Translating peptides into small molecules†
Gerd Hummel, Ulrich Reineke and Ulf Reimer
Jerini AG, Invalidenstraße 130, Berlin, 10115, Germany
First published on 24th August 2006
1 Peptides as drugs: the good, the bad and the ugly
Nature entrusts peptides with numerous tasks ranging from passing on messages as peptide hormones to priming the immune system, tuning metabolism and protein degradation or defending microorganisms as antibiotic agents. In addition, researchers have identified a variety of peptides with artificial biological functions that are not represented by naturally occurring molecules, e.g. receptor antagonists or inhibitors of protein–protein interactions. These peptides or their derivatives are valuable starting points for drug development and, indeed, a number of peptides or peptide-derived drugs have already made their way into the clinic.1 Compared to the standard properties of drug-like small molecules matching the “rule of five”, which are today's preferred formats, peptide-based drugs sometimes overcome the barrier of “undrugable targets” and are often extremely potent. Furthermore, many peptides are highly specific and do not accumulate in the body since they have short half-lives. As a consequence, peptides often show less systemic toxicity compared to small molecules.1 In addition, peptides are ideal, rapidly accessible lead structures for drug-target validation as well as further drug development due to their modular structure, variable presentation of different functional groups and ease of preparation, either chemically or biologically.Despite their favourable properties, peptide-based drugs are under-represented in the pharmaceutical market. This discrimination is usually due to their poor bioavailability, which sometimes necessitates non-oral administration or even special medical devices such as inhalers. Another related major disadvantage of peptides is their low metabolic stability due to proteolytic degradation. In addition, costs of goods for the drug substance are sometimes tremendous. Therefore, there is considerable interest to transform the “active principle” of biologically active peptides into small molecules with improved pharmacokinetic properties. In this chapter, we present an overview of ways to identify active peptides and how to tailor them for transformation into peptidomimetics or small molecules. This introduction is followed by descriptions of classical transformation approaches using stepwise substitutions of peptidic features. Conceptually different is the strategy in which peptides are used to generate pharmacophore models representing all necessary functional properties in the appropriate spacing and 3-D orientation. These pharmacophore models are then applied to in silico screens of small-molecule libraries to identify compounds that resemble the “active principle” of the starting peptides.
2 Origin of biologically active peptides
Organisms from all species produce a huge variety of peptides in order to respond to certain physiological or pathophysiological stimuli. Reflecting their many different functions, their molecular structures are extremely diverse, ranging from very short peptides such as the enkephalins to complex peptide hormones like insulin with 51 amino acids in two chains connected by two disulfide bonds.Also very similar to peptides occurring in nature are active peptides identified from the primary structure of larger proteins (knowledge- or sequence-based approach). This approach involves scanning the entire sequence of the protein with overlapping peptides, usually not longer than 15 amino acids (peptide scan), which are screened for interactions with a binding partner. The sequence common to the interacting peptides is the binding site2 and such peptides often inhibit the respective protein–protein interaction.
As an alternative to the knowledge or sequence-based approaches, many methods to identify bioactive peptides de novo have been developed. Biological display techniques such as phage display were described3 and widely applied very early on. Contemporary standard libraries of linear or cyclic peptides have a diversity of approximately 109 independent clones, meaning libraries with up to seven randomised positions can theoretically guarantee comprehensive coverage of the potential sequence repertoire. However, in vitro translation systems result in peptide libraries with even higher diversity, since coupling of the peptide with its mRNA is achieved in a cell-free system involving small particles of mRNA/peptide/ribosome or only mRNA–peptide complexes. In addition, no diversity-limiting transformation steps are required. Libraries with diversities from 1010 up to 1014 different peptides can be prepared using polysome or ribosome display4 or the PROfusion™ technology.5
Chemically prepared peptide libraries can be classified into three different types: (1) Multiple peptide synthesis techniques permit the preparation of hundreds or even thousands of individual peptides. Each sequence is known from its position during the, usually automated, synthesis process and large amounts of comparatively pure compounds are obtained. However, the feasible number of peptides is rather limited compared to the diversity achieved with biological libraries. In principle, all peptide-synthesis strategies can be applied to generate these libraries, including conventional solid or solution-phase procedures as well as array-based syntheses like the SPOT™ method.6 (2) Combinatorial library techniques generating mixtures of beads with one individual peptide each7 use a process called portion mixing or the “one-bead-one-peptide” approach.8 Depending on the bead size and reactor volume, up to 107 or even 108 peptides with natural as well as non-natural building blocks can be generated.7 The disadvantage of this technique is the need to identify the structures of the active compounds after screening using sophisticated but rather tedious coding and decoding or sequencing processes. (3) The sequence identification step can be circumvented by using combinatorial peptide libraries with randomised as well as defined positions.9 In this approach, the entire library is subdivided into a small number of peptide mixtures that have individual amino acids at certain positions: O1XXXXX, XO2XXXX, XXO3XXX, XXXO4XX, XXXXO5X and XXXXXO6(O = position with a defined amino acid, X = position with a mixture of amino acids). If the 20 naturally encoded amino acids are each used for the defined positions (O), this library comprises 120 separate mixtures that are screened for binding to the target. Subsequently, individual peptides representing all possible combinations of the most active amino acids at each position (positional scanning approach) are synthesised and screened. All randomised positions must be deconvoluted by an iterative process based on the results obtained with the starting library.10
3 General strategy for translating peptides into small molecules
The most commonly used strategy to transform peptides into non-peptides is outlined in Figure 1.11 | ||
| Fig. 1 Strategy for transforming peptides into small molecules. | ||
Once the primary structure of the biologically active peptide has been determined, the first step is to identify the smallest active fragment required for biological activity. This step involves preparing truncated peptides in which amino acids from the amino and carboxyl termini have been removed, one at a time. Subsequently, the influence of each individual amino acid on the biological activity is determined by systematically replacing each residue in the peptide with specific amino acids, such as alanine or D-amino acids. After the structure–activity relationship (SAR) of each amino acid in the peptide has been explored, the bioactive conformation is investigated by introducing constraints at various positions in the peptide to reduce its conformational flexibility. Whenever possible, all the effects of the introduced constraints should be analysed by biophysical methods. In the final step, the essential amino acid side chains are positioned on carefully selected non-peptide scaffolds to correspond with the derived model of the bioactive conformation. Additionally, the 3-D pharmacophore model can be used for virtual screening of compound libraries (see Section 5).
Although several examples exist in which peptides have been converted successfully into non-peptidic drug candidates, the process is difficult and there is no guarantee of success. The most common methods for the stepwise transformation of peptides into small molecules are presented in more detail below.
4 Tailoring peptide sequences for their translation into small molecules
Peptides to be transformed into small molecules should be as small and as rigid as possible. There is no rule to say which molecular weight leads to a successful transformation process, but as a rule of thumb, peptides with fewer than six residues are very good starting points, whereas peptides between 6–15 residues are challenging.Biologically active peptides often contain a well-defined core of key residues. In addition, these peptides include other dispensable positions, resulting from either the predefined peptide length used in the library design or evolutionary processes. In order to narrow down the peptide to the “active principle” or to minimise the molecular weight to facilitate peptide-based drug design, three different types of libraries are useful: (1) truncation libraries (synonyms: size scan, window scan) comprise peptides omitting one or more N-, C- or N- and C-terminal amino acids (Figure 2A). (2) Peptides from libraries of deletion analogues (Figure 2B) have one or more consecutive amino acid(s) deleted at all possible positions. (3) Compared to deletion libraries, combinatorial deletion libraries additionally cover peptides with two or more positions omitted independently throughout the sequence (Figure 2C). It should be noted that the number of peptide analogues covered by a combinatorial deletion library rapidly increases, depending on the number of deleted positions and the peptide length.
 | ||
| Fig. 2 Library design for analysis and optimisation of peptide length. | ||
Peptides are typically highly flexible, but should be as rigid as possible for the transformation process, for two reasons: (1) the binding-free energy of a peptide interacting with a binding partner can be improved, and even more important (2) a rigid conformation helps in generating a peptide-based pharmacophore model because functional groups that are important for activity are already prepositioned and can be assigned in three dimensions, preferably by NMR techniques.
Conformational flexibility can be reduced by introducing local and/or global constraints at various positions in the peptide. Local conformational constraints can be achieved by incorporating modified amino acids (D-, N-methyl, α-methyl, cyclic, α,β-dehydro, β-substituted amino acids), replacing the amide moiety by isosteres (CH=CH, CH2CH2, CH2NH, NHCO, CSNH, COCH2, CH2O, CH2S, SOCH2, CH(OH)CH2, etc.) and short-range cyclisations, either within a single amino acid (proline or proline mimetics) or between adjacent residues (Figure 3).
 | ||
| Fig. 3 Short-range cyclisations between adjacent amino acids. | ||
Global constraints are achieved by medium- or long-range cyclisations involving C- and N-termini, backbone-to-backbone, side chain-to-side chain or side chain–to-backbone cyclisations. Cyclisation can impose significant conformational restrictions on the peptide backbone and the location of attached side chains. The most common examples of side chain-to-side chain cyclisation include the formation of disulfide bridges between cysteine residues and the formation of lactam bridges between glutamic/aspartic acid and lysine residues (Figure 4).
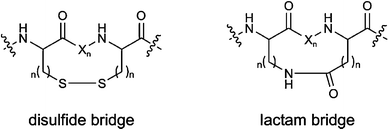 | ||
| Fig. 4 Examples of long-range cyclisations (X: amino acid). | ||
The binding conformation is usually unknown since structure determination of peptide–protein complexes by X-ray crystallography or NMR is time consuming and laborious. Furthermore, docking of peptides to binding partners in silico is one of the most complex modelling problems due to the tremendous intrinsic flexibility of the ligand. Therefore, a large number of cyclic peptide analogues have to be synthesised and screened to seek out the proper conformation of a biologically active peptide. A systematic approach is the “cyclisation scan” comprising all possible combinations of two cysteine residues within the starting peptide (Figure 5). A cyclisation scan by disulfide-bond formation via cysteine residues is the most easily implemented strategy. However, several other chemical cyclisation strategies can be similarly applied.
 | ||
| Fig. 5 Cyclisation scan. | ||
Additionally, secondary structure mimetics can be used to introduce constraints that may either have local or global effects. The secondary structure of a peptide is dominated by energetically favoured torsion angels ϕ, φ and ω, together with additional stabilizing factors such as hydrogen bonds and hydrophobic contacts. The most common secondary structures found in peptides and proteins are α-helices, β-sheets and reverse turns. Reverse turns have often been implicated as recognition elements for peptide–receptor interactions, and therefore introducing turn mimetics into peptides is an important strategy for probing bioactive conformations.12Reverse turns are classified according to the number of amino-acid residues involved as γ-turns (three amino acids), β-turns (four amino acids), α-turns (five amino acids) or π-turns (six amino acids).
The most common naturally occurring β-turn is usually defined as any tetrapeptide sequence, occurring in a non-helical region, in which the distance between Cα(i) and Cα(i + 3) is less than 7 Å.13 A β-turn is often stabilised by a hydrogen bond between the carbonyl function of residue i and the NH-group of residue i + 3 to give a 10-membered ring (Figure 6). The majority of the β-turn mimetics synthesised are dipeptide replacements for residues i + 1 and i + 2. A large number of β-turn mimetics have been reported in the literature;14 two examples are shown in Figure 6.

A γ-turn, which is a more rare reverse turn, is defined by a three-residue turn forming a seven-membered hydrogen bonded-ring between the carbonyl of the i residue and the amide NH of the i + 2 residue.17 Most of the γ-turn mimetics described in the literature consist of a six-or seven-membered heterocyclic-or carbocyclic-ring structure (Figure 7).

The introduction of conformational constraints should influence the backbone conformation without compromising any crucial side chain interaction with the receptor. A loss in activity after the introduction of a conformational constraint may either be caused by steric hindrance between the ligand and the receptor, due to the added constraining atoms, or by inability of the ligand to adopt the proper conformation. If activity is retained in the constrained analogue, the analogue is able to adopt the proper conformation, and binding is allowed in spite of the added restricting atoms.
Throughout the minimisation and conformational stabilisation process, the amino acid composition may have to be reoptimised to maintain activity and selectivity. This process is typically achieved by extensive synthesis and testing of amino acid substitution analogues. Systematic approaches are (1) amino-acid-substitution scans in which all positions of a peptide are substituted by one amino acid, e.g. alanine or proline and (2) complete substitutional analyses in which each position is substituted by a larger set of building blocks, such as all genetically encoded amino acids (Figure 8). Unnatural building blocks significantly increase the chemist's options for peptide optimisation and tailoring.
 | ||
| Fig. 8 Substitution scan and substitutional analysis. | ||
5 Transformation of peptide ligands into small molecules using computational approaches
The SAR of the constrained analogues together with the information obtained from biophysical studies (X-ray, NMR) and computational methods can be used, in an iterative process, to provide information about the receptor-bound and/or biologically active conformation. This model in turn can serve as the basis for a 3-D pharmacophore model. In the last step, topographic information from the 3-D pharmacophore model is used to position the amino acid side chains in the correct spatial arrangement on selected non-peptidic scaffolds or templates, such as small (five- to seven-membered) ring systems of defined stereochemistry. This process is well supported by the computational methods described in this section.Rational approaches take into account all available information about the target–ligand system in question to narrow down the chemical space of potential small molecule ligands. In the following section, we focus on some computational techniques that can support the transformation of peptidic ligands into small molecule ligands. Computational chemistry provides a number of techniques for this transformation, which can be divided into structure- and ligand-based techniques. Both categories rely on information about the structure of the target or the bound ligand. The so-called 2-D methods, which only take into account the topology of ligand molecules, have not proven to be very successful for the transformation of peptidic structures into small molecules.
Structure-based methods utilise information on the 3-D structure of the target protein. This information can be used as a template for docking experiments in which libraries of small molecules are fitted into the binding site of the target. The docking solutions are ranked by means of various scoring functions, leading to a hit list of potential binders.
Ligand-based design depends on structural information about the ligands. This ligand structure is either known from experimental techniques such as X-ray crystallography or NMR spectroscopy or can be deduced from other ligands with a known structure, such as proteins, or from SAR data and sequence information about the ligands. The ligand structure can be translated into a pharmacophore model, which is a 3-D representation of the ligand's functionalities or features important for the target–ligand interaction. This pharmacophore can be used for the virtual screening of huge libraries of small molecules. This virtual screening process leads to a relatively small number of hits, which are tested for activity. Pharmacophore-based virtual screening methods are standard tools in computational chemistry. Implementation of such algorithms is achieved using a number of commercially available programs such as Catalyst, CeriusII (both Accelrys), Unity (Tripos) or MOE (CCG), to name but a few. The key to making this approach feasible is by determining the structure of a bound peptide. Three starting situations are conceivable: detailed information on the active complex comprising the protein target and its peptidic ligand is available; the structure of the target protein and the sequence of one or more active peptidic ligands are known; or ligand sequences and detailed structure–activity data for these ligands are known.
An alternative approach in ligand-based design is to take the functional groups of the peptide that interact with the target and graft them onto a different, non-peptidic scaffold. The scaffold is chosen such that the groups adopt the appropriate 3-D relationship. The program CAVEAT was developed for this purpose, enabling the rapid search of 3-D molecular databases to identify templates that have bonds that adopt the correct orientation to serve as attachment points.20 This approach has been used in devising inhibitors of Factor Xa21 and antagonists of the bradykinin B2 receptor.22
A pharmacophore model can be constructed directly from the information available from the structure of a complex between a peptidic ligand and its target protein, which can reveal which interactions are productive and necessary for binding the ligand. Functionalities involved in the interaction pattern between the target and the ligand are called features. The spatial arrangement and the properties of these features are known from the structure of the complex. Typical pharmacophore features include: hydrophobes, ring centres, hydrogen-bond donors and acceptors and positive and negative ionisable functional groups. Mapping these features onto the ligand structure in the complex results in a pharmacophore model that can be readily used for virtual screening.
As an example, we describe the pharmacophore generation and virtual screening for the model-case thrombin. Due to the wealth of structural information about thrombin–inhibitor complexes, and the considerable number of known inhibitors, this protease is frequently used as a test case in computational chemistry. We use the coordinates of the bound inhibitor from the high-resolution crystal structure of a thrombin–D-Phe–Pro–Arg–chloromethyl ketone complex (pdb code 1ppb, resolution 1.92 Å, Figure 9A) as a template to generate a pharmacophore model. The four backbone atoms of the peptidic ligand involved in hydrogen bonds to thrombin are translated into one hydrogen-bond donor or three acceptor features in this model (Figure 9B). The phenylalanine aromatic ring in the peptide is overlaid with an aromatic ring feature and the peptide's proline ring is reflected by a hydrophobic feature. Finally, the guanidinium group of the peptidic arginine is represented as a positively ionisable feature in the pharmacophore model.
 | ||
Fig. 9 Virtual screening procedure based on the high-resolution (1.92 Å) complex crystal structure of thrombin and an inhibitory peptide. (A) Surface representation of the binding pocket of thrombin complexed with D-Phe–Pro–Arg–chloromethylketone. (B) Overlay of the bound structure of the thrombin ligand and the resulting pharmacophore (green: hydrogen-bond acceptors; violet: hydrogen-bond donor; brown: ring aromatic; blue: hydrophobic; red: positively ionizable). (C) Enrichment curve for 100 spikes of known thrombin inhibitors in a library of 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 260 molecules after virtual screening using the pharmacophore model shown in (B). 260 molecules after virtual screening using the pharmacophore model shown in (B). | ||
To test the performance of this rapidly generated pharmacophore model, we compiled a dataset of 10160 randomly chosen molecules from the catalogue of a commercial supplier of screening compounds (Maybridge). This set was spiked with 100 molecules described as thrombin inhibitors. In the subsequent virtual screening process, a conformational model of each compound in the screening library was fitted to the pharmacophore model. The resulting score is a measure of the quality of the overlap between the pharmacophore model in question and the respective library molecule. In this case, we used the Catalyst software package (Accelrys Inc.). After the screening procedure, the library was ranked according to the fitting scores. With our simple pharmacophore model, we were able to enrich almost 60% of the spiked thrombin inhibitors in the first 10% of the entire test library (Figure 9C). The performance of such pharmacophore models can be improved by including excluded volumes or shape information from the target molecule. Information about where a potential hit molecule might clash with the target protein and the shape of an interacting molecule can both be determined from the intricate structure of a target–peptide complex.
The thrombin–inhibitor example demonstrates a path from the bioactive 3-D structure of a peptide to small molecules. However, unambiguous structural information on the bioactive ligand conformation needed for such a transformation is only available in a limited number of cases. In most cases, additional complementary information has to be used to deduce the bioactive conformation of a bound ligand, and thus a pharmacophore model for virtual screening or design.
If a peptidic ligand presented to a target is a fragment of a protein with a known 3-D structure, or the structure of a different binding protein is known, these structures can help deduce the binding conformation of the peptidic ligand. For instance, this strategy was successful in screening for novel α4β1 (very late antigen-4, VLA-4) antagonists. A derivative of the tripeptide Leu–Asp–Val is known to bind tightly to α4β1. However, the bioactive conformation of this peptide is unknown. To deduce this conformation, the X-ray structure of the vascular cell adhesion molecule-1 (VCAM-1) was used.23 VCAM-1 is a natural ligand of α4β1. The tripeptide derivative was based on the X-ray crystal structure of the integrin-binding region of VCAM-1. After virtual design and screening of a library of 8624 molecules, 12 molecules were identified and synthesised as potential α4β1 antagonists. All of these molecules were active, with the most potent compound having an IC50 of 1 nM (Figure 10).
 | ||
| Fig. 10 Starting structure of the tripeptide derivative (left, IC50 = 0.6 nM) and resulting virtual screening hit (right, IC50 = 1.3 nM). | ||
Other approaches were successful even in the absence of any structural information about a complex or a ligand protein that could be used as a template. The main challenge in such cases is to cope with the inherent flexibility of peptidic molecules. A rapid exchange between different conformations is observed in the solution. However, depending on the sequence of the particular peptide, certain conformations can be stabilised. Such preferred conformations might be similar to the binding conformation in target–peptide complexes. NMR spectroscopy is an ideal technique for determining peptide conformation in solution. In addition, energetically favoured peptide conformations can also be calculated using different computational methods, and both NMR and computational methods have been used to estimate binding conformations of peptides. The probability of success increases when all available data concerning the SAR of the peptide family under investigation are taken into account. All possible introduced conformational constraints such as cyclisations or the incorporation of restraining amino acids such as imino acids or D-amino acids help to limit the vast conformational space of peptidic structures. Library strategies for the optimisation of peptide rigidity are described above.
The strategy is well illustrated by an example in which non-peptidic urotensin II receptor antagonists were identified based on the 11-amino acid cyclic peptide urotensin-II.24 The SAR of urotensin-II was evaluated by truncation libraries and Ala and D-amino acid scans. These libraries revealed that the residues WKY are key for peptide function and lead to a 200-fold less active cyclic hexapeptide Ac-CFwKYC-NH2 containing a D-amino acid. D-Configuration amino acids stabilise the formation of β-turn structures and thus introduce a conformational constraint into the flexible peptide. Consequently, although the solution structure of the full-length urotensin-II as measured by the NMR spectroscopy shows considerable conformational fluctuations, the conformation of Ac-CFwKYC-NH2 is very well defined. Both structures were used as templates for generating two pharmacophore models, each containing a positive ionisable and two hydrophobic aromatic features, reflecting the spatial arrangement of the side chains of the key residues wKC and WKC, respectively. The pharmacophore models were used for a virtual screen of the Aventis compound repository. Hit compounds were screened against the urotensin II receptor using a functional fluorometric imaging plate reader assay. For the urotensin II-derived pharmacophore, a hit rate of 2% was achieved with IC50 values ranging from 400 nM to 7 μM (Figure 11). The verified hits belong to six different scaffold classes.
 | ||
| Fig. 11 Starting fragment of the peptidic structure of urotensin II (left, EC50 of 2.5 nM for urotensin II) and hit structure from virtual screening (right, EC50 = 400 nM). | ||
This success contrasts with the screening results for the second pharmacophore model generated from the structurally better-defined cyclic hexapeptide. The hit rate of 0.2% in this screen is barely higher than the hit rates seen in high-throughput screens for G-protein-coupled receptor (GPCR) antagonists. This fact illustrates one problem with the ligand-based approach: the solution structure or a calculated low-energy structure does not necessarily reflect the bound conformation of the active peptide and may be a misleading starting point for pharmacophore generation and virtual screening. Therefore, selection of the “right” conformation is a critical step. This feature is further complicated by the fact that the binding conformation of active ligands is often not a low-energy conformation. An investigation of the conformational energies of complex crystal structures revealed an energetically non-favourable conformation for over 60% of the bound ligands.25 This factor is one of the reasons why computational assessment of binding conformations using straightforward techniques of simulated annealing or molecular dynamics is critical. When sampling the conformational space, the most important parameter for the simulation is the energy of the conformations, and normally, low energy conformations are selected. Moreover, due to the huge conformational space of peptides, a complete conformational sampling of this space for an oligopeptide is often not feasible.
In an elegant way, McDowell et al.26 circumvent both problems by introducing an ensemble molecular dynamics method. The idea is to use information from an ensemble of diverse flexible peptides interacting with a common site. The primary goal is then to identify only consensus conformations that represent a common spatial arrangement of shared binding features. Technically, this goal is achieved by a dynamics simulation of a collection of molecules, which are tethered together at their corresponding binding features, while ignoring non-bonded interactions between the molecules. This procedure only generates conformations from the entire conformational space where the constrained binding features within all peptides of an ensemble occupy a similar location in space. As a result, the conformational space of each peptide is considerably reduced by limiting it to the part, which overlaps with the corresponding conformational space of all peptides in the ensemble. The resulting structures do not represent low energy structures for a single peptide but for the entire ensemble, since the behaviour of one peptide constrains that of each other. The appropriate choice of tethering features requires a rigorous structure–activity analysis.
This approach was used to calculate the bioactive conformation of the Arg–Gly–Asp recognition sequence that inhibits the glycoprotein IIbIIIa–fibrinogen interaction, which is important for platelet aggregation.26 Based on this structural model, the compounds synthesised sharing a benzodiazepinedione scaffold showed efficient inhibition of platelet aggregation and oral bioavailability.27 The ensemble dynamics approach also helped generate a leukocyte functional antigen-1 (LFA-1) antagonist based on a discontinuous epitope of intercellular adhesion molecule-1 (ICAM-1) and cyclic peptides (Figure 12).28
 | ||
| Fig. 12 Superimposition of an LFA-1 antagonist on the discontinuous epitope of ICAM-1 (pdb-code 1iam, protein backbone shown as blue tube), which binds to LFA-1. Residues of the protein indicated in yellow contribute to LFA-1 binding and are mimicked perfectly by the small molecule. | ||
Structural information and computational methods have considerable potential in supporting the transformation of bioactive peptides into small molecules. Of major importance is combining all available information on the system of interest, including peptide SAR data, information on the consequences of mutations in the target on SAR, structural information from both the target protein and related proteins as well as the particular ligand and all other known ligands.
6 Conclusion
The design of “drug-like” small molecules from peptides is a good option for a number of different peptide–target interactions. This approach is especially well suited for target classes such as proteases and peptide or protein binding GPCRs, which are difficult to address using small molecules. However, although stepwise chemical transformation processes were successful in several cases, no general transformation strategies have emerged. In recent years, computational methods supported by information about the molecular structure of both targets and ligands have opened up new opportunities to overcome the common pitfalls of the transformation process. Even with these cutting-edge technologies, any transformation still remains a challenge. Several case studies have illustrated that the probability of success increases in parallel to the amount of knowledge about the system in question, such as SAR data, structural data, protein flexibility, binding kinetics and physiological context. The decreasing number of approved drugs produced by the pharmaceutical industry, accompanied by increasing expenses for research and development, demands alternative approaches to increase productivity. Peptide-based drug discovery may be one method to solve as yet unresolved problems.References
- A. Loffet, J. Pept. Sci., 2002, 8, 1 CrossRef CAS.
- H.M. Geysen, R.H. Meloen and S.J. Barteling, Proc. Natl. Acad. Sci. U. S. A., 1984, 81, 3998 CAS.
- J.K. Scott and G.P. Smith, Science, 1990, 249, 386 CrossRef CAS.
- L.C. Mattheakis, R.R. Bhatt and W.J. Dower, Proc. Natl. Acad. Sci. U. S. A., 1994, 91, 9022 CAS.
- R.W. Roberts and J.W. Szostak, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 12297 CrossRef CAS.
- H. Wenschuh, R. Volkmer-Engert, M. Schmidt, M. Schulz, J. Schneider-Mergener and U. Reineke, Biopolymers (Peptide Science), 2000, 55, 188 Search PubMed.
- A. Furka, in Combinatorial peptide and nonpeptide libraries, ed. G. Jung, VCH Verlagsgesellschaft, Weinheim, 1996, 111 Search PubMed.
- K. S. Lam, S. E. Salmon, E. M. Hersh, V. J. Hruby, W. M. Kazmierski and R. J. Knapp, Nature, 1991, 354, 82 CrossRef CAS.
- C. Pinilla, J. Appel, C. Dooley, S. Blondelle, J. Eichler, B. Dörner, J. Ostresh and R. A. Houghten, in Combinatorial peptide and nonpeptide libraries, ed. G. Jung, VCH Verlagsgesellschaft, Weinheim, 1996, p. 139 Search PubMed.
- R. A. Houghten, C. Pinilla, S. E. Blondelle, J. R. Appel, C. T. Dooley and J. H. Cuervo, Nature, 1991, 354, 84 CrossRef CAS.
- V. J. Hruby, Nat. Rev. Drug Discovery, 2002, 1, 847 CrossRef CAS.
- R. M. J. Liskamp, Recl. Trav. Chim. Pays-Bas, 1994, 113, 1 CAS.
- P. N. Lewis, F. A. Momany and H. A. Scheraga, Biochim. Biophys. Acta, 1973, 303, 211 CAS.
- V. J. Hruby and P. M. Balse, Curr. Med. Chem., 2000, 7, 945 CAS.
- M. Feigel, J. Am. Chem. Soc., 1986, 108, 181 CrossRef CAS.
- W. C. Ripka, G. V. De Lucca, A. C. Bach II, R. S. Pottorf and J. M. Blaney, Tetrahedron, 1993, 49, 3593 CrossRef CAS.
- M. Kahn, Synlett, 1993, 821 CrossRef CAS.
- W. F. Huffman, J. F. Callahan, D. S. Eggelston, K. A. Newlander, D. T. Takata, E. E. Codd, R. F. Walker, P. W. Schiller, C. Lemieux, W. S. Wire and T. F. Burks, in Peptides: Chemistry and Biology, Proceedings of the 10th American Peptide Symposium, ed. G. R. Marshall, ESCOM, New York, 1988, 105 Search PubMed.
- K. Brickmann, Z. Yuan, I. Sethson, P. Somfai and J. Kihlberg, Chem.–Eur. J., 1999, 5, 2241 CrossRef CAS.
- G. Lauri and P. A. Bartlett, J. Comput. Aided Mol. Des., 1994, 8, 51 CrossRef CAS.
- Y. Takano, M. Koizumi, R. Takarada, M. T. Kamimura, R. Czerminski and T. Koike, J. Mol. Graphics Modeling, 2003, 22, 105 Search PubMed.
- D. R. Artis, C. Brotherton-Pleiss, J. H. B. Pease, C. J. Lin, S. W. Ferla, S. R. Newman, S. Bhakta, H. Ostrelich and K. Jarnagin, Bioorg. Med. Chem. Lett., 2000, 10, 2421 CrossRef CAS.
- J. Singh, H. van Vlijmen, Y. Liao, W. C. Lee, M. Cornebise, M. Harris, I. H. Shu, A. Gill, J. H. Cuervo, W. M. Abraham and S. P. Adams, J. Med. Chem., 2002, 45, 2988 CrossRef CAS.
- S. Flohr, M. Kurz, E. Kostenis, A. Brkovich, A. Fournier and T. Klabunde, J. Med. Chem., 2002, 45, 1799 CrossRef CAS.
- E. Perola and P. S. Charifson, J. Med. Chem., 2004, 47, 2499 CrossRef CAS.
- R. S. McDowell, T. R. Gadek, P. L. Barker, D. J. Burdick, K. S. Chan, C. L. Quan, N. Skelton, M. Struble, E. D. Thorsett, M. Tischler, J. Y. K. Tom, T. R. Webb and J. P. Burnier, J. Am. Chem. Soc., 1994, 116, 5069 CrossRef CAS.
- R. S. McDowell, B. K. Blackburn, T. R. Gadek, L. R. McGee, T. Rawson, M. E. Reynolds, K. D. Robarge, T. C. Somers, E. D. Thorsett, M. Tischler, R. R. Webb and M. C. Venuti, J. Am. Chem. Soc., 1994, 116, 5077 CrossRef CAS.
- T. R. Gadek, D. J. Burdick, R. S. McDowell, M. S. Stanley, J. C. Marsters Jr., K. J. Paris, D. A. Oare, M. E. Reynolds, C. Ladner, K. A. Zioncheck, W. P. Lee, P. Gribling, M. S. Dennis, N. J. Skelton, D. B. Tumas, K. R. Clark, S. M. Keating, M. H. Beresini, J. W. Tilley, L. G. Presta and S. C. Bodary, Science, 2002, 295, 1086 CrossRef CAS.
Footnote |
| † This is Chapter 8 taken from the book Exploiting Chemical Diversity for Drug Discovery (Edited by M. Entzeroth and P. A. Bartlett) which is part of the RSC Biomolecular Sciences series. |
| This journal is © The Royal Society of Chemistry 2006 |