reaction intermediate.25 On the basis of these results, a plausible mechanism is proposed as follows. Initially, a La–OH species abstracts an α-proton of the donor to form a 1,3-diketonato La species. Next, an acceptor coordinate to vacant sites of the La species at the carbonyl oxygen to form a ternary La complex, followed by alkylation and hydrolysis to afford a Michael adduct along with regeneration of the La–OH species. Kinetic studies showed pseudo-zero-order dependence on 1a and first-order relationship with 2a. A kH/kD value of 1.1 was also observed in the intermolecular competitive reaction between 1e and acetylacetone-3,3-d2 (CH3COCD2COCH3) with 2a as a Michael acceptor. These results suggest that the alkylation might be a rate-determining step. The kinetic results are in sharp contrast with those of homogeneous La(OTf)3 catalysts, which follow first-order dependence on 1a and a zero-order relationship with 2a. We believe this phenomenon can be attributed to Brønsted basicity generated on the surface of the LaHAP catalyst.
O) band toward 1560 cm−1 in comparison to the free carbonyl group at around 1700 cm−1, suggesting an 1,3-diketonato La species is formed as a reaction intermediate.25 On the basis of these results, a plausible mechanism is proposed as follows. Initially, a La–OH species abstracts an α-proton of the donor to form a 1,3-diketonato La species. Next, an acceptor coordinate to vacant sites of the La species at the carbonyl oxygen to form a ternary La complex, followed by alkylation and hydrolysis to afford a Michael adduct along with regeneration of the La–OH species. Kinetic studies showed pseudo-zero-order dependence on 1a and first-order relationship with 2a. A kH/kD value of 1.1 was also observed in the intermolecular competitive reaction between 1e and acetylacetone-3,3-d2 (CH3COCD2COCH3) with 2a as a Michael acceptor. These results suggest that the alkylation might be a rate-determining step. The kinetic results are in sharp contrast with those of homogeneous La(OTf)3 catalysts, which follow first-order dependence on 1a and a zero-order relationship with 2a. We believe this phenomenon can be attributed to Brønsted basicity generated on the surface of the LaHAP catalyst.
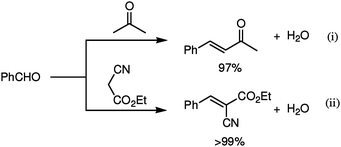 | ||
| Scheme 1 Carbon–carbon bond-forming reactions catalyzed by LaHAP. (i) LaHAP (2.0 mol%), benzaldehyde (0.5 mmol), acetone (2 mL), 60 °C, 37 h. (ii) LaHAP (2.0 mol%), benzaldehyde (1.5 mmol), ethyl cyanoacetate (1 mmol), toluene (2 mL), 60 °C, 24 h. | ||
Asymmetric Michael reaction catalyzed by TA-LaFAP
The extension to an asymmetric version of Michael reaction is a challenging task for synthetic organic chemists.26 Various chiral catalysts have been explored including cinchona alkaloids,27 alkoxide complexes,28 crown ether-metal alkoxide complexes,29 transition metal complexes,30 and bimetallic lanthanide complexes.31 Although remarkable progress has been achieved using these homogeneous catalysts, only a few heterogeneous ones are available to date, e.g. polymer-supported metal complexes,32 polymer-supported cinchona alkaloids,33 layered double hydroxides containing chiral organic guests,34 and MCM-41-grafted chiral amines.35Building upon the initial success of the present LaHAP catalyst, further investigations were conducted to develop an effective heterogeneous catalyst for the asymmetric Michael reaction. Our strategy focuses on the utilization of organic modifiers as chiral ligands, since we suppose the immobilized La species still have vacant coordination sites owing to their large ionic radius. Our procedure for the preparation of heterogeneous asymmetric catalysts is quite straightforward. To a stirred aqueous solution of 1 ∶ 1 of La(OTf)3 and (R,R)-tartaric acid (TA, 4a) for 30 min was added a fluoroapatite (FAP), Ca10(PO4)6F2, and then the mixture was further reacted for 24 h at 30 °C, affording the TA-LaFAP (La content: 0.36 mmol g−1) as a white powder. From energy-dispersive X-ray (EDX) analysis of the TA-LaFAP, the atomic ratio of La to TA was determined to be 1 ∶ 1 (La ∶ C = ca. 1 ∶ 4). The La K-edge XANES spectrum of the TA-LaFAP (C) was comparable with those of the LaHAP (A) and the LaFAP (B), as shown in Fig. 2. The absence of a peak in the vicinity of 3.7 Å in the FT-EXAFS suggested that no La–O–La contribution exists (C in Fig. 3). It is clear that the La species exist in a highly dispersed form having a +3 oxidation state.
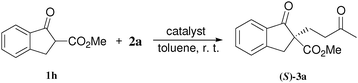
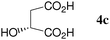
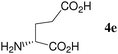
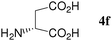
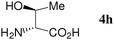
At first, several La catalysts and chiral organic ligands were investigated for the asymmetric Michael reaction of methyl 1-oxoindan-2-carboxylate (1h) with 2a in toluene at room temperature, as summarized in Table 4. It was found that the TA-LaFAP (La: 1.2 mol%) effectively served as a chiral heterogeneous catalyst to afford (S)-methyl 1-oxo-2-(3′-oxobutyl)-2-indanecarboxylate ((S)-3a) in 97% yield with a good enantioselectivity of 60% (entry 1). It is noteworthy that no enantioselectivity was observed by the combination of either parent FAP or La(OTf)3 with 4a (entries 2 and 3), showing that the immobilization of La species on the FAP surface is essential for attaining asymmetric catalysis. To the best of our knowledge, this is the first demonstration of an asymmetric reaction utilizing an apatite catalyst.36 The enantioselectivity achieved by the TA-LaFAP for the asymmetric Michael reaction of 1h with 2a was higher than those reported for other catalyst systems,37 such as the N,N′-dioxide-Sc(OTf)3 complex (39% ee),30f diaminodiol-CoII complex (38% ee),30c chiral amidine (8% ee),38 poly(cinchona alkaloid-co-acrylonitrile) (42% ee).33a This value is also comparable with those with polymer-supported quinine (65% ee),33b 1,1′-bi-2-naphthol-Na complex (57% ee),28 and quinine (56% ee).27a Enantioselectivity was dependent on the reaction temperature; when the reaction temperature was lowered to 10 °C, the yield significantly decreased with 40% ee (entry 4), whereas the reaction at a higher temperature of 40 °C resulted in decrease of enantioselectivity to 50% in spite of high chemical yield (entry 5). This nonlinear relationship between enantioselectivity and temperature has been reported previously for the Michael reaction catalyzed by MCM-41-grafted chiral amines.35 Interestingly, the use of the HAP instead of the FAP as a support gave a 30% ee with an opposite configuration (entry 6). In the case of (S,S)-tartaric acid (4b), (R)-3a was obtained with the same level of chemical yield and enantioselectivity as compared to those of 4a, revealing that the absolute configuration of the Michael adducts is determined by the employed ligand. Other dicarboxylic acid ligands such as (S)-malic acid (4c) and (R,R)-O,O′-dibenzoyltartaric acid (4d) favored excess formation of the (R)-isomer with moderate enantioselectivity (entries 8 and 9). The reaction that employed (S)-serine (4g) and (S)-threonine (4h) containing amino and monocarboxyl groups was less efficient (entries 12 and 13). As a consequence, it was found that the presence of dicarboxylic acids group showed beneficial effects on the enentioselectivity.
| Entry | Catalyst | Ligand | Yield (%)b | Ee(%)c | Confgn.d |
|---|---|---|---|---|---|
| a Reaction conditions: catalyst (1.2 mol%), 1h (0.5 mmol), 2a (0.75 mmol), toluene (2 mL), room temperature, 6 h, Ar atmosphere. b Determined by GC. c Determined by HPLC (Daisel Chiralpak AD-H). d Assigned by optical rotation. e La(OTf)3 (3.4 × 10−3 g, 1.2 mol%), 4a (8.6 × 10−4 g, 1.2 mol%). f 10 °C. g 40 °C. | |||||
| 1 | LaFAP |

|
97 | 60 | S |
| 2 | FAP | 18 | — | — | |
| 3e | La(OTf)3 | 29 | >1 | S | |
| 4f | LaFAP | 5 | 44 | S | |
| 5g | LaFAP | >99 | 50 | S | |
| 6 | LaHAP | >99 | 30 | R | |
| 7 | LaFAP |

|
98 | 59 | R |
| 8 | LaFAP |
|
>99 | 50 | R |
| 9 | LaFAP |

|
>99 | 46 | R |
| 10 | LaFAP |
|
>99 | 32 | R |
| 11 | LaFAP |
|
55 | 29 | S |
| 12 | LaFAP |

|
>99 | 19 | R |
| 13 | LaFAP |
|
>99 | 7 | S |
Next, the catalytic activities for the TA-LaFAP in the reaction of 1h with 2a were compared with various solvents using the TA-LaHAP. Fig. 5 shows the effect of solvents together with dielectric constant values (ε) as one of the criteria of polarity. Non-polar solvents with low dielectric constants such as toluene, n-hexane, and diethyl ether were good solvents among those examined, whereas polar solvents with higher dielectric constant values, such as methanol and DMF, gave poor results with respect to enantiomeric excess. The use of water afforded a completely racemic product despite offering high chemical yield. These results clearly suggest that specific interactions (e.g. hydrogen-bonding) between solvent and catalyst surface significantly influence the conformation of the transition state in the enantioselectivity-determining step. Vide supra, the decrease in enantioselectivity using TA-LaHAP could be ascribed to the negative effect of hydrogen-bonding between a surface hydroxyl group on the HAP and 4a.39 The critical role of hydrogen-bonding in the transition state was also observed in the amino acid-catalyzed asymmetric aldol reaction, in which addition of water to the reaction mixture severely decreased enantioselectivity.40
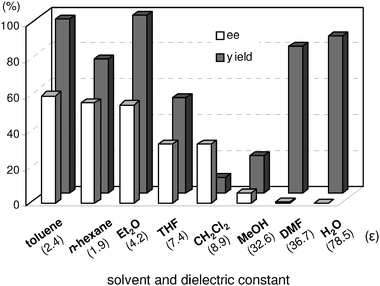 | ||
| Fig. 5 Solvent effects for asymmetric Michael reaction of 1h with 2a. Reaction was conducted with TA-LaFAP (1.2 mol% based on 1h), 1h (0.5 mmol), 2a (0.75 mmol), and toluene (2 mL) at room temperature for 6 h under Ar atmosphere. | ||
Another important parameter that affects the activity and enantioselectivity of the reaction is the choice of metal precursor. As shown in Fig. 6, we have evaluated the effect of RE(OTf)3 precursors having various ionic radii.41 It should be noted that catalytic performance varied significantly with ionic radius:42 the largest La3+ cation afforded the Michael adduct with the highest yields as well as enantioselectivity, while the use of smaller cations such as Y, Yb, and Sc gave unsatisfactory results in terms of both chemical and optical yields. The higher activities and enantioselectivities attained with larger cations might be attributed to increased coordination numbers, where the further binding of chiral organic modifiers and substrates to metals would be allowed to access a reasonable transition state even on the solid surface. In contrast, the decrease in ionic radii severely limits the interaction of chiral organic modifiers and substrates with metals because of the decrease in number of available vacant coordination sites.
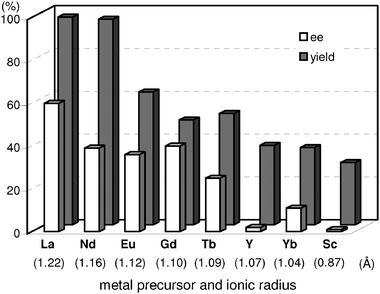 | ||
| Fig. 6 Effect of metal precursors (RE(OTf)3) for asymmetric Michael reaction of 1h with 2a. Reaction was conducted with catalyst (1.2 mol% based on 1h), 1h (0.5 mmol), 2a (0.75 mmol), and toluene (2 mL) at room temperature for 6 h under Ar atmosphere. Values in parentheses are ionic radii of 8-coordination for Sc3+ and 9-coordination for other metal cations (RE3+). | ||
The lifetime and leaching of active metal species into the reaction solution are crucial points to consider when chiral heterogeneous catalysts are employed for industrial and pharmaceutical applications.43 As shown in Fig. 7, the time course for the Michael reaction of 1h with 2a was monitored periodically. No induction period and degradation was observed in the reaction catalyzed by the TA-LaFAP. The enantioselectivity appeared to be independent of the conversion level and remained unchanged at around 60% during the reaction, suggesting that racemization of the Michael adduct did not take place. Upon completion of the Michael reaction, the TA-LaFAP catalyst was easily separated from the reaction mixture and could be reused with retention of its activity and enantioselectivity in the absence of an externally added chiral organic ligand; over 97% yields and 59% ee could be kept for at least three recycling experiments of the reaction between 1h and 2a. Again, ICP analysis of the filtrate could not detect the metal species. XAFS analysis revealed that no structural changes around the La3+ center were observed. It can be said that the catalyst deactivation, leaching, and product inhibition do not occur, and that the present asymmetric Michael reaction undoubtedly proceeds on the chiral heterogeneous apatite surface.
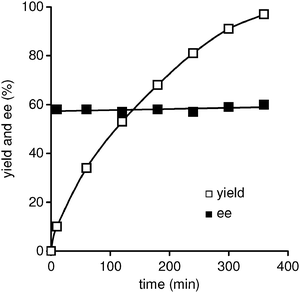 | ||
| Fig. 7 Time profile for the asymmetric Michael reaction of 1g with 2a catalyzed by TA-LaFAP. Reaction conditions: TA-LaFAP (0.016 g, 1.2 mol% based on 1g), 1g (0.5 mmol), 2a (0.75 mmol), toluene (2 mL), room temperature, 6 h, Ar atmosphere. | ||
Asymmetric induction
As mentioned above, preliminary screening of the chiral organic modifiers revealed that (R,R)-tartaric acid (4a) is the most suitable ligand to achieve high enantioselectivity, suggesting that dicarboxylic acids play an important role in the conformation of the enantio-differentiating complex on the solid surface. In the IR spectrum of the TA-LaFAP, the CO stretching vibration appeared at around 1630 cm−1, which is lower than that of the free 4a (1720 cm−1). Furthermore, it was found that (S)-malic acid (4c) and (R,R)-O,O′-dibenzoyltartaric acid (4d), which contain dicarboxylic acids, also act as good organic modifiers, as shown in Table 5. Upon consideration of the above observations as well as the results that the atomic ratio of La to TA was 1 ∶ 1 determined by EDX analysis, organic modifiers are thought to bond with the La species via coordination of two carboxylate groups in a bidentate manner. Although the precise transition state is unclear, the attainment of the desirable geometry might be directed by the hydrogen-bonding between OH groups of 4a and the substrate, in which the use of polar solvents with high dielectric constants values might prohibit this effective interaction, giving poor results with respect to enantiomeric excess.
Conclusions
We have developed a method for the design of nanostructured catalysts using apatites. Characterizations by physicochemical methods revealed the formation of a stable monomeric La phosphate complex on the surface of hydroxyapatite, which proved to be an efficient heterogeneous catalyst with superior activity for the Michael reaction. Further extension to an asymmetric version by the TA-LaFAP was also demonstrated. To the best of our knowledge, this is the first example of an asymmetric reaction achieved by an apatite catalyst. The features of the present apatite catalysts, such as facile preparation, robust structure, and easy recovery as well as simple recycling, are particularly important in the industrial synthesis of optically active compounds. Studies are ongoing to design novel asymmetric heterogeneous catalysts with enhanced enantioselectivity.Experimental
Materials
La(OTf)3 and (R,R)-tartaric acid were obtained from Tokyo Kasei Kogyo Co., Ltd. and used without further purification. Fluoroapatite (FAP), (NH4)2HPO4, and Ca(NO3)2·4H2O were purchased from Wako Pure Chemical Ind., Ltd. Solvents and all commercially available compounds were purified by standard procedures before use. Methyl 1-oxoindan-2-carboxylate (1h) was synthesized according to the literature procedure.44 All of the products are well known compounds and their identities were confirmed by comparison with IR, MS, and NMR spectra.Characterization
Powder X-ray diffraction patterns were recorded using Philips X’Pert-MPD with Cu Kα radiation. X-ray photoelectron spectroscopy was measured on a Shimadzu ESCA-KM using MgKα radiation. Energy-dispersive X-ray measurement was performed using a Philips EDAX DX-4 attached to a SEM. Infrared spectra were obtained with a JASCO FTIR-410. Elemental analysis was carried out using a Perkin Elmer 2400CHN. Inductively coupled plasma measurements were performed on a Nippon Jarrell-Ash ICAP-575 Mark II. X-ray absorption spectra were recorded in transmission mode at room temperature using a Si (311) monochromator on the BL01B1 station at SPring-8, JASRI, Harima, Japan (prop. No. 2005A0694-NXa-np). Data reductions were performed with a FACOM M-780 computer system of the Data Processing Center of Kyoto University. The Fourier transform was performed on EXAFS in the k range of 3.0–14 Å. The detailed procedure for data analysis is described elsewhere.45Syntheses and reactions
Acknowledgements
This work is supported by the Grant-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science, and Technology of Japan. We thank Dr Tomoya Uruga (JASRI) for XAFS measurement. T. H. thanks the JSPS Research Fellowships for Young Scientists.References
- (a) R. Anwander, Chem. Mater., 2001, 13, 4419 CrossRef CAS; (b) D. E. De Vos, M. Dams, B. F. Sels and P. A. Jacobs, Chem. Rev., 2002, 102, 3615 CrossRef CAS; (c) A. P. Wight and M. E. Davis, Chem. Rev., 2002, 102, 3589 CrossRef CAS; (d) C. Copéret, M. Chabanas, R. P. Saint-Arroman and J.-M. Basset, Angew. Chem., Int. Ed., 2003, 42, 156 CrossRef CAS.
- Q.-H. Fan, Y.-M. Li and A. S. C. Chan, Chem. Rev., 2002, 102, 3385 CrossRef.
- Z.-L. Lu, E. Lindner and H. A. Mayer, Chem. Rev., 2002, 102, 3543 CrossRef CAS.
- J. C. Elliott, Structure and Chemistry of the Apatites and Other Calcium Orthophosphates, Elsevier, Amsterdam, 1994 Search PubMed.
- (a) K. Yamaguchi, K. Mori, T. Mizugaki, K. Ebitani and K. Kaneda, J. Am. Chem. Soc., 2000, 122, 7144 CrossRef CAS; (b) K. Mori, K. Yamaguchi, T. Mizugaki, K. Ebitani and K. Kaneda, Chem. Commun., 2001, 461 RSC; (c) K. Mori, M. Tano, T. Mizugaki, K. Ebitani and K. Kaneda, New J. Chem., 2002, 26, 1536 RSC; (d) K. Mori, K. Yamaguchi, T. Hara, T. Mizugaki, K. Ebitani and K. Kaneda, J. Am. Chem. Soc., 2002, 124, 11572 CrossRef CAS; (e) K. Mori, T. Hara, T. Mizugaki, K. Ebitani and K. Kaneda, J. Am. Chem. Soc., 2003, 125, 11460 CrossRef CAS; (f) M. Murata, T. Hara, K. Mori, M. Ooe, T. Mizugaki, K. Ebitani and K. Kaneda, Tetrahedron Lett., 2003, 44, 4981 CrossRef CAS; (g) T. Hara, K. Mori, T. Mizugaki, K. Ebitani and K. Kaneda, Tetrahedron Lett., 2003, 44, 6207 CrossRef CAS; (h) K. Mori, T. Hara, T. Mizugaki, K. Ebitani and K. Kaneda, J. Am. Chem. Soc., 2004, 126, 10657 CrossRef CAS; (i) T. Hara, K. Mori, M. Oshiba, T. Mizugaki, K. Ebitani and K. Kaneda, Green Chem., 2004, 6, 507 RSC; (j) K. Mori, M. Oshiba, T. Hara, T. Mizugaki, K. Ebitani and K. Kaneda, Tetrahedron Lett., 2005, 46, 4283 CrossRef CAS; (k) K. Mori, Y. Mitani, T. Hara, T. Mizugaki, K. Ebitani and K. Kaneda, Chem. Commun., 2005, 3331 RSC.
- (a) K. Mikami, M. Terada and H. Matsuzawa, Angew. Chem., Int. Ed., 2002, 41, 3554 CrossRef CAS; (b) J.-C. G. Bűnzli and C. Piguet, Chem. Rev., 2002, 102, 1897 CrossRef; (c) K. Dehnicke and A. Greiner, Angew. Chem., Int. Ed., 2003, 42, 1340 CrossRef CAS.
- (a) G. W. V. Cave, C. L. Raston and J. L. Scott, Chem. Commun., 2001, 2159 RSC; (b) Organic Synthesis in Water, ed. P. A. Grieco, Blackie Academic and Professional, London, 1998 Search PubMed; (c) S. Kobayashi and K. Manabe, Acc. Chem. Res., 2002, 35, 209 CrossRef CAS.
- (a) J. O. Metzger, Angew. Chem., Int. Ed., 1998, 37, 2975 CrossRef CAS; (b) K. Tanaka and F. Toda, Chem. Rev., 2000, 100, 1025 CrossRef CAS.
- The situation for La species is in sharp contrast to our results for previously prepared Ru3+ (0.82 Å) 5a and Zn2+ (0.74 Å)5k exchanged hydroxyapatites via similar cation-exchange method under aqueous conditions, in which metal cation species replaced Ca2+ at site I.
- T. Yamamoto, T. Hatsui, T. Matsuyama, T. Tanaka and T. Funabiki, Chem. Mater., 2003, 15, 4830 CrossRef CAS.
- H. Sasai, T. Arai, Y. Satow, K. N. Houk and M. Shibasaki, J. Am. Chem. Soc., 1995, 117, 6194 CrossRef CAS.
- (a) M. E. Jung, in Comprehensive Organic Synthesis, ed. B. M. Trost and I. Fleming, Vol. 4, Pergamon, Oxford, 1991, p. 1 Search PubMed; (b) T.-L. Ho, Tactics of Organic Synthesis, Wiley, New York, 1994 Search PubMed.
- H. O. House, Modern Synthetic Reactions, 2nd edn., Benjamin, Menlo Park, Calif, 1972, p. 595 Search PubMed.
- (a) S. Pelzer, T. Kauf, C. van Wüllen and J. Christoffers, J. Organomet. Chem., 2003, 684, 308 CrossRef CAS; (b) For an excellent review on transition metal-catalyzed Michael reaction of 1,3-dicarbonyls, see: J. Christoffers, Eur. J. Org. Chem., 1998, 1259 Search PubMed.
- (a) E. Keller and B. L. Feringa, Tetrahedron Lett., 1996, 37, 1879 CrossRef CAS; (b) H. Kotsuki and K. Arimura, Tetrahedron Lett., 1997, 38, 7583 CrossRef CAS; (c) Y. Mori, K. Kakumoto, K. Manabe and S. Kobayashi, Tetrahedron Lett., 2000, 41, 3107 CrossRef CAS.
- M. Zahouily, Y. Abrouki, B. Bahlaouan, A. Rayadh and S. Sebti, Catal. Commun., 2003, 4, 521 CrossRef CAS.
- M. Zahouily, Y. Abrouki, A. Rayadh, S. Sebti, H. Dhimane and M. David, Tetrahedron Lett., 2003, 44, 2463 CrossRef.
- J. Christoffers, Chem. Commun., 1997, 943 RSC.
- A. C. Coda, G. Desimoni, P. Righetti and G. Tacconi, Gazz. Chim. Ital., 1984, 114, 417 CAS.
- The pKa values of other 1,3-dicarbonyl componds are as follows: 1a (10.5), 1b (11.5), 1c (7.8), 1d (10.1), and 1e (9.0).
- M. Bauer, T. Kauf, J. Christoffers and H. Bertagnolli, Phys. Chem. Chem. Phys., 2005, 7, 2664 RSC.
- J.-I. Tateiwa and A. Hosomi, Eur. J. Org. Chem., 2001, 1445 CrossRef CAS.
- (a) H. Hattori, Appl. Catal. A, 2001, 222, 247 CrossRef CAS; (b) Y. Ono, J. Catal., 2003, 216, 406 CrossRef CAS.
- Because of the weak Lewis acidity of the La3+ ion, homogeneous La(OR)3 complexes often act as a Brønsted base and catalyze a number of carbonyl reactions involving an enolate intermediate, see: (a) H. Sasai, T. Suzuki, S. Arai, T. Arai and M. Shibasaki, J. Am. Chem. Soc., 1992, 114, 4418 CrossRef CAS; (b) H. Sasai, T. Arai and M. Shibasaki, J. Am. Chem. Soc., 1994, 116, 1571 CrossRef CAS; (c) T. Dewa, T. Saiki and Y. Aoyama, J. Am. Chem. Soc., 2001, 123, 502 CrossRef CAS.
- (a) T. Kawabata, T. Mizugaki, K. Ebitani and K. Kaneda, J. Am. Chem. Soc., 2003, 125, 10486 CrossRef CAS; (b) T. Kawabata, M. Kato, T. Mizugaki, K. Ebitani and K. Kaneda, Chem. Eur. J., 2005, 11, 288 CrossRef.
- For general reviews of catalytic asymmetric Michael reactions, see: (a) E. N. Jacobsen, in Comprehensive Asymmetric Catalysis, ed. E. N. Jacobsen, A. Pfaltz and H. Yamamoto, Springer, Berlin, 1999 Search PubMed; (b) N. Krause and A. Hoffmann-Röder, Synthesis, 2001, 171 CrossRef CAS; (c) M. P. Sibi and S. Manyem, Tetrahedron, 2000, 56, 8033 CrossRef CAS.
- (a) H. Wynberg and R. Helder, Tetrahedron Lett., 1975, 31, 4057 CrossRef; (b) K. Hermann and H. Wynberg, J. Org. Chem., 1979, 44, 2238 CrossRef CAS.
- Y. Tamai, A. Kamifuku, E. Koshiishi and S. Miyano, Chem. Lett., 1995, 957 Search PubMed.
- D. J. Cram and G. D. J. Sogah, J. Chem. Soc., Chem. Commun., 1981, 625 RSC.
- (a) H. Brunner and H. Benedikt, Angew. Chem., Int. Ed. Engl., 1984, 23, 312 CrossRef; (b) G. Desimoni, P. Quadrelli and P. P. Righetti, Tetrahedron, 1990, 46, 2927 CrossRef CAS; (c) C. Botteghi, S. Paganelli, A. Schionato, C. Boga and A. Fava, J. Mol. Catal., 1991, 66, 7 CrossRef CAS; (d) M. Sawamura, H. Hamashima and Y. Ito, J. Am. Chem. Soc., 1992, 114, 8295 CrossRef CAS; (e) T. Suzuki and T. Torii, Tetrahedron: Asymmetry, 2001, 12, 1077 CrossRef CAS; (f) Y. Hamashima, D. Hotta and M. Sodeoka, J. Am. Chem. Soc., 2002, 124, 11240 CrossRef CAS; (g) M. Nakajima, S. Yamamoto, Y. Yamaguchi, S. Nakamura and S. Hashimoto, Tetrahedron, 2003, 59, 7307 CrossRef CAS.
- M. Shibasaki, H. Sasai and T. Arai, Angew. Chem., Int. Ed. Engl., 1997, 36, 1236 CrossRef.
- (a) S. Matsunaga, T. Ohshima and M. Shibasaki, Tetrahedron Lett., 2000, 41, 8473 CrossRef CAS; (b) T. Arai, Q.-S. Hu, A.-F. Zheng, L. Pu and H. Sasai, Org. Lett., 2000, 2, 4261 CrossRef CAS; (c) T. Sekiguti, Y. Lizuka, S. Takizawa, D. Jayaprakash, T. Arai and H. Sasai, Org. Lett., 2003, 5, 2647 CrossRef CAS; (d) Y. Hamashima, H. Takano, D. Hotta and M. Sodeoka, Org. Lett., 2003, 5, 3225 CrossRef CAS; (e) S. Takizawa, H. Somei, D. Jayaprakash and H. Sasai, Angew. Chem., Int. Ed., 2003, 42, 5711 CrossRef CAS.
- (a) N. Kobayashi and K. Iwai, J. Am. Chem. Soc., 1978, 100, 7071 CrossRef CAS; (b) M. Inagaki, J. Hiratake, Y. Yamamoto and J. Oda, Bull. Chem. Soc. Jpn., 1987, 60, 4121 CAS; (c) R. Alvarez, M.-A. Hourdin, C. Cavé, J. d’Angelo and P. Chaminade, Tetrahedron Lett., 1999, 40, 7091 CrossRef CAS.
- B. M. Choudary, B. Kavita, N. S. Chowdari, B. Sreedhar and M. L. Kantam, Catal. Lett., 2002, 78, 373 CrossRef CAS.
- A. Corma, S. Iborra, I. Rodríguez, M. Iglesias and F. Sánchez, Catal. Lett., 2002, 82, 237 CrossRef CAS.
- For recent reviews of heterogeneous asymmetric catalysts, see: (a) P. McMorn and G. J. Hutchings, Chem. Soc. Rev., 2004, 33, 108 RSC; (b) C. E. Song and S.-G. Lee, Chem. Rev., 2002, 102, 3495 CrossRef CAS.
- It was reported that higher enantioselectivity could be attained with other catalytic systems than that of the present TA-LaFAP catalysts, such as polymer-supported cinchona alkaloids (87% ee),33c cinchona alkaloids (76% ee),27b and chiral diamine Rh complex (75% ee).30e.
- H. Kotsuki, A. Sugino, H. Sakai and H. Yasuoka, Heterocycles, 2000, 53, 2561 CrossRef CAS.
- The existence of the hydroxyl group on the HAP surface was confirmed by UV-Vis spectroscopy. HAP was added to a toluene solution of aminopropyltriethoxysilane (APTS) and stirred at 50 °C for 2 h. After filtration and drying under vacuum, fluorescein isothiocyanate (FITC) hydrochloride was attached to the APTS-modified HAP in methanol solution. The UV-Vis spectrum of the FITC-modified HAP exbited the charge-transfer band at 460 nm, whereas the FAP modified by the same procedure did not show any absorption band in this region. The detailed procedure is described elsewhere, see: S. Ikeda, H. Nur, T. Sawadaishi, K. Ijiro, M. Shimomura and Ohtani, Langmuir, 2001, 17, 7976 Search PubMed.
- K. Sakthivel, W. Notz, T. Bui and C. F. Barbas III, J. Am. Chem. Soc., 2001, 123, 5260 CrossRef CAS.
- The use of La(NO)3·6H2O and LaCl3·7H2O as metal precursors instead of La(OTf)3 decreased both chemical and optical yields; La(NO)3·6H2O (77% yield, 55% ee), LaCl3·7H2O (59% yield, 44% ee).
- Size effects of rare earth metals on enantioselectivity have been observed in asymmetric reactions. For example: (a) H. Sasai, T. Suzuki, N. Itoh and S. Arai, Tetrahedron Lett., 1993, 34, 2657 CrossRef CAS; (b) D. A. Evans, S. G. Nelson, M. R. Gagné and A. R. Muci, J. Am. Chem. Soc., 1993, 115, 9800 CrossRef CAS; (c) S. E. Schaus and E. N. Jacobsen, Org. Lett., 2000, 2, 1001 CrossRef CAS; (d) T. Hamada, K. Manabe, S. Ishikawa, S. Nagayama, M. Shiro and S. Kobayashi, J. Am. Chem. Soc., 2003, 125, 2989 CrossRef CAS.
- I. W. C. E. Arends and R. A. Sheldon, Appl. Catal. A, 2001, 212, 175 CrossRef CAS.
- K. V. Emelen, T. D. Wit, G. J. Hoornaert and F. Compernolle, Tetrahedron Lett., 2002, 58, 4225.
- T. Tanaka, H. Yamashita, R. Tsuchitani, T. Funabiki and S. Yoshida, J. Chem. Soc., Faraday Trans., 1988, 84, 2987 Search PubMed.
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2006 |