A red-fluorescent substrate microarray for lipase fingerprinting†
Johann Grognux and Jean-Louis Reymond*
Department of Chemistry & Biochemistry, University of Berne, Freiestrasse 3, 3012 Berne, Switzerland. E-mail: jean-louis.reymond@ioc.unibe.ch; Fax: +41 31 631 80 57
First published on 24th August 2006
Abstract
A lipase substrate microarray was obtained by printing aliphatic C2–C12 monoesters of (5R)- and (5S)-3-(5,6-dihydroxyhexyloxy)benzaldehyde by reductive alkylation on amine-functionalized glass slides coated with bovine serum albumin and a short PEG linker. The microarray features 12 substrates and their 66 possible binary mixtures spotted in a 9 × 36 spot array. Lipase reactions are detected by chemoselective NaIO4-oxidation of the 1,2-diol hydrolysis product to form an aldehyde, which is then tagged with the red-fluorescent dye rhodamine B sulfohydrazide 1. Specific fingerprints are produced by active enzymes. These experiments provide the first example of lipase fingerprinting using microarrays.
Introduction
The detection of enzyme activities with specific substrates is an essential tool in biochemistry and biotechnology.1 Recording enzyme activities on multiple substrates simultaneously produces enzyme-specific activity fingerprints.2 Such fingerprints provide a basis for undertaking broad comparative functional studies of enzymes, which should lead to a refined understanding of structure–activity relationships, and might provide useful identification tools for routine work with enzymes. However unequivocal substrate families for fingerprinting are not available for most enzyme classes except for peptides and proteins for the analysis of protease3 and kinase specificity.4Herein we report the first microarray for lipase fingerprinting. Lipases are ubiquitous microbial enzymes of interest for biotransformations and as disease markers.5 Using simple microtiter-plate parallel assays for hydrolases,6 we recently showed that activity fingerprints measured with fluorogenic 1,2-diol monoesters allows us to classify lipases and esterases according to the acyl chain length dependence of reactivity.7 However the assay consumed relatively large quantities of both substrate and enzyme for each fingerprint and was difficult to implement in high-throughput.
Our lipase fingerprinting assay was improved in terms of throughput and reduced sample volume by using substrate cocktails analyzed by HPLC,8 or fluorogenic substrates adsorbed on silicagel plates.9 Nevertheless, both of these methods were limited to 10–30 substrate measurements per enzyme. To overcome this limitation, we turned our attention to microarray technology, which allows us to perform hundreds and even thousands of parallel measurements in arrayed micrometer-sized spots printed on the surface of glass slides.10 Microarray technology was recently implemented to measure enzyme activities in a variety of settings. Proteases have been profiled on microarrays using fluorogenic aminocoumarin amide peptides printed on glass slides,11 using soluble fluorogenic PNA-tagged peptide rhodamine-amides addressed to a DNA-microarray for product analysis,12 or by spotting protease substrates non-covalently followed by spraying of proteases.13 Kinases have been profiled with peptide microarrays.14 Microarray technology has also been used recently by attaching enzymes to the glass surface for testing with soluble activity-based probes,15 and for spotting various enzymes on slides coated with fluorogenic substrates.16 However, a single report of fluorogenic esterase substrates on microarrays showed no observable activity for these enzymes.17
The lipase fingerprinting microarray presented here uses substrates covalently linked to a glass surface and is based on a new, indirect tagging strategy for product detection (Scheme 1). Thus, aliphatic monoesters of surface tethered 1,2-diols are converted to free 1,2-diols upon enzyme hydrolysis. The 1,2-diol products are then detected by chemoselective oxidation with sodium periodate to form an aldehyde, which is marked by hydrazone formation with a fluorescent tag. This indirect tagging strategy is independent of the fluorophore, and was used here with the red fluorophore rhodamine B sulfohydrazide (1) compatible with standard microarray scanners. The aliphatic mono-esters of 1,2-diols used as substrate show negligible non-specific reactivity, in analogy to related fluorogenic substrates.18 This is critical for obtaining microarrays containing pure, product-free substrates. This would not be possible with standard fluorogenic lipase substrates consisting of labile esters of acidic phenols such as nitrophenol, umbelliferone, resorufin or fluorescein.19,20
 | ||
| Scheme 1 Principle of periodate-coupled lipase assay on microarray. | ||
Results and discussion
Lipase microarray substrates C2–C12 were designed in analogy to a substrate series used previously for enzyme fingerprinting and consisting in fluorogenic esters of aliphatic acids of increasing chain length.7 The microarray substrates featured a monoacylated 1,2-diol attached to an aromatic aldehyde via an ether linkage (Scheme 2). The aromatic aldehyde group would be used for coupling to an amine-functionalized glass slide by reductive alkylation. The reaction takes place under slightly acidic conditions (pH 4.5) allowing attachment of the prefunctionalized ester substrates without degradation of the enzyme-reactive group. Reductive alkylation has been used previously for covalent linkage of amine-functionalized oligonucleotides to aldehyde-coated microarrays.21 Here we placed the aldehyde group on the substrate after encountering difficulties in obtaining pure amine-functionalized ester substrates. | ||
| Scheme 2 Structure and synthesis of lipase substrates for microarrays. | ||
The substrates were prepared as follows (Scheme 2). Asymmetric dihydroxylation of 1-bromohex-5-ene 2 with AD-mix-α or AD-mix-β gave the optically enriched 1,2-diols (S)-3 and (R)-3, respectively. These were used to alkylate the sodium salt of 3-hydroxybenzaldehyde in DMF (dimethylformamide) to produce the corresponding ethers (R)-4 and (S)-4. The diols were then mono-esterified with aliphatic acyl chlorides of increasing chain length (C2, C4, C6, C8, C10, C12) to yield esters C2–C12 as either R or S enantiomers (12 substrates).
Rhodamine B sulfohydrazide 1 was selected as aldehyde staining reagent for its red fluorescent wavelength compatible with standard microarray scanners, although other dyes with similar properties are also available (Scheme 3).22 Hydrazone formation of aldehydes obtained by periodate cleavage of 1,2-diols is a well known tagging procedure in biology, for example for tagging gangliosides or sialyl residues with a dinitrophenyl group for recognition by antibodies.23 Reaction of rhodamine B sulfonyl chloride 5 with t-butyl carbazate gave the Boc-protected hydrazide 6, which was purified by preparative HPLC. The pure product 6 was then deprotected by treatment with trifluoroacetic acid to yield the free hydrazide reagent 1 as a trifluoroacetate salt. Solutions of hydrazide 1 were found to be sensitive to traces of acetone converting it to the corresponding hydrazone, therefore fresh solutions were prepared for each experiment.
 | ||
| Scheme 3 Synthesis of rhodamine sulfohydrazide 1. | ||
The lipase microarray assay was developed by iterative optimization of slide coating, substrate printing, enzyme incubation and tagging procedures. The best procedure found is described below with emphasis of the critical parameters influencing the assay.
Substrate microarrays were prepared starting from amine-functionalized glass slides (Scheme 4). The glass surface was first activated with bis-succinimidyl carbonate 7 and coated with bovine serum albumin (BSA) as described by MacBeath and Schreiber.24 The BSA-coated surface was then activated again with 7 and treated with 4,7,10-trioxa-1,13-tridecanediamine (TTD) to give a new amine-functionalized surface with a protein layer (BSA) and a short polyethylene glycol spacer arm for attachment of the enzyme substrates. The BSA–TTD double layer was the only surface compatible with lipase reactivity of the substrates as discussed below. Other amine-coated surfaces such as the slides themselves, slides coated with BSA alone, or slides coated with a double layer of TTD, gave no detectable enzyme reactivity in the subsequent assay. On the other hand, slides coated with a 4th generation PAMAM (polyamidoamine) dendrimer with 64 surface amines gave a surface on which all enzyme substrates were spontaneously hydrolyzed to the diol products after attachment.
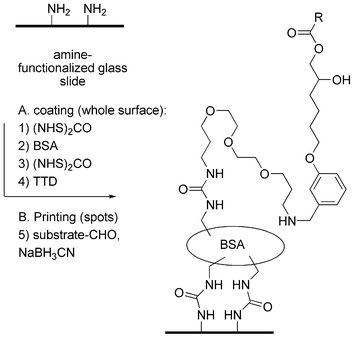 | ||
| Scheme 4 Slide coating and substrate printing procedure. | ||
Lipase substrates and reference products were then printed on the BSA-TTD coated slides. Substrate printing was optimized with racemic versions of product 4 and substrates C2–C10 printed in a 6 × 6 array. The optimal printing conditions involved overnight incubation of a 50 mM solution of the compounds with a molar equivalent of NaBH3CN in a 3 : 2 : 2 mixture of aqueous sodium acetate buffer pH 4.5, DMF and glycerol. This solvent mixture was reactive for reductive alkylation by ensuring the correct pH (acetate buffer), good solubility of the lipase substrates (DMF), and sufficient viscosity for good drop shape and slow evaporation (glycerol). Other solvent mixtures or ratios of the same components gave lower printing efficiency and poorer spot shape. Printing a lower substrate concentration (5 mM) gave only very faint spots in the subsequent assay and tagging procedure.
A 9 × 36, 324 spot array was printed for fingerprinting experiments using all 12 substrates (S)-C2-(S)-C12, (R)-C2-(R)-C12, and the reference products (S)-4 and (R)-4. In addition to printing the pure compounds at 50 mM, we also used all 66 possible binary combinations of substrates at 25 mM each, providing a total of 78 solutions for spotting. These solutions were distributed in a 384-well microtiter plate and a four-pin spotter was used to print the arrays defining six different blocks of 9 × 36 spots per slide. Each solution was printed in quadruplicate in each field to ensure statistical averaging of each data point. The slides were then thoroughly washed and stored in dry form at +4 °C for later use.
Microarray enzyme assays were carried out using the printed slides as follows. The blocks of 9 × 36 spots printed on the slides were delimited using a PAP-pen drawing a hydrophobic line to allow individual enzyme assays. Enzyme solutions (35 µL) were then applied manually to each block using a pipetter, and the slide was incubated for 2 h at 30 °C in a closed petri dish with water-saturated atmosphere to prevent evaporation. The slide was thoroughly rinsed and subjected to NaIO4-oxidation under optimized conditions of 1 mM aqueous NaIO4 for 30 min. After rinsing with water, tagging was carried out using 10 µM Rhodamine B sulfohydrazide 1 in PBS for 30 min. Higher concentrations of periodate or tag 1 did not increase staining intensity of the spots, but caused non-specific staining of the glass slide. The slides were then thoroughly washed with a sequence of 0.1% aqueous ammonium hydroxide, PBST–acetone 4 : 1, and finally DMF. The washing procedure was critical to avoid non-specific staining of the glass outside spots. Finally, each block delimiting a different enzyme reaction was analyzed using a microarray scanner (Fig. 1).
 | ||
| Fig. 1 Spot intensities of 9 × 36 spot lipase substrate microarrays after reaction with enzymes, oxidation with NaIO4, and tagging with rhodamine sulfohydrazide 1 (Scheme 1). Each field measures 1.35 × 0.35 cm. (A) Blank reaction with BSA, only the reference products (R)-4 and (S)-4 are marked. (B) Reaction with Hog pancreatic lipase 1 mg ml−1. (C) Reaction with Pseudomonas fluorescens type B lipase 1 mg ml−1. (D) Array layout. | ||
A series of lipases were tested for activity using the microarray. The assays were carried out in aqueous borate buffer pH 8.8 with 20% v/v DMSO. While all enzymes tested positive for activity using soluble fluorogenic versions of our substrates under these conditions, only very few of the enzymes displayed activity on the microarray as judged by their overall conversion across all substrates (Table 1). On the other hand the three most active enzymes, namely Candida antartica lipase (CAL), hog pancreatic lipase (HPL) and Pseudomonas fluorescens type B lipase (PSBL) retained their microarray activity upon dilution.
| Lipase | Code | 1000a | 100a | 10a | Substrate | Soln.b | Substrate |
|---|---|---|---|---|---|---|---|
| a Microarray assay conditions: 1000, 100 and 10 µg mL−1 enzyme in 20 mM aqueous borate pH 8.8 with 20% v/v DMSO, 2 h at 37 °C. Staining described in Scheme 3.b Initial reaction rate in pM s−1 for 100 µM solution of a fluorogenic 1,2-diol monoesters under the same conditions, data from ref. 7.c Data from ref. 18f. Esterases (pig liver esterase PLE1, PLE2, PLE3 and Thermoanaerobium brockii esterase TBE) showed no activity on microarrays. | |||||||
| Aspergillus aiger lipase, Fluka F62294 | ANL | — | — | 25'500 | C6 | ||
| Candida antartica lipase, Fluka F62299 | CAL | ++ | ++ | + | C2+C8 | 37'400 | C12 |
| Candida rugosa lipase, pur. Roche L3p | CRL1 | — | — | 55'200 | C12 | ||
| Candida rugosa lipase, Roche L3 | CRL2 | + | C10 | 52'700 | C8 | ||
| Chromobacterium viscosum lipoprotein lipase, Fluka F62333 | CVL | ++ | — | C6+C10 | 49'200 | C12 | |
| Hog pancreatic lipase, Fluka F62300 | HPL | ++ | ++ | — | C4 | 26'000 | C10 |
| Mucor javanicus lipase, Fluka F62304 | MJL | + | — | C8 | 28'600 | C12 | |
| Mucor miehei lipase, Fluka F62298 | MML | — | — | 22'600 | C10 | ||
| Pseudomonas cepacia lipase, Fluka F62309 | PCL2 | — | — | 40'700 | C12 | ||
| Pseudomonas fluorescens lipase, Fluka F62321 | PFL | — | — | 38'200 | C12 | ||
| Pseudomonas sp. B lipoprotein lipase, Fluka F62336 | PSBL | ++ | ++ | — | C2 | 53'200 | C12 |
| Pseudomonas sp. lipoprot lipase, Sigma L-9656 | PSL1 | + | C10 | 51'800 | C10c | ||
| Rhizopus arrhizus lipase, Fluka F62305 | RAL | + | — | C10 | 29'100 | C12 | |
| Rhizomucor miehei lipase, Fluka F62291 | RML | — | — | 30'100 | C12 | ||
The microarray fingerprints of the active lipases were analyzed in detail. Each data point was averaged over four individual spots, with generally less than 10% variability between spots. To facilitate visual analysis, the data were represented as a symmetrical 12 × 12 matrix showing relative conversion of each substrate or binary mixture in a yellow–red color scale (Fig. 2). Several fingerprints showed an apparent strong activity in spots printed with the shorter chain substrates C2 and C4 and their mixtures with other substrates. The strong reactivity of these short acyl chain substrates in the microarray contrasts to assays with soluble substrates, where lipases generally prefer longer chain aliphatic esters such as C12 substrates (Table 1). Interestingly, binary mixtures often showed stronger activities than the pure substrates, which might indicate variations in printing efficiencies between different mixtures, enzyme activation effects on mixed substrate surfaces, or a reduction of steric hindrance for access to the substrate. The stronger reactivity in substrate mixtures was beneficial to the experiment since it allowed us to detect enzymes that were only very weakly active with pure substrates on the microarray, for example CAL, CRL2 and RAL.
 | ||
| Fig. 2 Color-coded representation of lipase fingerprints on microarray. The color intensity of each substrate or binary mixture represents the averaged intensity over 4 spots on the microarray relative to the strongest reaction in the array. The percentage value for each enzyme gives the ratio of the averaged color intensity of the strongest reaction relative to the diol references. The data was generated from the microarrays as shown in Fig. 1. | ||
Fingerprinting the same enzyme several times but on different days produced similar fingerprints, as illustrated for the most reactive enzymes HPL, CAL and PSBL. The microarray fingerprints of the active lipases were analyzed for similarity by principal component analysis (PCA) to estimate the differentiation potential of the array (Fig. 3). PCA showed that the microarray could indeed differentiate between enzymes, while grouping repeated measurements of the same enzyme close to each other. HPL produced a characteristic fingerprint with very strong activity of (S)-C4 and (R)-C4 and all binary mixtures of these substrates. CAL and PSBL produced similar fingerprints with an overall strong reactivity of substrates and mixtures with short chain substrates (C2 and C4). A third group of enzymes with CVL, RAL, PSL1 and MJL was characterized by a somewhat stronger reactivity of intermediate chain length substrates C4–C8 over the other substrates. Finally CRL2 stood out by its overall weak reactivity.
 | ||
| Fig. 3 Principal component plot for microarray fingerprints of lipases. Enzymes are grouped according to clusters defined by Ward clustering on the basis of squared Euclidean distances. PC1 and PC2 represent 65% of the variance between the fingerprints. The fingerprints are shown in Fig. 2. | ||
Conclusion
The experiments above demonstrate the first substrate microarray for fingerprinting lipase activities. Products were detected by a new indirect tagging strategy involving periodate oxidation of 1,2-diol reaction products, followed by hydrazone formation between the generated aldehyde and rhodamine B sulfohydrazide 1 as a red-fluorescent tag, a method which is related to the cat-ELISA assays based on anti-product antibodies as product-selective reagents.25 Product detection by hydrazone formation is simpler than by product-specific antibodies, is applicable for any reaction producing 1,2-diols, 1,2-aminoalcohols, or directly aldehydes, and should be useful for a variety of enzymes other than lipases, such as acylases, phosphatases, or alcohol dehydrogenases.26 This tagging method was essential in the present lipase microarray to allow the use of stable aliphatic esters as substrates, and in rendering the enzyme assay independent of the fluorescent tag.While the periodate coupled tagging principle proved reliable for revealing the surface-tethered 1,2-diol products, we experienced difficulties in obtaining lipase reactivity on the microarray. Optimization of the coating procedure showed that the combination of BSA with a short PEG linker yielded stable yet lipase reactive microarrays, while other combinations gave either unreactive or unstable surfaces. Even under these optimized conditions, several enzymes showing good activity in solution were not active on the microarray, and only a limited number of lipases showed activity in our microarray assay. Nevertheless, two recent papers reported successful electrochemical assays of a lipase using a substrate attached to gold surfaces,27 providing independent evidence that solid-supported substrate are sometimes accepted by lipases. Further experiments to optimize the lipase substrate display chemistry for microarrays are underway.
Experimental section
Slide preparation
Amine derivatized slides (Genetix) are immersed in 30 mL of a solution of N,N-diisopropyl-ethylamine (DIEA, 0.51 mL, 3.0 mmol, 100 mM) and N, N′-disuccinimidyl carbonate (770 mg, 3.0 mmol, 100 mM) in DMF for 4 h, with gentle stirring. The slides are then successively washed with DMF and EtOH (2 × 10 min each), and dried by centrifugation. The activated slides were then immersed in 30 mL of a 1% BSA (W/V) in phosphate buffered saline (PBS, 160 mM NaCl, 10 mM phosphate pH 7.4) and gently stirred overnight. The slides were then washed with water, DMF and EtOH (2 × 10 min each), and dried by centrifugation. The BSA-coated slides were then subjected to activation with N,N′-disuccinimidyl carbonate as described above. Finally, the activated BSA slides were immersed in 30 mL of a solution of DIEA (1.54 mL, 9.0 mmol, 300 mM) and TTD (1.89 mL, 9.0 mmol, 300 mM) in DMF and gently stirred overnight. The slides were then successively washed with DMF and EtOH (2 × 10 min each), and dried by centrifugation.Substrate printing
Substrates were spotted on microarray slides using a QArraymini equipped with a Qu solid pins of 150 µm (ref. K2783) on HPLF head, on coated slides prepared from amine slides (ref. K2620), all provided by Genetix, New Milton, UK, at ambient humidity (no controller installed). All substrates and binary mixtures were spotted in quadruplicate at a concentration of 50 mM (25 mM for each substrate in the case of binary mixes) with 50 mM NaBH3CN in a solution of acetate buffer (200 mM, pH 4.5), glycerol and DMF (3 : 2 : 2). Spots are deposited on the slides with a pitch of 370 µm, and let for reaction overnight before washing with DMF and EtOH (2 × 15 min), and dried by centrifugation. Six fields were spotted per slide.Enzyme assays on microarrays
Each block on the microarray was surrounded with a hydrophobic border applied with a PAP-pen. The solution of enzyme (35 µL at the specified concentration, in 20 mM borate buffer pH 8.8, 20% DMSO V/V) was then applied to the array with a Pipetman (Gibson). The slide was then placed in a Petri dish with a piece of cotton-wool impregnated with water to limit evaporation of the enzyme solution by saturating the dish atmosphere, and incubated for 2 h at 30 °C. The slides were then washed for 2 × 15 min in PBST (PBS buffer pH 7.4, 0.05% Tween-20 V/V), rinsed with DMF and EtOH, and dried by centrifugation. A solution of NaIO4 (100 µM in water, 30 min) was then applied at 25 °C. After rinsing and drying, a solution of rhodamine sulfohydrazide 1 (10 µM in PBS) was applied for 30 min. The solution was rinsed away with deionized water, and washed by gentle stirring in successive solutions of NH4OH (0.1% in water), PBSTA (PBST and acetone, 4 : 1), and DMF for 15 min each. Slides were then rinsed with EtOH and dried by centrifugation.Microarray analysis
Microarrays were analyzed with a ScanArray 4000 (Packard Biochip Technology) with laser wavelength set at 488 nm and emission filter at 560 nm. Quantification was made with QuantArray 3.0 (same provider). Analysis was made with a laser power set to 100%.Acknowledgements
This work was supported by the University of Berne, the Swiss National Science Foundation, the COST program D16, and Protéus SA, Nîmes, France.References
- (a) Enzyme Assays: A Practical Approach, ed. Robert Eisenthal and Michael Danson, Oxford University Press, Oxford, UK, 2002 Search PubMed; (b) Enzyme Assays: Essential Data, ed. S. Gul, S. K. Sreedharan and K. Brocklehurst, John Wiley & Sons, 1998 Search PubMed; (c) M. T. Reetz, Angew. Chem., 2001, 113, 292 CrossRef; Angew. Chem., Int. Ed., 2001, 40, 284 Search PubMed; (d) D. Wahler and J.-L. Reymond, Curr. Opin. Chem. Biol., 2001, 5, 152 CrossRef CAS; (e) D. Wahler and J.-L. Reymond, Curr. Opin. Biotechnol., 2001, 12, 535 CrossRef CAS; (f) H. Lin and V. W. Cornish, Angew. Chem., Int. Ed., 2002, 41, 4402 CrossRef CAS; (g) F. H. Arnold and G. Georgiou, Methods Mol. Biol., 2003, 230, 213 Search PubMed; (h) J.-P. Goddard and J.-L. Reymond, Trends Biotechnol., 2004, 22, 363 CrossRef CAS; (i) J.-P. Goddard and J.-L. Reymond, Curr. Opin. Biotechnol., 2004, 15, 314 CrossRef CAS; (j) Enzyme Assays: High-throughput Screening, Genetic Selection and Fingerprinting, ed. J.-L. Reymond, Wiley-VCH, Weinheim, Germany, 2006 Search PubMed.
- (a) J.-L. Reymond and D. Wahler, ChemBioChem, 2002, 3, 701–708 CrossRef CAS; (b) J. Grognux and J.-L. Reymond, Enzyme Fingerprinting Assays for Hydrolases, in Enzyme Assays: High-throughput Screening, Genetic Selection and Fingerprinting, ed. J.-L. Reymond, Wiley-VCH, Weinheim, Germany, 2006, pp. 271–302 Search PubMed.
- (a) D. J. Maly, L. Huang and J. A. Ellman, ChemBioChem, 2002, 3, 16–37 CrossRef CAS; (b) J. Harris, Protease Substrate Profiling, in Enzyme Assays: High-throughput Screening, Genetic Selection and Fingerprinting, ed. J.-L. Reymond, Wiley-VCH, Weinheim, Germany, 2006, pp. 303–332 Search PubMed.
- (a) M. Uttamchandani, E. W. Chan, G. Y. Chen and S. Q. Yao, Bioorg. Med. Chem. Lett., 2003, 13, 2997–3000 CrossRef CAS; (b) S. Panse, L. Dong, A. Burian, R. Carus, M. Schutkowski, U. Reimer and J. Schneider-Mergener, Mol. Divers., 2004, 8, 291–299 Search PubMed; (c) N. Dephoure, R. W. Howson, J. D. Blethrow, K. M. Shokat and E. K. O'Shea, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 17940–17945 CrossRef CAS.
- (a) R. D. Schmidt and R. Verger, Angew. Chem., Int. Ed., 1998, 37, 1608 CrossRef; (b) M. T. Reetz, Curr. Opin. Chem. Biol., 2002, 6, 145 CrossRef CAS.
- (a) D. Wahler, F. Badalassi, P. Crotti and J.-L. Reymond, Angew. Chem., Int. Ed., 2001, 40, 4457 CrossRef CAS; (b) D. Wahler, F. Badalassi, P. Crotti and J.-L. Reymond, Chem.–Eur. J., 2002, 8, 3211–3228 CrossRef CAS; (c) A. M. F. Liu, N. A. Somers, R. J. Kazlauskas, T. S. Brush, F. Zocher, M. M. Enzelberger, U. T. Bornscheuer, G. P. Horsman, A. Mezzetti, C. Schmidt-Dannert and R. D. Schmid, Tetrahedron Asym., 2001, 12, 545–556 Search PubMed.
- J. Grognux and J.-L. Reymond, ChemBioChem, 2004, 5, 826–831 CrossRef CAS.
- (a) J.-P. Goddard and J.-L. Reymond, J. Am. Chem. Soc., 2004, 126, 11116–11117 CrossRef CAS; (b) R. Sicard, J.-P. Goddard, M. Mazel, C. Audiffrin, L. Fourage, G. Ravot, D. Wahler, F. Lefèvre and J.-L. Reymond, Adv. Synth. Catal., 2005, 347, 987–996 CrossRef CAS; (c) Y. Yongzheng and J.-L. Reymond, Mol. BioSyst., 2005, 1, 57–63 RSC.
- P. Babiak and J.-L. Reymond, Anal. Chem., 2005, 77, 373–377 CrossRef CAS.
- Y. Hu, M. Uttamchandani and S. Q. Yao, Comb. Chem. High Throughput Screening, 2006, 9, 203–212 CrossRef CAS.
- (a) C. M. Salisbury, D. J. Maly and J. A. Ellman, J. Am. Chem. Soc., 2002, 124, 14868–14870 CrossRef CAS; (b) D. N. Gosalia, C. M. Salisbury, J. A. Ellman and S. L. Diamond, Mol. Cell. Proteomics, 2005, 4, 626–636 Search PubMed.
- N. Winssinger, R. Damoiseaux, D. C. Tully, B. H. Geierstanger, K. Burdick and J. L. Harris, Chem. Biol., 2004, 11, 1351–1360 CrossRef CAS.
- (a) D. N. Gosalia and S. L. Diamond, Proc. Natl. Acad. Sci. U. S. A., 2003, 15, 8721–8726 CrossRef; (b) D. N. Gosalia, C. M. Salisbury, D. J. Maly, J. A. Ellman and S. L. Diamond, Proteomics, 2005, 5, 1292–1298 CrossRef CAS.
- (a) F. Toepert, T. Knaute, S. Guffler, J.-R. Pires, T. Matzdorf, H. Oschkinat and J. Schneider-Mergener, Angew. Chem., Int. Ed., 2003, 10, 1136–1140 CrossRef; (b) M. Schutkowski, U. Reineke and U. Reimer, ChemBioChem, 2005, 6, 513–521 CrossRef CAS.
- (a) R. Srinivasan, X. Huang, S. L. Ng and S. Q. Yao, ChemBioChem, 2006, 7, 32–36 CrossRef; (b) H. Schmidinger, H. Susani-Etzerodt, R. Birner-Gruenberger and A. Hermetter, ChemBioChem, 2006, 7, 527–534 CrossRef CAS; (c) D. P. Funeriu, J. Eppinger, L. Denizot, M. Miyake and J. Miyake, Nat. Biotechnol., 2005, 23, 622–627 CrossRef CAS.
- M. Uttamchandani, X. Huang, G. Y. J. Chen and S. Q. Yao, Bioorg. Med. Chem. Lett., 2005, 15, 2135–2139 CrossRef CAS.
- Q. Zhu, M. Uttamchandani, D. Li, M. L. Lesaicherre and S. Q. Yao, Org. Lett., 2003, 5, 1257–1260 CrossRef CAS.
- (a) F. Badalassi, D. Wahler, G. Klein, P. Crotti and J.-L. Reymond, Angew. Chem., Int. Ed., 2000, 39, 4067 CrossRef CAS; (b) D. Lagarde, H.-K. Nguyen, G. Ravot, D. Wahler, J.-L. Reymond, G. Hills, T. Veit and F. Lefevre, Org. Process. R. & D., 2002, 6, 441 Search PubMed; (c) E. M. Gonzalez-Garcia, J. Grognux, D. Wahler and J.-L. Reymond, Helv. Chim. Acta, 2003, 86, 2458 CrossRef CAS; (d) E. Nyfeler, J. Grognux, D. Wahler and J.-L. Reymond, Helv. Chim. Acta, 2003, 86, 2919 CrossRef CAS; (e) F. Badalassi, G. Klein, P. Crotti and J.-L. Reymond, Eur. J. Org. Chem., 2004, 2557 CrossRef CAS; (f) J. Grognux, D. Wahler, E. Nyfeler and J.-L. Reymond, Tetrahedron Asym., 2004, 15, 2981 Search PubMed; (g) Y. Yang, P. Babiak and J.-L. Reymond, Org. Biomol. Chem., 2006, 4, 1746–1754 RSC; (h) J.-L. Reymond, Food Technol. Biotechnol., 2004, 42, 265–269 Search PubMed.
- (a) G. G. Guibault and J. Hieserman, Anal. Chem., 1969, 41, 2006 CrossRef; (b) Y. Yang, P. Babiak and J.-L. Reymond, Helv. Chim. Acta, 2006, 89, 404–415 CrossRef.
- (a) M. Schmidt and U. T. Bornscheuer, Biomol. Eng., 2005, 22, 51 Search PubMed; (b) D. Gilham and R. Lehner, Methods, 2005, 36, 139 Search PubMed.
- (a) K. Lindroos, U. Liljedahl, M. Raitio and A. C. Syvanen, Nucleic Acids Res., 2001, 29, E69–9 CrossRef CAS; (b) M. Dufva, Biomol. Eng., 2005, 22, 173–184 Search PubMed.
- N. Panchuk-Voloshina, R. P. Haugland, J. Bishop-Stewart, M. K. Bhalgat, P. J. Millard, F. Mao, W. Y. Leung and R. P. Haugland, J. Histochem. Cytochem., 1999, 47, 1179–1188 Search PubMed.
- (a) S. Spiegel, A. Ravid and M. Wilchek, Proc. Natl. Acad. Sci. U. S. A., 1979, 76, 5277–5281 CrossRef CAS; (b) A. Ravid, A. Novogrodsky and M. Wilchek, Eur. J. Immunol., 1978, 8, 289–294 Search PubMed.
- G. MacBeath and S. L. Schreiber, Science, 2000, 289, 1760–1763 CAS.
- (a) D. S. Tawfik, B. S. Green, R. Chap, M. Sela and Z. Eshhar, Proc. Natl. Acad. Sci. U. S. A., 1993, 90, 373 CAS; (b) G. MacBeath and D. Hilvert, J. Am. Chem. Soc., 1994, 116, 6101 CrossRef CAS; (c) F. Benedetti, F. Berti, F. Massimiliano, M. Resmini and E. Bastiani, Anal. Biochem., 1998, 256, 67 CrossRef CAS; (d) P. Geymayer, N. Bahr and J.-L. Reymond, Chem.–Eur. J., 1999, 5, 1006 CrossRef CAS; (e) F. Taran, C. Gauchet, B. Mohar, S. Meunier, A. Valleix, P. Y. Renard, C. Creminon, J. Grassi, A. Wagner and C. Mioskowski, Angew. Chem., Int. Ed., 2002, 41, 124–127 CrossRef CAS.
- (a) G. Klein and J.-L. Reymond, Bioorg. Med. Chem. Lett., 1998, 8, 1113–6 CrossRef CAS; (b) G. Klein and J.-L. Reymond, Helv. Chim. Acta, 1999, 82, 400 CrossRef CAS.
- (a) W. S. Yeo and M. Mrkisch, Angew. Chem., Int. Ed., 2003, 42, 3121–3124 CrossRef CAS; (b) G. Valincius, I. Ignatjev, G. Niaura, M. Kazemekaite, Z. Talaikyte, V. Razumas and A. Svendsen, Anal. Chem., 2005, 77, 2632–2636 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedures and spectral data for all compounds, NMR-spectra for all compounds, and images of lipase fingerprints on microarrays. See DOI: 10.1039/b609275f |
| This journal is © The Royal Society of Chemistry 2006 |