Fluorescent reagents for in vitro studies of lipid-linked steps of bacterial peptidoglycan biosynthesis: derivatives of UDPMurNAc-pentapeptide containing D-cysteine at position 4 or 5
James A. Schoutena, Sangeev Baggaa, Adrian J. Lloydb, Gianfranco de Pascaleab, Christopher G. Dowsonb, David I. Roperb and Timothy D. H. Bugg*a
aDepartment of Chemistry, University of Warwick, Coventry, UK CV4 7AL. E-mail: T.D.Bugg@warwick.ac.uk; Fax: +44 (0)2476 524112; Tel: +44 (0)2476 573018
bDepartment of Biological Sciences, University of Warwick, Gibbet Hill Rd, Coventry, UK CV4 7AL
First published on 7th September 2006
Abstract
UDPMurNAc-L-Ala-γ-D-Glu-X-D-Ala-D-Ala (X = L-Lys or m-DAP) is the cytoplasmic precursor for the lipid-linked cycle of bacterial peptidoglycan biosynthesis, consisting of at least four enzymatic reactions, which are targets for antibacterial agents. Fluorescent derivatives of the UDPMurNAc-pentapeptide labelled at the 3rd, 4th, and 5th position of the peptide chain were prepared chemoenzymatically, in order to study the reactions catalysed by enzymes in this cycle. Derivatives labelled on the ε-amino group of the 3rd amino acid (N-dansyl, N-fluorescamine and N-phthalaldehyde) were prepared by chemical modification. Two methods were developed for preparation of analogues of UDPMurNAc-pentapeptide containing D-cysteine at position 4 or 5: either by MurF-catalysed ligation of the UDPMurNAc-tripeptide to synthetic D-Ala-D-Cys or D-Cys-D-Ala dipeptides; or by enzymatic synthesis of D-Ala-D-Cys by ligase VanD. D-Cys-containing UDPMurNAc-pentapeptides were labelled with pyrene maleimide, to give 4-pyrene and 5-pyrene labelled derivatives. The fluorescent UDPMurNAc-pentapeptides were processed as substrates by Escherichia coli MraY or E. coli membranes, giving 1.5–150-fold changes in fluorescence upon transformation to lipid intermediate I. Subsequent processing to lipid intermediate II gave rise only to small changes in fluorescence. Pyrene-labelled lipid intermediates I and II can be generated using Micrococcus flavus membranes, enabling the study of the later lipid-linked steps.
Introduction
Bacterial cell wall peptidoglycan biosynthesis is the target for several clinically used antibiotics, including the β-lactam class of penicillins and cephalosporins, and the vancomycin group of glycopeptides.1,2 The later stages of the biosynthetic pathway occur via a lipid-linked cycle of reactions in the cytoplasmic membrane, involving a lipid carrier, undecaprenyl phosphate, illustrated in Fig. 1. The first step, catalysed by translocase MraY (or translocase I), involves the reaction of the cytoplasmic precursor UDPMurNAc-L-Ala-γ-D-Glu-m-DAP-D-Ala-D-Ala (UDPMurNAc-pentapeptide) with undecaprenyl phosphate to form lipid intermediate I and UMP. The second step, catalysed by glycosyltransferase MurG (or translocase II), involves the transfer of a GlcNAc sugar from UDPGlcNAc to the C-4 hydroxyl of the MurNAc sugar, to form lipid intermediate II. In some bacteria, additional amino acids are then added to position 3 of lipid II by the FemA/MurMN class of ligases, e.g. (Gly)5 in Staphylococcus aureus, Ala-Ala or Ser-Ala in Streptococcuspneumoniae.3 Lipid II is then flipped from the cytoplasmic face to the exterior face of the membrane, presumably by an uncharacterised ‘flippase’. On the cell surface, lipid II is polymerised by the transglycosylase activity of Class A penicillin-binding proteins (PBPs) to form a glycan chain.4 The final step involves transpeptidation between the amino sidechain of the position 3 amino acid (Lys found in most Gram-positive bacteria, or meso-diaminopimelic acid found in most Gram-negative bacteria) and the D-alanine residue of a second strand, catalysed by the transpeptidase activity of Class A and Class B PBPs.4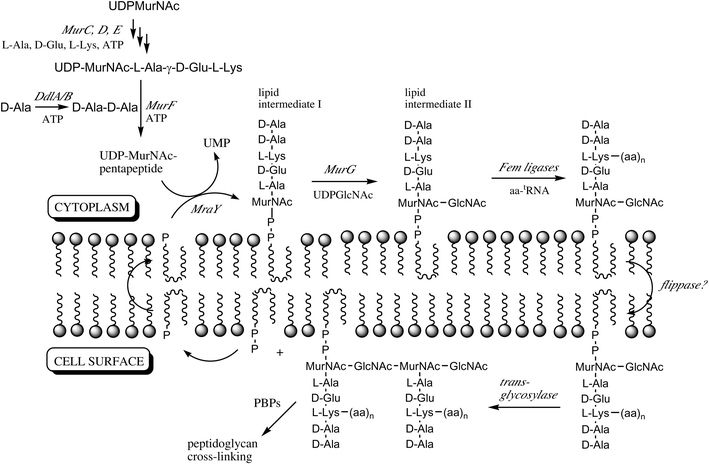 | ||
| Fig. 1 Lipid-linked steps of bacterial peptidoglycan biosynthesis, and late cytoplasmic steps referred to in this paper. The lipid carrier is undecaprenyl phosphate. PBP, penicillin binding protein. | ||
The lipid-linked steps are the cellular targets for a number of natural product antibiotics: translocase MraY is inhibited by nucleoside antibiotics mureidomycin A, liposidomycin B, and tunicamycin; cyclic peptide ramoplanin binds to lipid intermediates I and II, and inhibits MurG; the vancomycin group of glycopeptide antibiotics bind the –D-Ala-D-Ala terminus of lipid II; and the penicillin family of β-lactam antibiotics inactivate PBP's.1 However, only limited biochemical characterisation of these steps has been made, due to several factors: the difficulty of isolating advanced biosynthetic intermediates such as lipid I and lipid II; the lack of biochemical assays for the lipid-linked steps; and the fact that the steps are membrane-associated, involving lipid-linked intermediates and membrane-associated proteins.
We have previously developed a continuous fluorescence assay for the translocase I reaction, using a derivative of UDPMurNAc-pentapeptide labelled with a dansyl fluorophore at position 3, which we have used to study the kinetics of inhibition by mureidomycin A, liposidomycin B, and tunicamycin.5 This method has been extended to a high-throughput format for inhibitor screening by Stachyra et al.6 The N-dansylated lipid intermediate II has been prepared by Schwartz et al., and has been shown to give rise to fluorescence changes upon treatment with E. coli PBP-1B.7
The remaining lipid-linked steps have, to date, largely been studied through radiochemical or stopped, coupled assays. MurG has been assayed by monitoring the release of UDP,8 while the biochemical steps catalysed by transglycosylase and transpeptidation have been monitored only using radiochemical methods.9,10
The development of fluorescence-based assays for these steps would enable detailed enzymological studies; high-throughput screening for antibacterial agents; and fluorescence microscopy studies of bacterial cell wall assembly. The fluorescent analogues labelled at position 3 of UDPMurNAc-pentapeptide are of limited use for the study of the later lipid-linked steps, since the transpeptidation reaction and the FemAB/MurMN catalysed reactions both involve participation of the amino sidechain at position 3 as a nucleophile. In this paper we describe the preparation of a range of fluorescent UDPMurNAc-pentapeptides labelled at positions 3, 4, and 5 of the peptide chain, using the biosynthetic enzyme MurF (see Fig. 1) and a D-alanine-D-alanine ligase enzyme VanD of relaxed substrate specificity, and we report initial investigations into their enzymatic processing along the lipid-linked pathway.
Results
Preparation of N-modified UDPMurNAc-pentapeptides labelled at position 3
We5 and others6 have previously described the use of UDPMurNAc-L-Ala-γ-D-Glu-m-DAP(ε-dansyl)-D-Ala-D-Ala for assay of translocase I. In this study, three N-labelled derivatives were prepared, as shown in Fig. 2, for comparative studies. UDPMurNAc-L-Ala-γ-D-Glu-m-DAP-D-Ala-D-Ala was isolated as previously described from cultures of Bacillus subtilis W23,10 in 50–100 mg quantities. Three N-labelled fluorescent derivatives (N-dansyl, N-fluorescamine, and N-o-phthalaldehyde) were prepared by chemical modification of the meso-diaminopimelic acid (m-DAP) residue at position 3 of the pentapeptide sidechain of UDPMurNAc-pentapeptide. The N-dansyl derivative (λex 340 nm; λem 565 nm) was prepared by reaction with dansyl chloride in 0.25 M sodium carbonate pH 10.0, and was isolated after G15 gel filtration in 23% yield, and a sample purified further by HPLC. The N-fluorescamine derivative (λex 390 nm; λem 475 nm) was prepared by reaction with fluorescamine in acetone, and the N-OPA derivative (λex 340 nm; λem 455 nm) was prepared by reaction with ortho-phthalaldehyde and β-mercaptoethanol. The derivatives were stable to storage, and each was found to be a substrate for translocase MraY, as described below.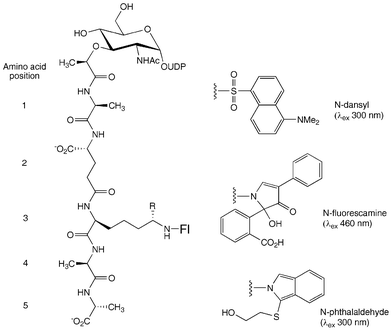 | ||
| Fig. 2 N-labelled fluorescent derivatives of UDPMurNAc-pentapeptide. R = H (Lys) or CO2H (m-DAP). | ||
Preparation of UDPMurNAc-pentapeptide analogues containing D-Cys at position 4 or 5
For studies of lipid-linked steps involving reaction of the amino substituent at position 3, derivatives labelled at position 3 would be of no use as substrates; therefore, a strategy for attachment of a fluorophore at position 4 or 5 has been developed. The D-Ala-D-Ala dipeptide at the end of the peptide chain is inserted enzymatically by ligase MurF, which catalyses the ATP-dependent condensation of UDPMurNAc-tripeptide with D-Ala-D-Ala. It is known from early studies by Neuhaus and Struve that the S. aureus MurF can accept alternative D,D-dipeptides as substrates, although showing some specificity for the C-terminal D-alanine.11 Furthermore, it is known that bacterial cell walls can be labelled by feeding growing bacterial cells with D-cysteine,12 which is presumably incorporated in place of D-alanine at position 4 or 5. Therefore, we examined whether a D-Ala-D-Cys or D-Cys-D-Ala dipeptide could be accepted by MurF, to make a UDPMurNAc-pentapeptide containing D-Cys at position 4 or 5, which could then be modified by a cysteine-specific reagent, as shown in Fig. 3.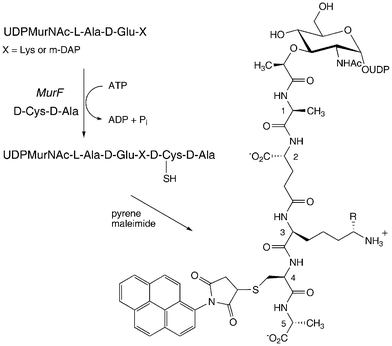 | ||
| Fig. 3 Preparation of 4-pyrene labelled UDPMurNAc-pentapeptide, using ligase MurF. R = H (Lys) or CO2H (m-DAP). | ||
Dipeptides D-Ala-D-Cys and D-Cys-D-Ala were prepared by solid phase peptide synthesis using Fmoc chemistry.13 Fmoc-D-Ala or Fmoc-D-Cys was coupled onto the Wang resin, and deprotected by treatment with 20% piperidine–DMF. Acetylation of unreacted sites was achieved with 1 : 1 : 8 acetic anhydride–DIPEA–DMF. A further round of coupling was followed by global deprotection in 95% TFA, 2.5% water, 2.5% triisopropylsilane. Free D-Ala-D-Cys and D-Cys-D-Ala dipeptides were isolated in yields of 15–25%.
UDPMurNAc-L-Ala-γ-D-Glu-m-DAP was prepared by D-cycloserine treatment of B. subtilis W23 cultures,14 in 5–10 mg quantities. Recombinant D-Ala-D-Ala adding enzymes MurF from E. coli (DAP-selective) and S. pneumoniae (Lys-selective) were expressed and purified to near homogeneity.
Assay of each MurF enzyme with D-Ala-D-Ala, D-Cys-D-Ala, and D-Ala-D-Cys established that each of the cysteine-containing dipeptides is accepted as a substrate (see Fig. 4). D-Cys-D-Ala is processed efficiently by MurF (see Fig. 4A), at approximately a 50% rate compared with D-Ala-D-Ala, while D-Ala-D-Cys is converted 20-fold more slowly (see Fig. 4B). These observations are consistent with the reportedly higher specificity of MurF for the C-terminal position of the dipeptide.11
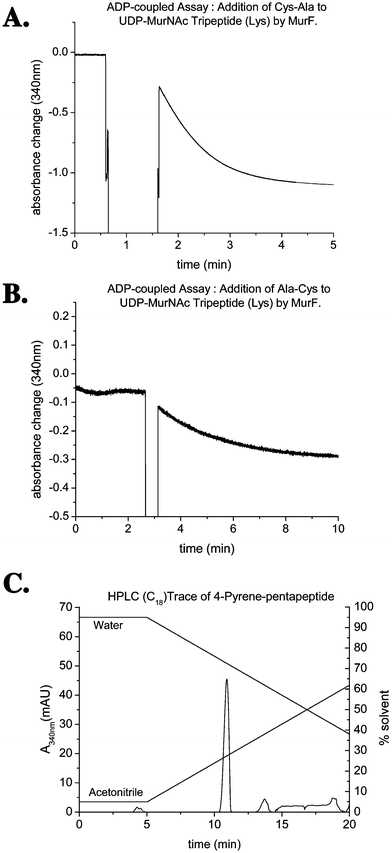 | ||
| Fig. 4 A. Conversion of D-Cys-D-Ala by MurF, monitored by ADP release continuous assay. B. Conversion of D-Ala-D-Cys by MurF, monitored by ADP release continous assay. C. HPLC purification of 4-pyrene labelled UDPMurNAc-pentapeptide. | ||
Analysis of the MurF conversion of D-Cys-D-Ala with UDP-MurNAc-tripeptide(DAP) by MonoQ anion exchange FPLC showed the formation of a new peak at retention time 29.7 min, which showed a positive reaction with thiol-selective reagent DTNB, and gave the expected molecular mass by electrospray mass spectrometry ([M − H]− 1180.0). It was found that decomposition of the thiol-containing UDPMurNAc-pentapeptides occurred over a time period of 12–24 h, giving rise to a species of the same mass which did not give a positive reaction with DTNB. Treatment with reducing agents such as sodium borohydride, followed by treatment with DTNB at pH 8, also gave no positive reaction, indicating that disulfide formation was not responsible for this behaviour. One possible explanation is an intramolecular cyclisation of the free thiol onto the uracil C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C, via a conjugate addition reaction, a known reaction for uridine-containing compounds bearing amine substituents attached to C-5′.15
C, via a conjugate addition reaction, a known reaction for uridine-containing compounds bearing amine substituents attached to C-5′.15
Therefore, modification of the free thiol by pyrene maleimide was carried out shortly after the formation of the D-Cys-labelled UDPMurNAc-pentapeptide by MurF. The MurF-catalysed reaction of UDPMurNAc-tripeptide with D-Cys-D-Ala was performed in 50 mM Hepes buffer pH 7.6, followed by treatment with pyrene maleimide after 1 h, and gave an isolated yield of only 3%.
It was found that MurF conversions could be carried out in 50 mM MES buffer pH 6.0 at approximately a 50% rate, compared to pH 7.6, thus reducing the possibility of any base-catalysed side reactions. In addition, labelling was initiated after just 20 min, resulting in a significantly improved yield of 30%. The 4-pyrene labelled UDPMurNAc-pentapeptide(Lys) was isolated by C18 reverse phase HPLC (see Fig. 4C), and was characterised by negative ion electrospray mass spectrometry (m/z 738.2, (M − 2H)2−). The 5-pyrene labelled compound was prepared following the same method in the presence of D-Ala-D-Cys instead of D-Cys-D-Ala. The yield for this reaction was substantially lower than observed under the modified conditions for the 4-pyrene derivative (∼3%), suggesting greater susceptibility to base-catalysed side reactions.
Use of ligase VanD for incorporation of D-Cys
An alternative enzymatic method for preparation of D-Ala-D-Cys has also been developed. It is known that VanA-type ligases involved in high-level vancomycin resistance preferentially make D-Ala-X dipeptides and depsipeptides, rather than the normal D-Ala-D-Ala dipeptide.16 Recombinant VanD from Enterococcus faecium17 was found to accept D-Cys as a substrate: incubation of VanD with 10 mM D-Cys, 1 mM D-Ala, and 1 mM ATP, was found to give a single dipeptide product. Analysis of this product by electrospray mass spectrometry established that it contained one residue of D-alanine and one residue of D-cysteine ([MH]+ 193.1). Labelling of the N-terminal amino acid by reaction with 1-fluoro-2,4-dinitrobenzene, followed by acid hydrolysis of the labelled dipeptide, released DNP-Ala as the N-terminal residue, identifying the product as D-Ala-D-Cys.Incubation of 10 mM D-Cys, 1 mM D-Ala with VanD, E. coli MurF, UDPMurNAc-tripeptide (DAP) and ATP gave the modified UDPMurNAc-pentapeptide containing D-Cys at position 5, which was immediately treated with pyrene maleimide. The 5-pyrene labelled UDPMurNAc-pentapeptide product, shown in Fig. 5, was isolated by MonoQ anion exchange FPLC (retention time 30.0 min). The isolated product was fluorescent (λex 340 nm, λem 400, 510 nm), and was characterised by electrospray mass spectrometry ([M − 3H]3− 506.0). Since this procedure was carried out on a 0.1 µmole scale, it is not possible to quote an isolated yield, but the product peak isolated by FPLC was the major species observed at 280 nm, indicating an efficient reaction.
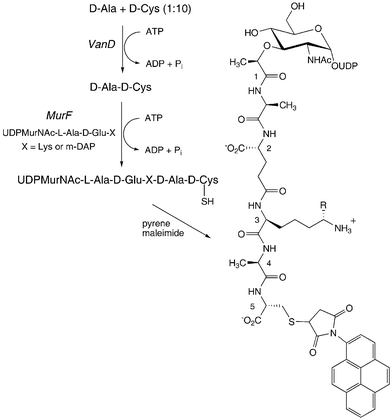 | ||
| Fig. 5 Preparation of 5-pyrene labelled UDPMurNAc-pentapeptide using ligase VanD. R = H (Lys) or CO2H (m-DAP). | ||
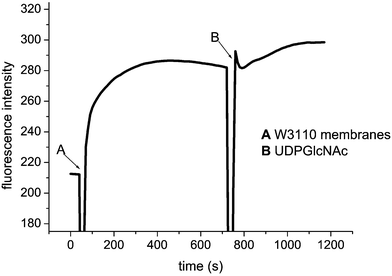 | ||
| Fig. 6 Fluorescence changes observed upon treatment of 4-pyrene labelled UDPMurNAc-pentapeptide by E. coli membranes. | ||
Enzymatic processing by translocase MraY
Incubation of the three N-modified UDPMurNAc-pentapeptides with overexpressed, solubilised E. coli MraY in 1% Triton X-100 gave 6-fold, 3-fold and 2-fold increases in fluorescence intensity respectively, while smaller fluorescence changes were observed using 1% CHAPS as detergent (see Table 1). Thus, each of the modified UDPMurNAc-pentapeptides is accepted as a substrate for translocase I, as noted previously for the N-dansyl derivative, and most sensitive fluorescence changes in the MraY-catalysed reaction are observed using the N-dansyl derivative.| Initial Fl | +MraY/1% Triton X-100 | +MraY/1.5% CHAPS | |
|---|---|---|---|
| N-dansyl | 7.7 | 45.7 | 47.5 |
| N-fluorescamine | 3.0 | 9.4 | 3.4 |
| N-OPA | 7.3 | 15.3 | 8.0 |
The lipid I products of the MraY-catalysed reaction of the N-labelled analogues with undecaprenyl phosphate were isolated by extraction into 1 : 1 chloroform–methanol. Analysis by reverse phase HPLC showed the formation of a new fluorescent peak at retention time 21 min. This peak was isolated, and was re-incubated with fresh MraY and UMP, giving a linear decrease in fluorescence, due to the reverse reaction catalysed by MraY. The formation of dansyl-UDPMurNAc-pentapeptide was observed by HPLC, indicating that the extracted lipid I remains biologically active.
Processing of fluorescent substrates by E. coli membrane-bound enzymes
The processing of three N-labelled UDPMurNAc-pentapeptides and two S-labelled UDPMurNAc-pentapeptides, and UDPGlcNAc, by a particulate membrane fraction from E. coli was examined by fluorescence spectroscopy, by addition of E. coli W3110 membranes, in 50 mM Tris pH 7.5, containing 10 mM β-mercaptoethanol and 1 mM MgCl2. For each of the three N-labelled substrates, large and rapid fluorescence changes were observed (see Table 2), giving 10–150-fold increases in fluorescence, followed by a gradual decrease (presumably due to quenching). Using the two S-pyrene labelled substrates, time-dependent increases in fluorescence were also observed, 2–3-fold in magnitude.| λex(nm) | λem (nm) | Initial Fl | Max Fl | tmax (s) | |
|---|---|---|---|---|---|
| N-dansyl | 340 | 540 | 1.0 | 11.4 | 10 |
| N-OPA | 340 | 420 | 2.0 | 315 | 90 |
| N-FLA | 390 | 495 | 2.0 | 90 | 50 |
| 4-S-pyr | 340 | 500 | 40 | 67 | 600 |
| 5-S-pyr | 340 | 500 | 40 | 90 | 700 |
Largest fluorescence changes were observed using the N-OPA derivative, in which the time-dependent increase and decrease in fluorescence could clearly be observed. With this derivative, a series of control experiments were carried out, in order to examine what biochemical processes were responsible for the changes in fluorescence. The size of the fluorescence increase (252.7 Fl units) was reduced by 43% when half the amount of protein was added (145.9 Fl units), and no fluorescence change was observed in a control with no added protein, indicating that the change in fluorescence is due to an enzyme-catalysed process. Reaction in the absence of UDPGlcNAc gave a similar fluorescence change (289.2 Fl units), implying that the majority of the observed fluorescence change was caused by the formation of lipid I, catalysed by MraY. In a two-stage assay, addition of membranes without UDPMurNAc, until maximum fluorescence at 100 s, gave an increase of 242.8 Fl units; at this point 1 mM UDPGlcNAc was added, and an additional small increase of 21 Fl units was observed, indicating that a small additional increase in fluorescence is accompanied by the MurG reaction, but that <10% change in fluorescence is observed between lipid intermediates 1 and 2 in this case. Analysis of the reaction mixtures by thin layer chromatography on silica, eluting with EtOH–H2O–AcOH (4 : 2 : 1), revealed the conversion of OPA-UDPMurNAc-pentapeptide (Rf 0.20) to OPA-lipid I (Rf 0.90), but no evidence of polymerisation to form peptidoglycan (Rf 0.0).
Similar two-stage experiments were carried out using the 4- and 5-pyrene-labelled UDPMurNAc-pentapeptides (Fig. 6). Using the 4-S-pyrene labelled substrate, a rapid increase in fluorescence was observed upon addition of E. coli membranes (without UDPGlcNAc), and upon addition of UDPGlcNAc a further increase in fluorescence was observed, corresponding to 25% of the initial increase, indicating that 4-S-pyrene-lipid I is processed by MurG, either more efficiently or with a greater fluorescence change than for the N-labelled derivatives.
In order to confirm that conversion to lipid intermediates I and II was taking place, authentic samples of labelled lipid I and II were needed. Breukink et al. have published a procedure using membranes from Micrococcus flavus to generate lipid intermediates I and II, which they have applied to several substrates, including N-dansyl UDPMurNAc-pentapeptide.18 Using the procedure of Breukink et al., a sample of 4-pyrene UDPMurNAc-pentapeptide was converted either into lipid I (in the absence of UDPGlcNAc) or lipid II (in the presence of UDPGlcNAc). As shown in Fig. 7, discrete fluorescent spots were observed by thin layer chromatography, corresponding to fluorescent lipid I and II, with a small amount of residual pyrene-lipid I present in the second lane. Analysis of the incubations of 4-pyrene labelled UDPMurNAc-pentapeptide with E. coli membranes showed the formation of the same fluorescent spots, corresponding to 4-pyrene-lipid I and 4-pyrene lipid II. Faint spots at lower Rf were also observed, suggesting limited polymerisation was occurring, but no baseline spot was observed, corresponding to mature peptidoglycan.
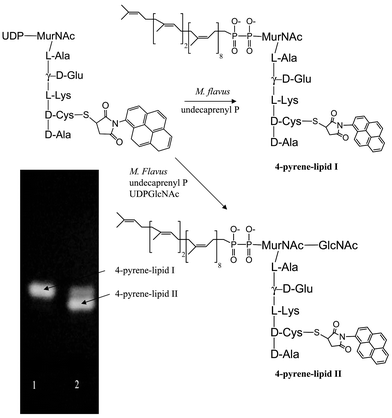 | ||
| Fig. 7 Preparation of 4-pyrene labelled lipid intermediates I and II using M. flavus membranes. | ||
Conclusions
The lipid-linked steps of bacterial peptidoglycan biosynthesis are targets for several known antibiotics, and are potential targets for the development of new antibacterial agents, but there are currently limited methods to study these steps in vitro. In this paper we have developed a new strategy for fluorescent labelling of the UDPMurNAc-pentapeptide precursor, by incorporation of D-Cys at position 4 or 5 of the pentapeptide sidechain. We have shown that both D-Cys-D-Ala and D-Ala-D-Cys are accepted as substrates by ligase MurF, though with a 10-fold preference for D-Cys-D-Ala. The Cys-containing UDPMurNAc-pentapeptide can be labelled with N-pyrene maleimide, to give a fluorescent derivative. The 4-pyrene and 5-pyrene labelled UDPMurNAc-pentapeptides are both accepted as substrates by the first step of the lipid cycle, catalysed by MraY, giving substantial changes in fluorescence. Subsequent conversion to lipid intermediate II catalysed by MurG is observed, although with only small subsequent change in fluorescence. We have separately used the N-dansyl UDPMurNAc-pentapeptide, in combination with a fluorescent UDPGlcNAc analogue, to develop a fluorescence resonance energy transfer (FRET) assay for MurG;19 further studies using this FRET assay will be reported in due course.The availability of lipid II intermediates labelled at positions 4 and 5 allows the study of their reaction with the later lipid-linked steps, catalysed by ligases MurMN and the transglycosylase/transpeptidase reactions of the penicillin-binding proteins. Although preliminary experiments with E. coli membranes suggest only limited polymerisation, the bulky pyrene maleimide group may hinder the processing of these substrates. The thiol sidechain of D-Cys allows the attachment of a wide range of functional groups in these positions, therefore a number of possible strategies for study of the latter steps via kinetic assays and microscopy are now feasible. We have recently demonstrated that lipid intermediates I and II can be inserted into tethered lipid bilayers,20 enabling a variety of physical methods for in vitro studies of peptidoglycan biosynthesis.
Experimental
Materials
UDPMurNAc-L-Ala-γ-D-Glu-m-DAP and UDPMurNAc-L-Ala-γ-D-Glu-m-DAP-D-Ala-D-Ala were isolated from antibiotic-treated cultures of B. subtilis W23, as previously described.5,14 UDPMurNAc-L-Ala-γ-D-Glu-L-Lys was prepared enzymatically, using recombinant Pseudomonas aeruginosa MurA, MurB, MurC, MurD, and MurE enzymes, expressed and purified as described by El Zoeiby et al.21 Undecaprenyl phosphate was purchased from Larodan Chemicals. All other chemicals and biochemicals were purchased from Sigma-Aldrich Ltd.Recombinant E. coli MurF was expressed and purified as described previously.14 Recombinant S. pneumoniae MurF and recombinant E. faecium VanD17 were gifts of Dr D. Roper.
Analytical FPLC anion exchange chromatography was carried out on a MonoQ HR 5/5 column, eluting with a gradient of 0.01–0.05 M (0–20 min) then 0.05–1 M (20–70 min) ammonium acetate pH 7.5 at flow rate 0.5 ml min−1, monitoring at 254 nm.
Synthesis of N-dansyl UDPMurNAc-pentapeptide (DAP)
UDPMurNAc-pentapeptide (containing DAP, 5 mg, 4.2 µmol) was dissolved in 0.25 M sodium carbonate solution at pH 10.0 (2 mL), with 50% aqueous acetone (0.7 ml). 4 aliquots of dansyl chloride (4 mg, 7.4 µmol) were added at 20 min intervals, acetone being removed after the last addition by rotary-evaporation concentrating the sample to ∼1 ml. Dansyl-UDPMurNAc-pentapeptide was separated from dansic acid by its passage down a gel filtration Sephadex G15 column (74 × 1 cm), eluting with water, monitoring at 260 and 315 nm. Fractions containing dansylated material were pooled and lyophilised overnight to give 1.34 mg of a yellow powder (23% yield). Data: fluorescence λex 340 nm, λex 500 nm. HPLC retention time 6.8 min. m/z (ES-MS −ve ion) 714.1 ((M − 2H)2−), predicted 713.7.Preparation of OPA-UDPMurNAc-pentapeptide (DAP)
A solution of ortho-phthalaldehyde (75 µl) and β-mercaptoethanol (75 µl) in ethanol (225 µl) was mixed with 0.05 M boric acid (1.0 ml, pH 9.5) for 2 min. 750 µl of the mixture was added to UDPMurNAc-pentapeptide (5.5 mg, 4.6 µmol), and incubated for 30 min at room temperature. The reaction mixture was purified by C18 reverse-phase HPLC, flow rate 0.8 ml min−1 in isocratic MeOH (100%). The product peak at 9.2 min was collected and lyophilised to give a yellow powder (1.1 mg, 22%). Data: fluorescence λex 340 nm, λex 485 nm. m/z (FAB-MS −ve ion) 1383.4 ((M − H)−), predicted 1383.2.Synthesis of N-fluorescamine UDPMurNAc-pentapeptide (DAP)
UDPMurNAc-pentapeptide (containing DAP, 4.0 mg, 3.4 µmol) was added to a solution of fluorescamine (0.5 g, 1.8 mmol) and triethylamine (150 µl) in acetone (1 ml). The mixture was purified by C18 reverse-phase HPLC in isocratic MeCN–0.1% TFA (flow rate 0.8 ml min−1, monitoring at 254 nm). The peak at 11.4 min was collected and lyophilised to yield 1.1 mg of an orange powder (22%). Fluorescence λex 390 nm, λex 475 nm. m/z (ES-MS −ve ion) 735.9 ((M − 2H)2−), predicted 735.5.Solid phase synthesis of D-Ala-D-Cys and D-Cys-D-Ala
Solid phase peptide synthesis was carried out using a polystyrene resin functionalised with a Wang linker (CN Biosciences). In a typical procedure (e.g.: D-Ala-D-Cys) Wang resin (200 mg, 0.186 mmol loading) was swollen in DMF (5 ml) for 2 h. PyBOP (483 mg, 0.93 mmol, 5 eq), hydroxybenzotriazole (126 mg, 0.93 mmol, 5 eq), N-Fmoc-S-Trityl-D-Cysteine (586 mg, 0.93 mmol, 5 eq) in anhydrous DMF (2 ml) and Diisopropylethylamine (307 µl, 1.86 mmol, 10 eq) were then added to the resin and stirred for 90 min. The resin was drained and washed with 10 volumes of DMF followed by 10 volumes of dichloromethane. The coupling reaction was then repeated a second time. At this stage any unreacted sites on the resin were capped using acetic anhydride (0.25 ml) and diisopropylethylamine (0.25 ml) in DMF (2 ml) for 10 min. After rinsing with DMF and dichloromethane as before, the resin-bound cysteine derivative was then Fmoc-deprotected by the addition of 20% piperidine–DMF (2 × 5 ml) for 10 min. After a further rinsing, PyBOP (483 mg, 0.93 mmol, 5 eq), hydroxybenzotriazole (126 mg, 0.93 mmol, 5 eq), N-Fmoc-D-Alanine (290 mg, 0.93 mmol, 5 eq) in anhydrous DMF (2 ml) and diisopropylethylamine (307 µl, 1.86 mmol, 10 eq) were then added to the resin and stirred for 90 min. This reaction was repeated once more and capping and Fmoc-deprotection were carried out as described above. Fully deprotected D-Cys-D-Ala was generated by the addition of 95% TFA, 2.5% water, 2.5% triisopropylsilane (5 ml). After 2 h the cleavage solution was drained from the resin, dried in vacuo, and triturated with dried diethyl ether. The remaining solid precipitate was dissolved in 0.1% TFA and applied to a SAX cation exchange cartridge (Varian). The column bound material was washed with 0.1% TFA and then eluted in 0.35% ammonia solution. Fractions containing product were identified by Ellman's reagent and pooled and lyophilised to afford a pale yellow solid (9.3 mg, 0.048 mmol, 26%). ESMS positive ion [MH]+ 193.06 calculated, [MH]+ 193.10 detected. Synthesis of D-cysteinyl-D-alanine was carried out using the analogous procedure to the above.Kinetic assays with ligase MurF
Assays were carried out using a Cary 1 spectrophotometer and a 200 µl quartz cuvette. A typical assay contained 50 mM Hepes buffer pH 7.6, 10 mM MgCl2, 1 mM DTT, 50 mM KCl, 1.25 mM ATP, 1.25 mM phosphoenolpyruvate, 0.4 mM NADH and pyruvate kinase/lactate dehydrogenase (Roche, 20 µg). Finally MurF (15 µg), UDP-MurNAc tripeptide (0.2 mM) and the appropriate cysteine dipeptide (0.1 mM) were added. The reaction was followed at 340 nm (ε = 6.2 × 103 M−1 cm−1).Preparation of 4- and 5-pyrene labelled UDP-MurNAc pentapeptide derivatives
A typical procedure is as follows, for preparation of the 4-pyrene labelled derivative. In a 1.0 ml total volume, a solution of 50 mM MES buffer pH 6.0, 10 mM MgCl2, 10 mM phosphoenolpyruvate, 5 mM ATP, 1 mM DTT and 200 µM UDP-MurNAc-tripeptide(Lys) was degassed for 10 min by gentle bubbling of nitrogen gas. Under nitrogen, D-Cys-D-Ala (500 nmol, 2.5 eq) was added, followed by S. pneumoniae MurF (150 µg) and pyruvate kinase (Roche, 250 µg). The reaction was then stirred at room temperature under nitrogen for 25 min, before addition of N-1-pyrene maleimide (6 mg, 20 µmol, 100 eq) in 200 µl of DMSO. After three hours, the supernatant was filtered twice through Centricon 10 spin columns to remove proteins. The combined filtrate was then lyophilized overnight. The white solid was then dissolved in water and purified by HPLC on a Phenomenex prodigy ODS column (flow rate 0.8 ml min−1) using a gradient of 5% acetonitrile–water to 100% acetonitrile over a period of 20 min. The fluorescent product eluting at 10.9 min was collected and lyophilised to afford a colourless solid (56 nmol, 28%), ESMS −ve [M − 2H]2− 738.2. The 5-pyrene labelled derivative was prepared using the same protocol, using D-Ala-D-Cys (2.5 µmol, 12.5 eq). Typical yields were substantially lower for this reaction (6 nmol, 3%).Preparation of 5-pyrene labelled UDP-MurNAc-pentapeptide using ligase VanD
Incubations (total volume 300 µl) contained: 10 mM Tris buffer pH 8.6, 100 mM MgCl2, 2 mM ATP, 0.5 mM UDPMurNAc-tripeptide(DAP) (150 nmoles), 1 mM D-cysteine, 0.1 mM D-alanine, 5 µM VanD, and 7 units MurF. After incubation at 4 °C for 1 h, N-pyrene maleimide (200 µl of 0.4 mg ml−1 stock solution in 10 mM Hepes buffer pH 7.0) was added. After a further 1 h incubation, samples were centrifuged (13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm) for 2 min, then applied directly to a MonoQ anion exchange FPLC column (HR 5/5), eluting with 10 mM ammonium acetate (pH 7.5) at 0.5 mL min−1 for 10 min. A gradient of 0.01–0.5 M ammonium acetate (pH 7.5) was then applied over 20 min, then the column was eluted with 1.0 M ammonium acetate for a further 5 min. The fluorescent product (retention time 30 min) was collected and lyophilised to give the 5-pyrene labelled UDPMurNAc-pentapeptide, fluorescence λex = 340 nm, λem = 377 nm; m/z (ES−) 506.0 (M − 3H)3−, calc. 506.4.
000 rpm) for 2 min, then applied directly to a MonoQ anion exchange FPLC column (HR 5/5), eluting with 10 mM ammonium acetate (pH 7.5) at 0.5 mL min−1 for 10 min. A gradient of 0.01–0.5 M ammonium acetate (pH 7.5) was then applied over 20 min, then the column was eluted with 1.0 M ammonium acetate for a further 5 min. The fluorescent product (retention time 30 min) was collected and lyophilised to give the 5-pyrene labelled UDPMurNAc-pentapeptide, fluorescence λex = 340 nm, λem = 377 nm; m/z (ES−) 506.0 (M − 3H)3−, calc. 506.4.Kinetic assay of fluorescent UDPMurNAc-pentapeptides with E. coli MraY
E. coli DH5α membranes of pJYF3C overexpressing the mraY gene22 were solubilised in detergent containing buffer (50 mM Tris pH 7.5, 1 mM MgCl2, 2 mM β-mercaptoethanol, 10% glycerol and 1.5% CHAPS) and shaken for 1 h at 4 °C. Any unsolubilised material was removed by centrifugation (105g, 30 min) giving a final protein concentration of 4 mg ml−1. Fluorescence analysis was performed on a Hitachi F-2000 Spectrofluorimeter at room temperature.Undecaprenyl phosphate (10 µl of 5 mg ml−1 solution in 1 : 1 chloroform–isopropanol) was added into a 1.5 ml plastic Eppendorf, and solvents removed with a stream of nitrogen. The solubilised MraY (60 µl) was then added, followed by 150 µl buffer (Tris 200 mM, pH 7.5, KCl 100 mM and MgCl2 50 mM), and 80 µl water. After thorough mixing with a glass pipette, the assay mixture was transferred to a 300 µl fluorescence cuvette. The addition of 10 µl of stock fluorescent substrate (0.4 mM) initiated the reaction. The wavelength parameters were set to monitor the fluorophore under investigation.
Isolation of N-dansyl-lipid I
Following the MraY assay, as described above, lipid I products were extracted into 1 : 1 chloroform–methanol (90 µl). Analysis by C18 reverse phase HPLC, eluting with 100% methanol at 0.8 ml min−1, gave a new peak at retention time 21.5 min, which was isolated. Re-incubation of this material with MraY, using the above assay conditions, was followed by fluorescence spectroscopy and HPLC.Treatment of fluorescent UDPMurNAc-pentapeptide derivatives with E. coli membranes
E. coli W3110 inner membranes were prepared as described above, and were suspended in 50 mM Tris buffer pH 7.5 containing β-mercaptoethanol (10 mM) and MgCl2 (1 mM), to give a protein concentration of 30 mg ml−1. Assays were carried out at 25 °C, in a 300 µl total volume, and were initiated by addition of fluorescent substrate. In a typical assay, a solution of 50 mM Tris buffer pH 8.0, 0.1% Triton, 1 mM UDPGlcNAc, 80 µg ml−1 undecaprenyl phosphate, 50 µM unlabelled UDP-MurNAc pentapeptide and 1 µM of fluorescent UDPMurNAc-pentapeptide was used. The solution was equilibrated for 10 min before addition of W3110 membranes to final concentration of 0.8 mg ml−1. The reaction was followed by fluorescence emission detection for a period of 10–50 min. Excitation and emission wavelengths as follows: dansyl, 340/565 nm; fluorescamine, 390/475 nm; OPA, 340/455 nm; pyrene, 340/377 nm. In two stage experiments, the above assay was carried out without 1 mM UDPGlcNAc, which was then added after maximum fluorescence was reached.Preparation of 4-pyrene lipid I and II
The procedure used was based upon that described by Breukink et al.18 in a total volume of 50 µl. For the lipid II derivative, undecaprenyl phosphate (100 µg, 113 nmol, 11 eq) was dissolved in a solution of 50 mM HEPES at pH 7.6, 1 mM MgCl2, 1% Triton X-100, 4 mM UDP-GlcNAc and 200 µM 4-pyrene-UDP-MurNAc pentapeptide. The reaction was initiated by the addition of M. flavus vesicles (500 µg), and was stirred slowly for 3 h at room temperature. Fluorescent lipid product was extracted into n-butanol, dried under nitrogen and analysed by silica thin layer chromatography using a chloroform : methanol : water : ammonia (88 : 48 : 10 : 1) mobile phase. The preparation of 4-pyrene-lipid I was carried out as described above in the absence of the UDP-GlcNAc co-substrate.Acknowledgements
This work was supported by the European Commission (contract no. LSHM-CT-2003-503335) COBRA network, an EPSRC studentship (to S.B.), and a BBSRC studentship (to G.P.). We would like to thank Prof. R. Levesque (Quebec) for the gift of clones for MurA-F, and Dr N. Woodford (PHLS) for the gift of the VanD clone.References
- T. D. H. Bugg, in Comprehensive Natural Products Chemistry, ed. M. Pinto, Elsevier Science Ltd., Oxford, 1999, vol. 3, pp. 241–294 Search PubMed.
- J. van Heijenoort, Nat. Prod. Rep., 2001, 18, 503 RSC.
- J. M. Ghuysen, Annu. Rev. Microbiol., 1991, 45, 37 CrossRef CAS; C. Goffin and J. M. Ghuysen, Microbiol. Mol. Biol. Rev., 1998, 62, 1079 CAS.
- S. Rohrer and B. Berger-Bachi, Antimicrob. Agents Chemother., 2003, 47, 837 CrossRef CAS.
- P. E. Brandish, M. Burnham, J. T. Lonsdale, R. Southgate, M. Inukai and T. D. H. Bugg, J. Biol. Chem., 1996, 271, 7609 CrossRef CAS; P. E. Brandish, K. Kimura, M. Inukai, R. Southgate, J. T. Lonsdale and T. D. H. Bugg, Antimicrob. Agents Chemother., 1996, 40, 1640 CAS.
- T. Stachyra, C. Dini, P. Ferrari, A. Bouhss, J. van Heijenoort, D. Mengin-Lecreulx, D. Blanot, J. Biton and D. Le Beller, Antimicrob. Agents Chemother., 2004, 48, 897 CrossRef CAS.
- B. Schwartz, J. A. Markwalder, S. P. Seitz, Y. Wang and R. L. Stein, Biochemistry, 2002, 41, 12552 CrossRef CAS.
- L. Chen, H. B. Men, S. Ha, X. Y. Ye, L. Brunner, Y. N. Hu and S. Walker, Biochemistry, 2002, 41, 6824 CrossRef CAS.
- B. Chandrakala, B. C. Elias, U. Mehra, N. S. Umapathy, P. Dwarakanath, T. S. Balganesh and S. M. De Sousa, Antimicrob. Agents Chemother., 2001, 45, 768 CrossRef CAS.
- M. D. F. S. Barbosa, H. O. Ross, M. C. Hillman, R. P. Meade, M. G. Kurilla and D. L. Pompliano, Anal. Biochem., 2002, 306, 17 CrossRef CAS.
- F. C. Neuhaus and W. G. Struve, Biochemistry, 1965, 4, 120 CrossRef CAS.
- M. A. De Pedro, J. C. Quintela, J.-V. Höltje and H. Schwarz, J. Bacteriol., 1997, 179, 2823.
- E. Atherton and R. C. Sheppard, Solid Phase Peptide Synthesis: A practical approach, Oxford University Press, Oxford, UK, 1989 Search PubMed.
- D. J. Miller, S. M. Hammond, D. Anderluzzi and T. D. H. Bugg, J. Chem. Soc., Perkin Trans. 1, 1998, 131 RSC.
- K. Isono and T. Azuma, Chem. Pharm. Bull., 1972, 20, 193 CAS.
- T. D. H. Bugg, S. Dutka-Malen, M. Arthur, P. Courvalin and C. T. Walsh, Biochemistry, 1991, 30, 2017 CrossRef CAS.
- L. M. Dalla Costa, P. E. Reynolds, H. A. P. H. M. Souza, D. C. Souza, M. F. I. Palepou and N. Woodford, Antimicrob. Agents Chemother., 2000, 44, 3444 CrossRef.
- E. Breukink, H. E. van Heusden, P. J. Vollmerhaus, E. Swiezewska, L. Brunner, S. Walker, A. J. Heck and B. de Kruijff, J. Biol. Chem., 2003, 278, 19898 CrossRef CAS.
- J.-J. Li and T. D. H. Bugg, Chem. Commun., 2004, 182 RSC.
- M. J. Spencelayh, Y. Cheng, R. J. Bushby, T. D. H. Bugg, J.-J. Li, P. J. F. Henderson, J. O'Reilly and S. D. Evans, Angew. Chem., Int. Ed., 2006, 45, 2111 CrossRef CAS.
- A. El Zoeiby, F. Sanschagrin, P. C. Havugimana, A. Garnier and R. C. Levesque, FEMS Microbiol. Lett., 2001, 201, 229 CrossRef.
- A. J. Lloyd, P. E. Brandish, A. M. Gilbey and T. D. H. Bugg, J. Bacteriol., 2004, 186, 1747 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2006 |