Protein and small molecule microarrays: powerful tools for high-throughput proteomics
Mahesh
Uttamchandani
a,
Jun
Wang
b and
Shao Q.
Yao
*abc
aDepartment of Biological Sciences, National University of Singapore, 3 Science Drive 3, Singapore 117543
bDepartment of Chemistry, National University of Singapore, 3 Science Drive 3, Singapore 117543. E-mail: chmyaosq@nus.edu.sg; Fax: +65 6779 1691; Tel: +65 6874 1683
cNUS MedChem Program of the Office of Life Sciences (OLS), National University of Singapore, Singapore 117543
First published on 25th November 2005
Abstract
Advances in genomics and proteomics have opened up new possibilities for the rapid functional assignment and global characterization of proteins. Large-scale studies have accelerated this effort by using tools and strategies that enable highly parallel analysis of huge repertoires of biomolecules. Organized assortments of molecules on arrays have furnished a robust platform for rapid screening, lead discovery and molecular characterization. The essential advantage of microarray technology is attributed to the massive throughput attainable, coupled with a highly miniaturized platform—potentially driving discovery both as an analytical and diagnostic tool. The scope of microarrays has in recent years expanded impressively. Virtually every biological component—from diverse small molecules and macromolecules (such as DNA and proteins) to entire living cells—has been harnessed on microarrays in attempts to dissect the bewildering complexity of life. Herein we highlight strategies that address challenges in proteomics using microarrays of immobilized proteins and small molecules. Of specific interest are the techniques involved in stably immobilizing proteins and chemical libraries on slide surfaces as well as novel strategies developed to profile activities of proteins on arrays. As a rapidly maturing technology, microarrays pave the way forward in high-throughput proteomic exploration.
 Mahesh Uttamchandani | Mahesh Uttamchandani (b. 1979) graduated with a first class honours in Microbiology from the National University of Singapore in 2003. A Defence Science and Technology Agency scholar, he joined the DSO National Laboratories as a research engineer in 2004 and is presently pursuing his PhD in Biology under the supervision of Professor Shao Q. Yao. His research interests include combinatorial chemistry, chemical biology and proteomics. |
 Jun Wang | Jun Wang was born in Guangshui, China in 1979. He studied Chemistry at Wuhan University in China, where he received his bachelor’s degree in 2003. He subsequently worked one year for Scinopharm Kunshan Biomedical Co, Ltd as an R&D scientist, mainly focusing on the synthesis of drug intermediates. He joined Professor Shao Q. Yao’s group at NUS in 2004 as a postgraduate student. The projects he is currently undertaking involve the design and synthesis of potent and selective matrix metalloprotease inhibitors, and site-specific labelling of metalloproteases inside the cell. |
 Shao Q. Yao | Professor Shao Q. Yao (b. 1970) grew up in China. He graduated with a bachelor’s degree in Chemistry from Ohio State University in Columbus, Ohio, USA in 1993, and subsequently obtained his PhD in organic & bioorganic chemistry from Purdue University, USA in 1998 with Professor Jean Chmielewski. He conducted his post-doctoral research in the fields of bioorganic chemistry, functional genomics & molecular biology first at the University of California at Berkeley, USA, then at the Scripps Research Institute, USA, both under the supervision of Professor Peter G. Schultz. He came back to Asia and joined the National University of Singapore in 2001 and currently holds a joint appointment between the Department of Chemistry and the Department of Biological Sciences. His research interests include combinatorial chemistry, chemical biology, proteomics and bioimaging. |
Introduction
Over the last decade, microarrays have transformed the life science research landscape. Novel applications of microarrays ranging from expression profiling1 and mapping interaction networks to molecular fingerprinting and ligand discovery,2 have significantly impacted both basic and applied spheres of research. Creative ideas by biologists, chemists and engineers alike are propelling this interdisciplinary technology to interesting new levels. The success of microarrays has inspired a growing following of scientists to take on high-throughput, discovery-driven research, drawing impetus towards accelerated information assimilation and knowledge growth.Though the technology was first introduced as miniaturized DNA assemblies on chips, it was not long before further pioneering efforts made it possible to sequester small molecules3 and subsequently proteins4 in addressable grids for rapid analysis. Now it has also become feasible to examine a host of other biomolecules, including membrane proteins,5 carbohydrates6 and peptides,7 as well as complex structures like tissues8 and even live cells on arrays,9 providing a tremendous opportunity for screening and large-scale analysis. It is remarkable that so many diverse applications have stemmed from a single technology platform. With microarrays, this is attributed to significant advantages rooted in miniaturization, parallelization and automation. Breaking away from antecedent microtiter-based (or well-based) assays, microarrays offer a flat reaction surface that ameliorates the washing and incubation steps, while providing a significantly higher sample density. This design concept makes it convenient to undertake thousands of assays quickly and cheaply by effectively reducing the amount of often precious reagents required to perform highly informative experiments. The commercial availability of complementary research infrastructure is equally important in catalyzing interest and outreach of this technology platform, making it readily accessible to any researcher interested and willing.10
Every molecular class displayed on arrays presents distinctive challenges while offering unique opportunities.11 Small molecules and protein microarrays have witnessed tremendous growth in recent years with significant technical and conceptual improvements made towards library creation and immobilization formats. These developments, together with novel advances herein described, have set the foundation for these platforms to eventually take on more routine applications in discovery and diagnostics.12
Protein microarray
The first report of using protein arrays for protein–protein interaction, ligand binding and biochemical investigations was by MacBeath and Schreiber in 2000.4 Though the variety of proteins tested was small, it dispelled initial scepticism of proteins losing their activities when covalently immobilized on glass surfaces—a critical consideration when studying proteins on arrays (in this case aldehyde-derivatized slides were used). Proteins are complex molecules, thus comprehensive global analysis of proteins in a parallel format is no trivial task. It was thus a significant developmental milestone when Snyder's group reported the yeast proteome array in 2001,13 where 5800 yeast open reading frames were expressed and presented on a single glass slide for large-scale proteomic analysis. The protein collection was individually purified and tethered via a hexahistidine motif onto nickel-coated slides before being screened simultaneously for interactions with calmodulin. This resulting microarray has been commercialized (Yeast ProtoArray™, Protometrix, Invitrogen) and since been used for a variety of applications.14 Since these seminal works, protein microarrays have generally seen major developments in two major aspects, in terms of immobilization methods for anchoring huge repertoires of proteins and expanding areas of applications using novel strategies. These will be discussed in the following sections.Array fabrication strategies
Immobilizing proteins stably onto chips is the first and most fundamental step in any successful protein microarray venture. Factors such as molecular orientation, immobilization chemistry and protein stability are key considerations that govern how the proteins are presented for parallel analysis and screening.15 Some groups have sought the use of “capture” agents like aptamers or antibodies to assemble proteins as microarrays. Though the involvement of an intermediary molecule avoids direct covalent immobilization, this approach introduces concerns of cross-reactivity of the intermediary scaffold itself and binding stability, problems that are avoided with direct covalent tethering. The latter strategy could, however, be more vulnerable to loss of protein function upon immobilization, a phenomenon peculiarly difficult to circumvent on any support base, but is generally minimized in microarrays by selecting suitable immobilization strategies, appropriate buffering conditions for spotting and low temperature storage of printed slides.Recent developments have expanded the microarray “toolbox”, providing a plethora of options depending on the downstream screening requirements. This has included a variety of chemistry introduced to immobilize proteins (some of which may have equal applicability to small molecules, including peptides), as well as strategies for self-assembly and in situ microarray creation. Immobilization chemistry takes on two typical formats, either regioselective immobilization which results in non-uniformly oriented proteins (for example using aldehyde surfaces which may bind to any available amine group in a protein), or site-specific immobilization which orientates all proteins uniformly (for example by His-tag immobilization).
Our group recently developed a versatile purification and tagging approach for proteins using inteins (i.e. spliceable protein motifs).16 Proteins were first expressed with a C-terminal intein and chitin-binding domain for affinity capture on chitin columns. The proteins were then treated with a cysteine–biotin conjugate, which triggered the intein cleavage to release the protein from support and simultaneously tagged it with biotin. This intein-mediated biotinylation approach provides a feasible strategy for purifying large numbers of proteins in a scalable format for high-throughput immobilization onto avidin-coated glass surfaces.16 The stability of the avidin–biotin linkage is an additional feature that represents a highly stable and extremely strong tether for array creation. We further demonstrated our intein-mediated strategy could be readily applied to biotinylate proteins both in live cells (bacteria and mammalian cells) as well as in established cell-free protein expression methods.17 This demonstrates the versatility of the strategy in preparing proteins for immobilization on protein microarrays.
An alternative strategy has since been developed that uses inteins in the introduction of N-terminal cysteine containing proteins that can chemoselectively react (by native chemical ligation) with thioester groups on derivatized slides (Fig. 1A).18 The same concept has been applied in reverse, by introducing the thioesters on the C-terminus in a protein (also by intein cleavage) for reaction with cysteine-derivatized surfaces, thereby anchoring proteins through their carboxy terminus (Fig. 1A).19 Compared to the previous strategy,16 both these methods mediate covalent attachment of proteins onto treated slide surfaces. These strategies also share the advantage of employing small tags that minimally perturb the overall protein architecture, presenting them close to their native state for interaction and binding assessments. Tirell and colleagues have developed a capture strategy by exploiting heterodimeric leucine zipper pairs.20 Proteins to be immobilized were fused with the ZR domain as an affinity tag for capture on slides coated with the complementary ZE capture domain (Fig. 1B). The strategy was successfully demonstrated using glutathione-S-transferase (GST) and green fluorescent protein.
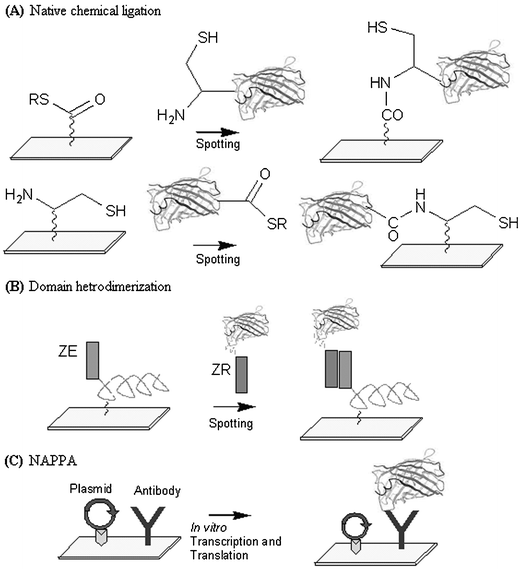 | ||
| Fig. 1 Various strategies developed for fabricating protein microarrays. (A) Covalent attachment using native chemical ligation.18, 19 (B) Leucine zipper domain hetrodimerization.20 (C) Nucleic acid programmable protein array.24 | ||
Lahiri et al. introduced a method to immobilize membrane proteins on arrays,5 thereby expanding the scope of protein microarrays beyond soluble proteins. Membrane monolayers were generated using vesicular solutions of egg-yolk phosphatidyl choline (PC) with dihexadecanoylphosphatidylethanolamine or 4 ∶ 1 mixtures of dipalmitoyl PC ∶ dimyristyl PC on γ-aminopropylsilane (GAPS)-coated slides. Three G-protein coupled receptors (GPCRs) were spotted on these membrane arrays which were found to localize stably on this lipid support and were accordingly presented for ligand binding assessments. The group recently developed alternative porous glass substrates (also coated with GAPS) as a more robust surface for probing functional interactions of GPCRs.21
Further approaches to generate protein arrays have emerged from extensions of DNA-based approaches. For example, Weng et al. tethered in vitro translated proteins with their coding mRNAs, and studied these assemblies on slides printed with complementary nucleotide sequences.22 This strategy was shown to localize the protein conjugates to predefined “addresses” by simple hybridization. It was also demonstrated that the relative amount of immobilized proteins could be directly controlled by varying the concentration of the capture oligonucleotides spotted. This strategy, termed PROfusion™ technology, adopts traditional DNA microarray strategies for the provision of protein microarrays by self-assembly. Choi et al. devised an alternative strategy also using DNA surfaces by exploiting the GAL4 DNA binding domain to generate fusion proteins for immobilization onto slides coated with the target dsDNA sequence (that binds with the GAL4 domain selectively, with a low dissociation constant in the nanomolar range).23
Ramachandran et al. have since taken the strategy a step further by immobilizing a variety of plasmids (cross-linked using ultraviolet light to psoralen–biotin) that code for target proteins together with a C-terminal GST epitope.24 During the printing process, anti-GST antibodies were co-immobilized together with avidin and the biotinylated plasmids onto predefined locations on the array. Proteins were expressed by subjecting the array surface to in vitro transcription and translation, allowing each protein to be immobilized in situ through the GST tag (Fig. 1C). Cross reactivity between spots was shown to be negligible by using suitable spotting densities as well as other optimized conditions. The strategy, termed nucleic acid programmable protein array (NAPPA) enables long-term storage of the stable DNA microarrays, which can be readily converted, when required, into active protein microarrays.
Applications
Protein microarrays are highly informative tools that have been used for high-throughput interaction studies with various biomolecules including proteins, DNA and small molecules as well as in biochemical investigations of protein activity for functional annotation and characterization.Mapping protein interactions
There is much potential in screening whole proteome microarrays for a variety of different purposes. It provides a unique window into the ensemble of proteins present in an organism for parallel analysis; offering huge opportunities in screening potential drug interactions as well in detecting post-translational modifications that regulate protein behaviour. The yeast proteome array has already been successfully shown to probe for novel interacting partners of calmodulin as well as in the discovery of phospholipid binding proteins.12 It has also been employed to study the specificity of eleven commercial polyclonal and monoclonal antibodies against yeast proteins, as well as three antibodies commonly used against specific epitopes (haemagglutinin, FLAG and myc) allowing thorough examination for cross-reactivities.25 As expected, the monoclonal antibodies exhibited higher specificity than their polyclonal counterparts; whilst among the polyclonal antibodies, those targeting peptide motifs had the highest relative specificity.The yeast proteome array was also used to identify potential targets of a small molecule that suppresses the growth inhibition of rapamycin.26 The biotinylated molecule was screened on the yeast proteome array and revealed two potential target proteins, Tep1p and Nir1p, that associate with phosphatidylinositides, providing insight to how the pathway might be regulated. This study clearly demonstrates the utility of screening small molecules against protein arrays, especially in revealing the mechanism of action of drug candidates.
In an attempt to probe gene regulation, Hall et al. described a hybrid approach using both a protein microarray and the chromatin immunoprecipitation (ChIP-chip) method to map novel protein–DNA interactions.27 Briefly, fluorescently labelled genomic DNA was screened on the yeast proteome array to shortlist over 200 putative DNA-binding proteins. Eight of these proteins, that had not been previously known to bind DNA, were selected for further investigation using the ChIP-chip strategy. By subjecting the DNA immunoprecipitated from the shortlisted proteins to conventional DNA microarray analysis on an intergenic chip, the DNA sequences targeted by these proteins were conveniently unravelled. To further expand the prospect of proteomic profiling on arrays, a human proteome array containing 3000 proteins has recently been made available (Invitrogen), opening up even greater avenues for research.
DNA interactions with immobilized proteins have also been used to map p53 binding interactions with the GADD45 promoter.28 The simultaneous presentation of an array of p53 variants enabled rapid functional characterization of the p53 protein in relation to its polymorphic forms. The array was also screened for MDM2 interactions as well as for p53 phosphorylation by casein kinase II.
Protein arrays have also been used in more specialized applications. 49 coiled-coil strands of human leucine zipper transcription factors were screened on a microarray for interactions.29 Letarte et al. have used protein microarrays for epitope mapping of human leukocyte membrane protein (CD200).30 A similar strategy was also employed by Poetz et al. in the detailed characterization of binding profiles of recombinant Fab fragments.31 Protein microarrays have also been used to identify 14-3-3 kinase interactions with tuberin, proteins involved in phosphotidylinositol 3′-kinase signalling.32 A microarray was prepared with candidate protein domains and motifs, including 14-3-3 proteins, and screened against phosphorylated and non-phosphorylated tuberin peptides. Estrogen receptors have also been immobilized on aldehyde surfaces for ligand binding assays.33
Global protein profiling
Our group has developed a strategy that enables the rapid analysis and discovery of different classes of proteins in a microarray.34 By taking advantage of fluorescently labelled, mechanism-based suicide inhibitors (targeting serine hydrolases, phosphatases and cysteine proteases), we were able to selectively identify and label proteins immobilized on the microarray in an activity-dependent manner. The strategy was tested with twelve representative enzymes and the probes were shown to exclusively label the targeted classes of enzymes (immmobilized regioselectively on epoxy treated slides). Not only did this demonstrate that enzymatic activity is preserved when proteins are immobilized on the surface, but it also furnished unique opportunities in enzyme characterization and functional annotation in a high-throughput fashion using the microarray. We have recently extended this approach to the fingerprinting of protein activities on microarrays using a novel repertoire of activity-based probes that are able to evaluate protease specificity irrespective of class.35Building on this approach, Eppinger et al. have developed a strategy for kinetic determination of enzyme activity on microarrays.36 Papain, a cysteine protease, was immobilized on N-hydroxysuccinimide (NHS)-activated hydrogel slides and reacted under a variety of concentrations and for different periods of time with a fluorescently labelled suicide inhibitor against the enzyme. The fluorescent data obtained were normalized and analyzed to obtain kinetic constants of the enzymatic reaction. The strategy was further explored to determine the inhibition constants of known inhibitors of papain, by subjecting titrated mixtures of these inhibitors with the immobilized enzyme. The same group has further employed this microarray strategy to profile a group of six cathepsins, an important class of cysteine proteases, against a seven known inhibitors, obtaining inhibition constants which were in good agreement with those from traditional approaches.37
The ability to perform such activity-based profiling studies of enzymes in whole proteomes has also been recently demonstrated. Sieber et al. have used an antibody array platform to capture enzymes of interest from complex proteomes that were pretreated with fluorescently tagged activity-based probes, forging enzyme profiling in a complex cellular milieu with the advantage of high-throughput deconvolution (by capture) and analysis on microarrays.38 Both serine proteases and metalloproteases were screened using this approach. However, even as the range of activity-based probes is being expanded greatly to cater to almost every enzyme class (potentially enabling multiple enzyme classes to be profiled simultaneously), the bottleneck in applying this strategy lies in the availability of high-quality antibodies for studying every protein of interest. Apart from applications in enzyme profiling, Jung and Stephanopoulos have developed an alternative application of enzyme arrays for pathway optimization.39 By immobilizing enzyme mixtures in different proportions in spots on the array, the authors showed that it was possible to optimize various biosynthetic pathways in a high-throughput manner, including the five-step pathway for trehalose biosynthesis. This strategy provides clear opportunities for industrial biosynthesis applications.
Outlook
New directions and creative ideas are fuelling exciting methodologies in deploying and using protein microarrays. Some of these interesting concepts will be discussed in this section, offering a sense of things to come as well as in describing frontier technologies that will benefit from further development.Several groups are building capabilities that will soon allow for even greater miniaturization in protein microarrays. Gu et al. created “nano-wells” using conductive atomic force microscope (AFM) lithography, of diameters and depths of 91 ± 6 nm and 1.31 ± 0.12 nm respectively, within oligo(ethylene glycol) monolayers assembled on silicon surfaces.40 Avidin was successfully immobilized in these wells and detected using fluorescently labelled biotin. His-tagged ubiquitin and thioredoxin have also been immobilized in nano-wells using dip-pen nanolithography on nickel surfaces, without the requirement of an applied electric field.41
Surface plasmon resonance (SPR)-based approaches offer label-free measurement of protein interactions, providing a useful complement to fluorescence-based methods which currently dominate in microarray detection and analysis.42 Usui-Aoki et al. were able to perform 400 real-time antibody interaction measurements simultaneously on a specialized protein chip and detector.43 Ha et al. have also used spectral SPR sensors to analyze multiple proteins on arrays.44 An alternative strategy has utilized self-assembled monolayers on a gold surface together with MALDI-TOF MS for label-free detection of protein interactions on biochips.45
In a creative synergy of electrospray ionization coupled with array creation, Ouyang et al. have developed a method to deposit proteins separated by mass spectrometry onto microarrays.46 The technique, termed ion-soft landing, was used to separate and deposit four proteins, namely cytochrome c, lysozyme, insulin and apomyoglobin while retaining their biological activities on gold surfaces. This method is significant in that it is able to selectively isolate proteins in a mixture and presents them for parallel analysis on microarrays. The strategy has also been applied to arraying small molecules.47
Small molecule microarray
Small chemical molecules provide efficacious handles in the selective perturbation of protein functions. This property makes small molecules viable drug candidates, and rapid evaluation of repertoires of small molecules for biological activity is thus the fundamental first step in drug discovery. Development of robust screening tools, including small molecule microarrays (SMM), is fundamental for the pursuit of efficacious drug candidates and biologically relevant compounds. Discovery of molecules that modulate protein functions provides unique insights into various cellular mechanisms, allowing biological pathways to be better targeted and controlled. Modern synthetic approaches complement the vast diversity and complexity of natural products by contributing libraries of compounds for parallel analysis on microarrays. The massive number of compounds now available through large-scale solid phase synthesis and combinatorial chemistry is making it necessary to develop even more robust screening capabilities.SMM was first introduced by MacBeath and Schreiber in 1999.4 The same group has pioneered the diversity-oriented synthesis (DOS) approaches; capturing even larger realms of chemical space for simultaneous screening on SMM.48 The one-bead–one-compound (OBOC) combinatorial approach has further catalyzed library generation capabilities and has also been successfully applied to SMM.49
Library design for array-based screening
It would be ideal to develop immobilization strategies that could immobilize any molecule without concern over binding compatibility, orientation and surface properties. However, until such a global solution materializes, functional group selection remains a key consideration in library design and synthesis. Specifically, the small molecules should possess specific affinity handles that react predictably and chemoselectively with the corresponding functional groups on the substrate surface. As progress is made in synthetic strategies in line with different types of sophisticated immobilization chemistry, we may one day be able to immobilize any compound conveniently in an array format for high-throughput screening. Already a greater attention to this problem has given rise to major improvements, spearheading progress both in expanding the varieties of chemistry available for molecule immobilization as well as generating technologies to sequester both chemical and natural product libraries on microarrays.Several strategies for developing chemical libraries compatible with SMM immobilization have been developed. Germeroth et al. prepared a SMM in which the small molecule library was synthesized directly on the substrate itself.50 1,3,5-Triazines were directly synthesized on cellulose or polypropylene membranes by SPOT synthesis.51 A similar strategy was also reported by Blackwell and colleagues in the use of the Ugi four-component reaction to construct small molecule arrays on cellulose membranes.52 In contrast, the majority of chemical libraries used in SMM's are synthesized on other platforms before immobilization on the microarray. A clear example of this route is the DOS libraries developed and exploited by Schreiber et al. for array-based applications.53–56 One study reported a library comprised 3780 structurally unbiased 1,3-dioxane small molecules synthesized by split-pool synthesis using a “one-bead–one-stock solution” approach.53 This was used in the dissection of a signalling pathway mediated by glucose by identifying ligands of the protein Ure2p.54 Another development saw the synthesis of 6336 phenol-containing fused bicycles and tetracycles from a six-step stereoselective synthesis.49 This strategy afforded products with between two to four rings and up to 6 stereocenters. These compounds were immobilized on diazobenzilidene-functionalized glass surfaces that captured phenol and other groups of comparable acidity. The microarrays were screened against Cy-5 labelled calmodulin and to afford 16 putative hits, revealing the strongest ligand that bound with a Kd of 0.12 ± 0.03 µM.
Hydroxyl-functionalized molecules have also been immobilized on chlorinated glass slides.55 Three libraries were combined to provide 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 396 compounds immobilized on a single array. One class of the compounds was generated through the 1,3-dioxane synthetic pathway, whilst the two other libraries were made from biaryl scaffolds using DOS.56 The arrays were screened against Hap3p (a component of a yeast transcription factor complex) and visualized using Cy-5 labelled anti-GST antibody. Of the two hits obtained from the array screens, only one was found to truly bind to Hap3p (with a dissociation constant of 5.03 µM), whilst the other was discovered to bind GST. This ligand was further found to inhibit Hap3p in vivo. Further modifications of this molecule provided a more potent ligand against Hap3p with a Kd of 0.33 µM.
396 compounds immobilized on a single array. One class of the compounds was generated through the 1,3-dioxane synthetic pathway, whilst the two other libraries were made from biaryl scaffolds using DOS.56 The arrays were screened against Hap3p (a component of a yeast transcription factor complex) and visualized using Cy-5 labelled anti-GST antibody. Of the two hits obtained from the array screens, only one was found to truly bind to Hap3p (with a dissociation constant of 5.03 µM), whilst the other was discovered to bind GST. This ligand was further found to inhibit Hap3p in vivo. Further modifications of this molecule provided a more potent ligand against Hap3p with a Kd of 0.33 µM.
We have developed a SMM using 1,3,5 triazines as this scaffold has demonstrated desirable bioactivity.57 Unlike typical one bead-one stock DOS-based approaches earlier described, the parallel synthetic strategy adopted enabled the identity of each small molecule to be known a priori, facilitating immediate post-screening identification of hits. The library synthesis incorporated diverse linkers, taking into account of the linker effect in overall molecular potency. This further enabled the direct use of these molecules in solution-based assays, without any modification or linker removal.58 After immobilization through an amine-containing linker onto N-hydroxysuccinimide derivatized slides, the molecules were screened with human IgG for the discovery of safe and cost-effective small molecule alternatives of Protein A and Protein G for industrial antibody purification and production. The best hits obtained were subjected to SPR analysis, affording the tightest binder with a Kd of 2.02 µM.
Schultz and co-workers have developed a unique approach in generating libraries for SMM through the use of peptide nucleic acids (PNAs).59 The PNA moiety was used to encode the ligand identity and at the same time bears the unique advantage of its synthetic flexibility, resistance to degradation within a biological milieu, and most importantly its ability to hybridize with a complementary strand of DNA. The PNA-conjugated ligand libraries were incubated with the target while a DNA-based microarray was used in the rapid deconvolution of the positive hits. The PNA libraries were synthesized using established procedures.60
Array fabrication strategies
Several methods have been described for the development of small molecule microarrays, including covalent immobilization, photoactivatable cross-linking and in situ synthesis. As with protein microarrays, immobilization is critical in SMM preparation as it determines the way in which the molecules are oriented for interaction with targets. Depending on the requirements, a homogenous orientation may be preferable, as in enzyme characterization. However in the case of epitope binding, it may be desirable to randomly immobilize the small molecules so as to expose every available facet for interaction.Covalent immobilization
There have already been a variety of established chemistries for the covalent attachment of molecules onto arrays. Methods using aldehyde, NHS and epoxy surfaces to capture amines, native chemical ligation as well as other more specialized binding chemistry have already been mentioned in earlier sections so will not be repeated herein. In this section we focus on more recently introduced strategies for the covalent capture of small molecules onto microarrays.Waldmann et al. have established a highly chemoselective method for attachment of compounds on an array.61 The strategy exploits the Staudinger ligation that couples azide-functionalized molecules with a phosphane-derivatized glass substrate (Fig. 2A). This method has the desirable advantage in that it utilizes a pair of reactive functional groups that are biologically inert. This strategy is compatible with water and oxygen and is also suitable in a wide variety of solid-phase synthetic strategies.
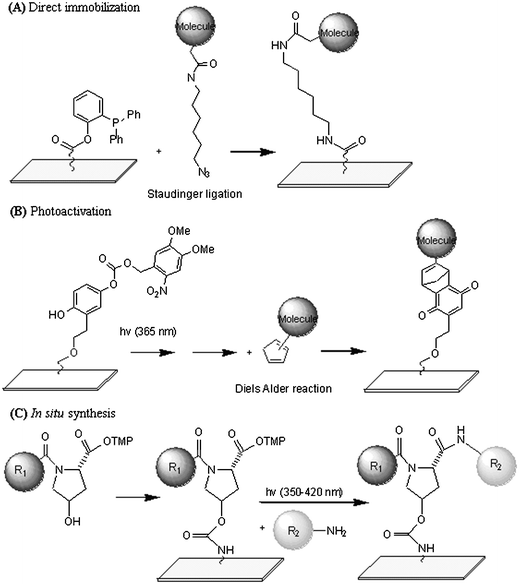 | ||
| Fig. 2 Various strategies developed for fabricating SMM’s. (A) Covalent immobilization using Staudinger ligation.61 (B) Photoactivation strategy followed by capture.63 (C) Sequential molecular assembly in situ.65 | ||
Increasing the density of molecules that may be immobilized within the same spotting surface could effectively improve the sensitivity of array-based applications. It was with this aim that Puskas and co-workers used dendrimer linkers to greatly increase immobilization efficiency with reduced backgrounds. A variety of different surfaces were developed for the covalent immobilization of small molecules (as well as nucleic acids and proteins) through acrylic and epoxy reactive surfaces.62
Photoactivation strategies
Mrksich et al. assembled alkanethiolate monolayers (SAM) consisting of nitroveratryloxycarbonyl (NVOC)-protected hydroquinone groups on gold-coated substrates.63 By site-directed UV irradiation using a photomask, the NVOC group was selectively liberated to generate hydroquinone, which underwent spontaneous chemical or electrochemical oxidation to give benzoquinone. This reaction renders the surface reactive to cyclopentadiene-tagged ligands (Fig. 2B). The technique was found to be amenable to techniques such as AFM and near field scanning optical microscopy (NSOM), making further miniaturization of the format possible.Kanoh and colleagues have used a photoinduced cross-linking reaction to anchor small molecules on a microarray using a strategy that allows multiple facets of the molecule to be exposed for interaction.64 A photoactivatable diazirin linker was coated on the slide surface. Following UV irradiation, the resulting reactive species generated on the surface reacts with proximate small molecules in a functional group-independent manner, leading to small molecule immobilization. This thus eliminates the need for specific functional groups amongst library members and is therefore well suited for immobilizing naturally derived repertoires of compounds.
In situ synthesis
There have been recent developments that enable the direct synthesis of small molecule microarrays in situ. Belshaw et al. have introduced orthogonal safety-catch protecting groups based on the trimethoxyphenacyl group.65 These groups were used for the maskless light-directed synthesis of a 2 × 2 small molecule microarray that may be broadly extended for the synthesis of even larger libraries (Fig. 2C). A similar strategy was developed using peptoids (oligomers of N-substituted glycines). Kodadek and colleagues immobilized MeNPOC-protected glycolic acid onto chemically modified slides through an ester linkage. Irradiation with UV light activated specific regions of the slide’s exposed hydroxyl groups which were then activated with tosyl chloride followed by displacement with a primary amine.66 The stepwise method of microarray synthesis in this manner facilitates the rapid creation of large molecular diversities with a only few synthetic steps.67 It is also possible to automate the entire procedure, reducing the time taken for array creation. The spot density may also be further enhanced, expanding greatly the number of compounds that may be screened simultaneously on a single glass slide.Applications
Small molecule microarrays are well established tools in discovering ligands for virtually any target. In addition to the various examples earlier described,53–56 Schreiber et al. synthesized 18000 enantioenriched and skeletally diverse 1,3-dioxanes for evaluation on array.68 This represents the first report of stereochemically diverse sets of small molecules to be immobilized and screened on SMM. Each member of the library was chemically encoded. Subsets of this library were immobilized by diisocyanate capture and screened against calmodulin. Patterned incorporation of heptamethyleneimine elements in the molecular scaffold was found to bear importance in binding to calmodulin. Several ligands with low micromolar dissociation constants against the protein were subsequently identified. A large assortment of small molecules immobilized on a microarray also offers a new way of analyzing proteins. Mihara and colleagues used an array of fluorescent labelled looped peptides to obtain “protein fingerprints” against a diverse set of proteins.69 Using a large library of peptoids, Muralidhar and Kodadek obtained unique binding “fingerprints” against three proteins, namely GST, maltose-binding protein and ubiquitin.70 Specifically, 7680 octameric peptoids were synthesized in situ and presented on a maleimide-functionalized glass slide. Upon screening with the target proteins, unique and reproducible fluorescence readouts, or fingerprints, of proteins were generated across all the small molecule features on the SMM. The identity of the small molecules of the array were not known a priori due to the synthetic strategy used, necessitating a big deconvolution effort to identify the molecules at every address on the array (such information is useful in mapping protein–ligand interactions for example). Nevertheless, these reports both demonstrate the valuable throughput of SMM as a chemical genetic tool and showcase its importance in fuelling discovery-driven research.
Characterizing enzymes using SMM
Moving beyond the study of non-covalent biochemical interactions on SMM, several groups have reported strategies to fingerprint and characterize proteins based on their enzymatic activities.70–76 The methods typically employ fluorogenic coumarin derivatives comprising an enzyme recognition domain. In the conjugated state, the small molecule is virtually non-fluorescent; but upon enzymatic recognition and catalysis, the coumarin moiety is liberated, restoring its fluorescence for easy detection. In one report, small molecule probes were successfully designed against four different enzyme classes, namely epoxide hydrolases, esterases, proteases and phosphatases and were immobilized in a microarray format for parallel analysis against their representative enzymes.71 The significant advantage of this approach is the label-free analysis of proteins, setting the foundation towards high-throughput profiling of disease states using SMM as a potential diagnostic tool. In a similar study, Ellman and colleagues have developed fluorogenic peptide substrate arrays for studying protease activity.72Another creative approach by Diamond and Gosalia saw the use of nanolitre droplets as microreactors for testing small molecule inhibition against a variety of caspases.73 352 small molecules were first printed in spots of glycerol which remained hydrated, such that the molecules were freely suspended in solution. Subsequently, sequential aerosolized application of a caspase and a fluorogenic substrate enabled parallel evaluation of the inhibition potency against all 352 small molecules through direct fluorescence analysis. This strategy revealed a caspase inhibitor that showed high potency against all three of the caspase isoforms screened. The greatest advantage offered by this strategy is in performing highly parallel solution-based assays on microarrays at extremely low reaction volumes (1.6 nL per spot). However, the necessary use of a large amount of an organic solvent such as glycerol or DMSO (up to 50% in a reaction) limits the applicability of this strategy as most enzymes are not active under these conditions. The strategy has since been further explored in profiling serine proteases using coumarin-based peptide substrate libraries.74
To circumvent the need for using excessive organic solvents in an enzymatic reaction, our group, as well as others,75 have reported the creation of surfaces that are now able to profile enzymes rapidly in a microarray format. Enzymes were spotted in spatially addressable, segregated droplets (appropriately buffered without the use of glycerol) on the slides coated with fluorogenic substrates. We tested this system using 37 enzymes and were able to obtain fluorescence readouts of proteases and phosphatases across temporal and stoichiometric dimensions concurrently on a single microarray surface.76 Angenedt and colleagues also performed such multiplexed assays in subnanolitre volumes, and were able to obtain a detection sensitivity of up to 35 enzyme molecules in a single droplet.77 This “nanodroplet” strategy thus materializes the promise of the microarray in providing a miniaturized, high-throughput screening method, applicable not only to the profiling of proteins, but also in rapid screening of small molecule libraries for potent inhibitors.
SMM applications involving DNA microarrays
The process of creating sizable arrays, involving first the preparation of libraries of different molecules and second the addressable display of these compounds on slides for parallel screening, has invariably been the greatest bottleneck of any small molecule microarray endeavor. One solution to this rides on the now routinely used and commercially available oligonucleotide microarray, making it cost-effective and attractive to augment this platform with large-scale small molecule screenings, with a DNA hybridization-based deconvolution approach for identifying small molecule interactions of interest.The PNA approach has also been applied towards enzyme profiling.77,78 A library of 192 rhodamine-based fluorogenic protease substrates was prepared by split-pool combinatorial synthesis with PNA tags.78 These substrates were used to profile the proteolytic activity of individual proteases as well as those present in clinical blood samples. Dust mite extracts were also screened for proteolytic activity and a Derp 1 protease isolated was further screened for inhibitors using the PNA-encoded small molecule libraries.79
Our group has pioneered the use of activity-based small molecule probes to target multiple enzymes expressed with the coding mRNAs in a ribosome display format, followed by high-throughput isolation and identification of subclasses of proteins with desired activities using a decoding DNA microarray.80 The use of ribosome display is advantageous as it enables in vitro expression of a large pool of proteins in a cell-free format, with the incorporation of a unique mRNA tag for every member in the protein library. The library was incubated with a biotinylated activity-based small molecule probe captured on streptavidin beads. Following PCR amplification of mRNAs associated with the isolated proteins, their identity was easily revealed, in parallel, by hybridization on the decoding DNA microarray. The strategy was tested against the protein tyrosine phosphatase family, where the four targeted enzymes were shown to be cleanly isolated and identified on a DNA microarray from a mixture of nearly 400 other proteins. This approach has furnished a proteomic tool with great promise in genome-wide profiling of various classes of proteins en masse.
Phage libraries have also been used to genetically encode small molecules. Walsh and co-workers have reported a method of tethering phages to small molecules for potential high-thoughput chemical screening.81 Sfp phosphopanthe-theinyl transferase was used to covalently link small molecules monovalently onto the peptidyl carrier protein expressed on M13 phage. Five small molecules were conjugated to different phages (each with a unique genetic identity) and the small molecules, enriched from a typical biopanning experiment against selected target proteins, were readily detected and identified using the DNA microarray.
Outlook
New strategies have recently been introduced that expand the scope and application of small molecule microarrays. A creative extension by Neri et al. has demonstrated the ability to screen multivalent assemblies of small molecules against targets of interest.82 The technology uses libraries of organic molecules tethered to oligonucleotide tags that contain both a code to uniquely identify each small molecule as well as a domain to mediate self-assembly of the library by hybridization (Fig. 3A). Elegantly, constructing chemical libraries in this format exponentially expands the molecular scaffolds that can be generated and screened from a single defined library, greatly enriching its size and application. Binders with sub-micromolar dissociation constants were isolated against bovine carbonic anhydrase and human serum albumin using this strategy. The strategy is well suited to the identification of chelated small molecules that could target multipocketed enzymes such as kinases, phosphatases and proteases.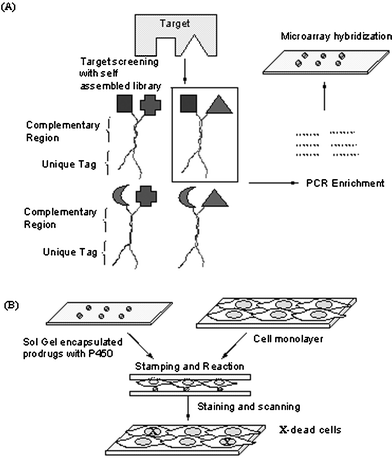 | ||
| Fig. 3 Novel strategies in applying SMM. (A) Self-assembling chemical libraries.82 (B) Metabolizing enzyme toxicity assay on microarrays.85 | ||
The most time-consuming step of any array-based application is in the preparation and synthesis of diverse sets of samples for screening. Langer and co-workers have been able to address this by simply printing combinations of 25 different acrylate, diacrylate, dimethylacrylate and triacrylate monomers on pHEMA-coated slides that were polymerized, in situ, to afford 576 different acrylate polymers on the array.83 Long-wave UV irradiation initiated polymerization and the entire process was carried out in an oxygen-free atmosphere. The concatenated small molecules were screened for differential ability in supporting embryonic stem cell attachment and proliferation.
Other exciting new developments have also involved the rapid screening of small molecules against mammalian cells.83, 84 Small chemicals were impregnated in discs of biodegradable polymers and assembled in a microarray, upon which cells were seeded.84 As the polymer degraded, the compounds were released into the cells localized above the spots in a controlled manner. The strategy was successfully demonstrated through the administration of toxic compounds as well as in the application of a 70-member library for synthetic lethal screening on the array. Clark et al. have also developed a cell-based assay to evaluate the toxicity of prodrugs when reacted with immobilized P450 on arrays.85 The P450 and small molecule drugs were encapsulated in sol–gels following application of cell monolayers, which were later assessed for lethal effects through appropriate staining and conventional microarray scanning (Fig. 3B). These promising strategies thus offer novel methods for performing high-throughput cell based assays for analyzing small molecule libraries for potential therapeutic applications.
Conclusions
Within just a short five years since inception, the many advances made in protein and small molecule microarrays have brought these platforms to the forefront of high-throughput research. Recent work has paved the way towards meaningful new areas in which these technologies may be further exploited. As the technology increases in popularity, remarkable insights will be gained into the pathways and biological processes mediated by proteins and small molecules. This knowledge may then be channeled to the development of improved targets for medicine. Further developments should see microarrays taking on applications in bedside diagnosis as well as in defining biologically useful hits more effectively and in vivo approaches. Notwithstanding, the implications of protein and small molecule microarrays have already been many and important, and are likely to be even more noteworthy in the future.Acknowledgements
Funding support described by the authors' lab in this manuscript was provided by National University of Singapore (NUS) and the Agency for Science, Technology and Research (A*STAR) of Singapore.References
- M. Schena, D. Shalon, R. W. Davis and P. O. Brown, Science, 1995, 270, 467 CrossRef CAS.
- G. Y. J. Chen, M. Uttamchandani, R. Y. P. Lue, M. Lesaicherre and S. Q. Yao, Curr. Top. Med. Chem., 2003, 3, 705 Search PubMed; J. LaBaer and N. Ramachandran, Curr. Opin. Chem. Biol., 2005, 9, 14 CrossRef CAS; K. Y. Tomizaki, K. Usui and H. Mihara, ChemBioChem, 2005, 6, 782 CrossRef CAS; D. Wilson and S. Nock, Angew. Chem., Int. Ed., 2003, 42, 494 CrossRef CAS; D. S. Yeo, R. C. Panicker, L. P. Tan and S. Q. Yao, Comb. Chem. High Throughput Screening, 2004, 7, 213 CAS.
- G. MacBeath, A. N. Koehler and S. L. Schreiber, J. Am. Chem. Soc., 1999, 121, 7967 CrossRef CAS.
- G. MacBeath and S. L. Schreiber, Science, 2000, 289, 1760 CAS.
- Y. Fang, A. G. Frutos and J. Lahiri, J. Am. Chem. Soc., 2002, 124, 2394 CrossRef CAS.
- I. Shin, J. W. Cho and D. W. Boo, Comb. Chem. High Throughput Screening, 2004, 7, 565 CAS; K. S. Ko, F. A. Jaipuri and N. L. Pohl, J. Am. Chem. Soc., 2005, 127, 13162 CrossRef CAS.
- D. H. Min and M. Mrksich, Curr. Opin. Chem. Biol., 2004, 8, 554 CrossRef CAS.
- J. Kononen, L. Bubendorf, A. Kallioniemi, M. Barlund, P. Schraml, S. Leighton, J. Torhost, M. J. Mihatsch, G. Sauter and O. P. Kallioniemi, Nat. Med., 1998, 4, 844 CrossRef CAS.
- J. Ziauddin and D. M. Sabatini, Nature, 2001, 411, 107 CrossRef.
- A. Dove, Nat. Methods, 2005, 2, 709 CrossRef CAS.
- M. Uttamchandani, D. P. Walsh, S. Q. Yao and Y. T. Chang, Curr. Opin. Chem. Biol., 2005, 9, 4 CrossRef CAS.
- S. Bodovitz, T. Joos and J. Bachmann, Drug Discovery Today, 2005, 10, 283 CrossRef CAS.
- H. Zhu, M. Bilgin, R. Bangham, D. Hall, A. Casamayor, P. Bertone, N. Lan, R. Jansen, S. Bidlingmaier, T. Houfek, T. Mitchell, P. Miller, R. A. Dean, M. Gerstein and M. Snyder, Science, 2001, 293, 2101 CrossRef CAS.
- M. G. Smith, G. Jona, J. Ptacek, G. Devgan, H. Zhu, X. Zhu and M. Snyder, Mech. Ageing Dev., 2005, 126, 171 CrossRef CAS.
- J. S. Merkel, G. A. Michaud, M. Salcius, B. Schweitzer and P. F. Predki, Curr. Opin. Biotechnol., 2005, 16, 447 CrossRef CAS.
- M. L. Lesaicherre, Y. P. R. Lue, G. Y. J. Chen, Q. Zhu and S. Q. Yao, J. Am. Chem. Soc., 2002, 124, 8768 CrossRef CAS.
- R. Y. P. Lue, G. Y. J. Chen, Y. Hu, Q. Zhu and S. Q. Yao, J. Am. Chem. Soc., 2004, 126, 1055 CrossRef CAS.
- A. Girish, H. Sun, D. S. Y. Yeo, G. Y. J. Chen, T.-K. Chua and S. Q. Yao, Bioorg. Med. Chem. Lett., 2005, 15, 2447 CrossRef CAS.
- J. A. Camarero, Y. Kwon and M. A. Coleman, J. Am. Chem. Soc., 2004, 126, 14730 CrossRef CAS.
- K. Zhang, M. R. Diehl and D. A. Tirell, J. Am. Chem. Soc., 2005, 127, 10136 CrossRef CAS.
- Y. Hong, B. L. Webb, H. Sui, E. J. Mozdy, Y. Fang, Q. Wu, L. Liu, J. Beck, A. M. Ferrie, S. Raghavan, J. Mauro, A. Carre, D. Mueller, F. Lai and J. Lahiri, J. Am. Chem. Soc., 2005, 127, 15350 CrossRef.
- S. Weng, K. Gu, P. W. Hammond, P. Lohse, C. Rise, R. W. Wagner, M. C. Wright and R. G. Kuimelis, Proteomics, 2002, 2, 48 CrossRef CAS.
- Y. S. Choi, S. P. Pack and Y. J. Yoo, Biochem. Biophys. Res. Commun., 2005, 329, 1315 CrossRef CAS.
- N. Ramachandran, E. Hainsworth, B. Bhullar, S. Eisenstein, B. Rosen, A. Y. Lau, J. C. Walter and J. LaBaer, Science, 2004, 305, 86 CrossRef CAS.
- G. A. Michaud, M. Salcius, F. Zhou, R. Bangham, J. Bonin, H. Guo, M. Snyder, P. F. Predki and B. I. Schweitzer, Nat. Biotechnol., 21, 1509 Search PubMed.
- J. Huang, H. Zhu, S. J. Haggarty, D. R. Spring, H. Hwang, F. Jin, M. Snyder and S. L. Schreiber, Proc. Natl. Acad. Sci. USA, 2004, 101, 16594 CrossRef CAS.
- D. A. Hall, H. Zhu, T. Royce, M. Gerstein and M. Snyder, Science, 2004, 306, 482 CrossRef CAS.
- J. M. Boutell, D. J. Hart, B. L. J. Godber, R. Z. Kozlowski and J. M. Blackburn, Proteomics, 2004, 4, 1950 CrossRef CAS.
- J. R. Newman and A. E. Keating, Science, 2003, 300, 2097 CrossRef CAS.
- M. Letarte, D. Voulgaraki, D. Hatherly, M. Foster-Cuevas, N. J. Sounders and A. N. Barclay, BMC Biochem., 2005, 6, 2 Search PubMed.
- O. Poetz, R. Ostendorp, B. Brocks, J. M. Schwenk, D. Stoll, T. O. Joos and M. F. Templin, Proteomics, 2005, 5, 2402 CrossRef CAS.
- M. Y. Liu, S. Cai, A. Espejo, M. T. Bedford and C. L. Walker, Cancer Res., 2002, 62, 6475 CAS.
- S. H. Kim, A. Tamrazi, K. E. Carlson, J. R. Daniels, I. Y. Lee and J. A. Katzenellenbogen, J. Am. Chem. Soc., 2004, 126, 4754 CrossRef CAS.
- G. Y. J. Chen, M. Uttamchandani, Q. Zhu, G. Wang and S. Q. Yao, ChemBioChem, 2003, 4, 336 CrossRef CAS.
- R. Srinivasen, H. Xuan, S. L. Ng and S. Q. Yao, ChemBioChem, 2006 DOI:10.1002/cbic200500340.
- J. Eppinger, D. P. Funeriu, M. Miyake, L. Denizot and J. Miyake, Angew. Chem., Int. Ed., 2004, 43, 3806 CrossRef CAS.
- D. P. Funeriu, J. Eppinger, L. Denizot, M. Miyake and J. Miyake, Nat. Biotechnol., 2005, 23, 622 CrossRef CAS.
- S. A. Sieber, T. S. Mondala, S. R. Head and B. F. Cravatt, J. Am. Chem. Soc., 2004, 126, 15640 CrossRef CAS.
- G. Y. Jung and G. Stephanopoulos, Science, 2004, 304, 428 CrossRef.
- J. Gu, C. M. Yam, S. Li and C. Cai, J. Am. Chem. Soc., 2004, 126, 8098 CrossRef CAS.
- J.-M. Nam, S. W. Han, K. B. Lee, X. Liu, M. A. Ratner and C. A. Mirkin, Angew. Chem., Int. Ed., 2004, 43, 1246 CrossRef CAS.
- V. Kanda, J. K. Kariuki, D. J. Harrison and M. T. McDermott, Anal. Chem., 2004, 76, 7257–62 CrossRef CAS.
- K. Usui-Aoki, K. Shimada, M. Nagano, M. Kawai and K. Hisashi, Proteomics, 2005, 5, 2396 CrossRef CAS.
- J. S. Yuk, J.-W. Jung, S.-H. Jung, J.-A. Han, Y.-M. Kim and K.-S. Ha, Biosens. Bioelectron., 2005, 20, 2189 CrossRef CAS; J. S. Yuk, S. H. Jung, J. W. Jung, D. G. Hong, J. A. Han, Y. M. Kim and K. S. Ha, Proteomics, 2004, 4, 3468 CrossRef CAS.
- W.-S. Yeo, D.-H. Min, R. W. Hsieh, G. L. Greene and M. Mrksich, Angew. Chem., Int. Ed., 2005, 44, 2.
- Z. Ouyang, Z. Takats, T. A. Blake, B. Gologan, A. J. Guymon, J. M. Wiseman, J. C. Oliver, V. J. Davisson and R. G. Cooks, Science, 2003, 301, 1351 CrossRef CAS.
- T. A. Blank, Z. Ouyang, J. M. Wiseman, Z. Takats, A. J. Guymon, S. Kothari and R. G. Cooks, Anal. Chem., 2004, 76, 6293 CrossRef CAS.
- P. A. Clemons, A. N. Koehler, B. K. Wagner, T. G. Sprigings, D. R. Spring, R. W. King, S. L. Schreiber and M. A. Foley, Chem. Biol., 2001, 8, 1183 CrossRef CAS.
- D. Barnes-Seeman, S. B. Park, A. N. Koehler and S. L. Schreiber, Angew. Chem., Int. Ed., 2003, 42, 2376 CrossRef CAS.
- D. Scharn, H. Wenschuh, U. Reineke, J. Schneider-Mergener and L. Germeroth, J. Comb. Chem., 2000, 2, 361 CrossRef CAS.
- R. Frank, Tetrahedron, 1992, 48, 9217 CrossRef CAS.
- Q. Lin, J. C. O'Neill and H. E. Blackwell, Org. Lett., 2005, 7, 4455 CrossRef.
- H. E. Blackwell, L. Perez, R. A. Stavenger, J. A. Tallarico, E. C. Eatough, M. A. Foley and S. L. Schreiber, Chem. Biol., 2001, 8, 1167 CrossRef CAS; S. M. Sternson, J. B. Louca, J. C. Wong and S. L. Schreiber, J. Am. Chem. Soc., 2001, 123, 1740 CrossRef CAS.
- F. G. Kuruvilla, A. F. Shamji, S. M. Sternson, P. J. Hergenrother and S. L. Schreiber, Nature, 2002, 416, 653 CrossRef CAS; O. Kwon, S. B. Park and S. L. Schreiber, J. Am. Chem. Soc., 2002, 124, 13402 CrossRef CAS; D. R. Spring, Org. Biomol. Chem., 2003, 1, 3867 RSC.
- A. N. Koehler, A. F. Shamji and S. L. Schreiber, J. Am. Chem. Soc., 2003, 125, 8420 CrossRef CAS.
- D. R. Spring, S. Krishnan, H. E. Blackwell and S. L. Schreiber, J. Am. Chem. Soc., 2002, 124, 1354 CrossRef CAS; R. A. Stavenger and S. L. Schreiber, Angew. Chem., Int. Ed., 2001, 40, 3417 CrossRef CAS.
- M. Uttamchandani, D. P. Walsh, S. M. Khersonsky, X. Huang, S. Q. Yao and Y. T. Chang, J. Comb. Chem., 2004, 6, 862 CrossRef CAS; H. E. Park, S. Y. Lee, H. Y. Ahn, J. C. Shin, Y. T. Chang and Y. A. Joe, J. Appl. Pharmacol., 2003, 11, 85 Search PubMed.
- H. S. Moon, E. M. Jacobson, S. M. Khersonsky, M. R. Luzung, D. P. Walsh, W. N. Xiong, J. W. Lee, P. B. Parikh, J. C. Lam, T. W. Kang, G. R. Rosania, A. F. Schier and Y. T. Chang, J. Am. Chem. Soc., 2002, 124, 11608 CrossRef CAS; J. T. Bork, J. W. Lee and Y. T. Chang, Tetrahedron Lett., 2003, 44, 6141 CrossRef CAS; J. T. Bork, J. W. Lee, S. M. Khersonsky, H. S. Moon and Y. T. Chang, Org. Lett., 2003, 5, 117 CrossRef CAS.
- N. Winnsinger, S. Ficarro, P. G. Schultz and J. L. Harris, Proc. Natl. Acad. Sci. USA, 2002, 99, 11139 CrossRef CAS; N. Winnsinger, J. L. Harris, B. J. Backes and P. G. Schultz, Angew. Chem., Int. Ed., 2001, 40, 3152 CrossRef CAS.
- J. S. Kong, S. Venkatraman, K. Furness, S. Nimkar, T. A. Shepherd, Q. M. Wang, J. Aube and R. P. Hanzlik, J. Med. Chem., 1998, 41, 2579 CrossRef CAS; T. J. Caulfield, S. Patel, J. M. Salvino, L. Liester and R. Labaudiniere, J. Comb. Chem., 2000, 2, 600 CrossRef CAS; C. Walsh, Tetrahedron, 1982, 38, 871 CrossRef CAS.
- M. Kohn, R. Wacker, C. Peters, H. Schroder, L. Soulere, R. Breinbauer, C. M. Niemeyer and H. Waldmann, Angew. Chem., Int. Ed., 2003, 42, 5830 CrossRef.
- L. J. Hackler, G. Dorman, Z. Kele, L. Urge, F. Durvas and L. G. Puskas, Mol. Diversity, 2003, 7, 25 Search PubMed.
- W. S. Dillmore, M. N. Yousaf and M. Mrksich, Langmuir, 2004, 20, 7223 CrossRef CAS.
- N. Kanoh, S. Kumashiro, S. Simizu, Y. Kondoh, S. Hatakeyama, H. Tashiro and H. Osada, Angew. Chem., Int. Ed., 2003, 42, 5584 CrossRef CAS.
- A. Shaginian, M. Patel, M.-H. Li, S. T. Flickinger, C. Kim, F. Cerrina and P. J. Belshaw, J. Am. Chem. Soc., 2004, 126, 16704 CrossRef CAS.
- S. Li, D. Bowerman, N. Marthandan, S. Klyza, K. J. Luebke, H. R. Garner and T. J. Kodadek, J. Am. Chem. Soc., 2004, 126, 4088 CrossRef CAS.
- S. P. Fodor, J. L. Read, M. C. Pirrung, L. Stryer, A. T. Lu and D. Solas, Science, 1991, 251, 767 CrossRef CAS.
- J. C. Wong, S. M. Sternson, J. B. Louca, R. Hong and S. L. Schreiber, Chem. Biol., 2004, 11, 1279 CrossRef CAS.
- M. Takahashi, K. Nokihara and H. Mihara, Chem. Biol., 2003, 10, 53 CrossRef CAS.
- R. Muralidhar and T. Kodadek, Proc. Natl. Acad. Sci. USA, 2005, 102, 12672 CrossRef.
- Q. Zhu, M. Uttamchandani, D. B. Li, M. L. Lesaicherre and S. Q. Yao, Org. Lett., 2003, 5, 1257 CrossRef CAS.
- S. M. Salisbury, D. Maly and J. Ellman, J. Am. Chem. Soc., 2002, 124, 14868 CrossRef CAS.
- D. N. Gosalia and S. L. Diamond, Proc. Natl. Acad. Sci. USA, 2003, 100, 8721 CrossRef CAS.
- D. N. Gosalia, C. M. Salisbury, D. J. Maly, J. A. Ellman and S. L. Diamond, Proteomics, 2005, 5, 1292 CrossRef CAS.
- P. Babiak and J.-L. Reymond, Anal. Chem., 2005, 77, 373 CrossRef CAS.
- M. Uttamchandani, X. Huang, G. Y. J. Chen and S. Q. Yao, Bioorg. Med. Chem. Lett., 2005, 15, 2135 CrossRef CAS.
- P. Angenendt, H. Lehrach, J. Kreutzberger and J. Gloker, Proteomics, 2005, 5, 420 CrossRef CAS.
- N. Winssinger, R. Damoiseaux, D. C. Tully, B. H. Geierstanger, K. Burdick and J. L. Harris, Chem. Biol., 2004, 11, 1351 CrossRef CAS.
- J. Harris, D. E. Mason, J. Li, K. W. Burdick, B. J. Backes, T. Chen, A. Shipway, G. Van Heeke, L. Gough, A. Ghaemmaghami, F. Shakib, F. Debaene and N. Winnsinger, Chem. Biol., 2004, 11, 1361 CrossRef CAS.
- Y. Hu, G. Y. J. Chen and S. Q. Yao, Angew. Chem., Int. Ed., 2005, 44, 1048 CrossRef CAS.
- J. Yin, F. Liu, M. Schinke, C. Daly and C. T. Walsh, J. Am. Chem. Soc., 2004, 126, 13570 CrossRef CAS.
- S. Melkko, J. Scheuermann, C. E. Dumelin and D. Neri, Nat. Biotechnol., 2004, 22, 568 CrossRef CAS.
- D. G. Anderson, S. Levenberg and R. Langer, Nat. Biotechnol., 2004, 22, 863 CrossRef CAS.
- S. N. Bailey, D. M. Sabatini and B. R. Stockwell, Proc. Natl. Acad. Sci. USA, 2004, 101, 14144 CrossRef.
- M.-Y. Lee, C. B. Park, J. S. Dordick and D. S. Clark, Proc. Natl. Acad. Sci. USA, 2005, 102, 983 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2006 |