Optimizing the affinity and specificity of proteins with molecular display
A. M.
Levin
a and
G. A.
Weiss
*ab
aDepartments of Chemistry, University of California, Irvine, CA 92697, USA. E-mail: gweiss@uci.edu
bMolecular Biology and Biochemistry, University of California, Irvine, CA 92697, USA
First published on 8th November 2005
Abstract
Affinity maturation of receptor–ligand interactions represents an important area of academic and pharmaceutical research. Improving affinity and specificity of proteins can tailor potency for both in vivo and in vitro applications. A number of different display platforms including phage display, bacterial and yeast display, ribosome display, and mRNA display can optimize protein affinity and specificity. Here, we will review the advantages and disadvantages of these molecular display methods with a focus on their suitability for protein affinity maturation.
 Aron Levin | Aron Levin was born in 1979 in Memphis, Tennessee. He received his BA in Biochemistry from the University of Kansas in 2001 and his PhD in Chemistry from the University of California, Irvine in 2005. At U.C. Irvine, he worked with Prof. Gregory Weiss to examine receptor–ligand interactions using phage display. He is currently a postdoc at Stanford University. |
 Gregory Weiss | Gregory Weiss is an Assistant Professor at the University of California, Irvine in the Departments of Chemistry, Molecular Biology and Biochemistry. Weiss received his PhD in Chemistry and Chemical Biology from Harvard University in 1997. As a postdoctoral fellow at Genentech, he was awarded National Research Service Award fellowship from the NIH. The recipient of a Young Investigator Award from the Arnold and Mabel Beckman Foundation, he joined the faculty at U.C. Irvine in 2000. His laboratory applies combinatorial techniques for engineering and dissecting protein function. |
Introduction
The goal of affinity maturation is to strengthen protein binding to a particular ligand. Ultimately, affinity maturation can produce high affinity proteins with specific functions for use as therapeutic or research reagents.1–10 For example, biomedical applications ranging from disease diagnostics to gene therapy often require improved affinity, specificity, or both for therapeutic uses. Molecular display techniques provide powerful tools for tailoring protein affinity and specificity.The invention of phage display demonstrated display of a polypeptide on the surface of filamentous phage.11 This coupling of genotype (the encapsulated DNA encoding the displayed protein) and phenotype (the displayed protein) allowed affinity enrichment selections from large libraries with millions to trillions of polypeptide variants.12 Bacterial display offered the first cell surface display system, in this case with proteins displayed on the surface of E. coli.13–16 The advent of cell-surface display introduced the advantages of quantitative screening by fluorescence-activated cell sorting (FACS). Yeast display offers the advantages of cell-surface display, but using an eukaryotic organism, which is well suited for the display of extracellular mammalian proteins.17
Cell-free protein engineering techniques apply an in vitro transcription/translation (IVTT) reaction for the display of proteins on ribosomes18 or mRNA.19 DNA display, another IVTT method, has not been reported in affinity maturation of a protein–ligand interaction, and is not reviewed here.20,21 The primary advantage of the cell-free techniques is the ability to create potentially huge libraries with up to 1014 different polypeptide variants, since the library size is not limited by cellular transformation efficiencies necessary for construction of cell or viral surface display libraries.10 All molecular display technologies can provide high-quality protein libraries, which can be subjected to in vitro selection methods to yield affinity-matured proteins. Depending on the protein of interest and its starting affinity, some molecular display methods can be more useful than others. For example, one might begin a protein discovery program by first using the large library sizes of phage display or mRNA display for lead discovery, then applying a cell-surface display method for final affinity maturation.
The molecular display methods described here are subject to some caveats. For example, one important consideration is “background” binding between the display system and the target protein. Such deleterious binding can prevent isolation of the highest affinity ligands by corrupting selections with overwhelming numbers of false positives. To pre-empt this problem, the first step of any molecular display project should examine background binding to guide choice of the appropriate molecular display scaffold. Next, development of selection conditions requires some insight about the stability of the target protein. Finally, selection and screening conditions should test for specificity. Conditions for screening individual variants obtained from the selection should minimally examine binding to the blocking agent used to compete for non-specific interactions. When optimizing specificity, screening conditions can also compare target with off-target binding.
To obtain high affinity binders from molecular display, library selections can feature either thermodynamic or kinetic control (Fig. 1). For thermodynamic control selections (also called “affinity selections”), a biotinylated target ligand at a concentration below the desired Kd is mixed with the molecular display library. During equilibration, the highest affinity receptor can theoretically out-compete other receptor variants for binding to the ligand. Members of the library binding to the biotinylated ligand are then captured with streptavidin attached to a solid support, such as magnetic beads or a microtiter plate. For kinetic control (also called “off-rate selections”), the library initially binds to a biotinylated ligand. Then, unbiotinylated, competitor ligand is added for a set amount of time before streptavidin capture. Longer competition times can select variants with slower off-rates.22
 | ||
| Fig. 1 Two methods for selecting affinity-matured receptors displayed on the surface of phage. A) Thermodynamic control or “affinity-based” selections allow phage-displayed receptors to compete for a limited amount of biotinylated ligand. B) In kinetic control or “off-rate-based” selections, phage-displayed receptors are first saturated with biotinylated ligand, and then mixed with competing non-biotinylated ligand. The two approaches are applicable to all molecular display methods. | ||
Since, to our knowledge, affinity maturation by all relevant molecular display techniques has not been reviewed recently, many older articles in addition to newer ones are included here. This review is organized with commonly used virus- and cell-based molecular display methods of phage, bacterial and yeast display first, followed by the cell-free molecular display methods of ribosome and mRNA display. By examining selected examples of successful affinity maturation projects from each system, effective strategies for successful affinity maturation can be described.
Phage display
Phage display, the oldest and most commonly applied molecular display technique, has been used to display and affinity mature a wide range of proteins. In one early example of affinity maturation, human growth hormone (hGH) variants with high affinity to hGH binding protein (hGHbp) were developed as potential treatments for acromegaly.23,24 Lowman and Wells prepared phage particles displaying hGH variants as fusions to the phage minor coat protein P3. Phage with variants of hGH displayed were subjected to several rounds of in vitro selection to isolate variants binding to solid phase-immobilized hGHbp. The selection procedure yielded affinity-enhanced hGH variants from five different libraries of mutant hGH proteins. Combining the substitutions provided a final hGH variant that bound hGHbp with 380-fold stronger affinity (Kd ≈ 0.9 pM) than wild-type hGH.25,26 The selection strategy largely decreased the off-rate with little change in the hGH on-rate, as measured by surface plasmon resonance (SPR), demonstrating the power of monovalent phage display on P3 for improving off-rates. Additionally, the strategy of combining mutations from separate libraries to construct high-affinity protein variants has proven very useful for other projects.27–29 The success of this approach illustrates two fundamental principles of protein engineering. First, the energetic contributions from individual amino acid substitutions are largely additive. This rule holds true in the absence of unusual conformational rearrangements or charge–charge pairs at the interface.30 Thus, multiple substitutions can often be combined to yield a super mutant with the net binding energy of all substitutions. Second, there is no requirement for particular substitutions to be introduced early, midway, or late in the process of affinity maturation. These principles provide a firm foundation for applying molecular display to affinity maturation.Another early example of affinity maturation with phage display applied bovine pancreatic trypsin inhibitor (BPTI) for inhibition of the serine protease human neutrophil elastase.31 An efficacious inhibitor of HNE was proposed as treatment for a hereditary disorder resulting in decreased expression of an HNE inhibitor. Using multivalent display of BPTI on phage, Roberts, Ladner, and co-workers produced a small library of only 1000 different variants of BPTI. The results, however, were phenomenal as an affinity enhancement over wild-type binding to HNE of over three million-fold was obtained through a mutant with a 1 pM dissociation constant. This experiment used a modified M13 phage genome with up to five copies per phage of BPTI displayed as fusions to P3. This example demonstrates success with a multivalent phage display system. More importantly, the results demonstrate that selections for enhanced binding affinity can also identify improved enzyme inhibitors.32
One very successful approach to affinity maturation switches between multivalent and monovalent display using a phagemid system. Phagemid vectors for phage display feature a single open reading frame encoding the displayed protein fused to an anchoring coat protein. In this common format, the fusion protein is packaged into phage particles upon co-infection with a helper phage, which provides all components necessary for phage assembly and infectivity. The phagemid system provides at least two important advances over the phage genome display format. First, the phagemid system is exceptionally stable, and easy to propagate. Second, better control over display levels is possible through choice of the anchoring coat protein. For example, displaying a library of peptides or small proteins on the M13 major coat protein, P8, can usually achieve high copy, multivalent display. A naïve library of ligands displayed in a multivalent format can leverage avidity or “velcro” effects to identify an initial, often weak or medium affinity ligand. Upon identification of the initial ligand, the protein can next be displayed on the M13 phage minor coat protein, P3, for typically monovalent display and further display-based optimization. Monovalent display can allow selections based on affinity, thus avoiding avidity effects.33–35
Methods for regulating specific genes in humans, viruses, and plants could improve prospects for gene therapy. Zinc finger domains have been explored as artificial transcription factors for controlling gene transcription.36–38 Isalan, Choo, and co-workers described an elegant affinity maturation of phage-displayed zinc fingers for transcriptional control. As in the previously described example of hGH affinity maturation, mutations from multiple zinc finger libraries were combined to yield the re-engineered transcription factor. The zinc finger affinity maturation featured two complementary three-finger libraries displayed on P3 with one-and-a-half zinc fingers from each library randomized. Thermodynamic control selections with limiting concentrations of target DNA were followed by combination of the selected mutations. The resultant zinc fingers were tested for binding to a nine base-pair DNA sequence in the HIV-1 promoter, a demanding target. The approach successfully generated DNA-binding proteins with low nanomolar Kd and high DNA sequence specificity.28
In one elegant example, affinity maturation was used to modify not the displayed protein, but the molecular display scaffold itself. To develop an anchoring phage coat protein for C-terminal phage display, libraries of linkers to a poly-His tag were selected for optimization of display.39,40 The resultant linker for the P8 C-terminus allows display of polypeptides with free C-termini, in contrast to the standard N-terminal display used in essentially all other phage display experiments. Next, Laura, Sidhu, and co-workers displayed a library of random heptapeptides linked to the C-terminus major coat protein P8 by an affinity-matured 13-residue linker. With a free C-terminus, the displayed peptides were selected for binding to the Erbin PDZ domain. PDZ domains mediate protein–protein interactions in the cell by binding the free C-termini of various proteins. Having identified Erbin PDZ ligands by C-terminal phage display, the next series of selections focused on affinity maturation to obtain ligands with high affinity and specificity. In this series of experiments, affinity maturation played two roles—optimizing the molecular display system and, more conventionally, for tailoring binding affinity.
Examples of affinity maturation with N-terminal phage display can apply focused mutagenesis methods to improve the efficiency of the affinity maturation process. Homolog shotgun scanning has been used to simultaneously affinity mature proteins and provide structure activity relationship data by making subtle homologous amino acid substitutions.41 In the first example of homolog shotgun scanning applied to affinity maturation, a short (20 residue) peptide was examined for binding to elongation factor-Tu (EF-Tu).42 The EF-Tu ligand was first selected from a very large naïve phage display library with >1011 different peptides. Next, a smaller, more focused library, exploring substitutions of wild-type or a close homolog in every position of the EF-Tu ligand, was synthesized to dissect and optimize sidechain requirements for binding to EF-Tu. In this two-step approach to discovering peptide ligands, the first library provided a broad snapshot of chemical space, encoded by a series of NNS codons (which includes all 20 amino acids and the TAG stop codon)43 to uncover the initial lead peptide (Fig. 2). The second library, using homolog shotgun scanning, optimized the sidechain functionalities of the lead peptide. This approach mimics medicinal chemistry, whereby a lead compound is refined with small changes to optimize the ligand fit for its receptor. Further demonstrating the value of homolog shotgun scanning, the technique has been used to affinity mature engrailed homeodomain for binding to a specific DNA sequence.29
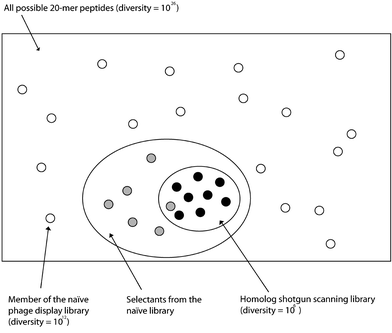 | ||
| Fig. 2 A two-step method for discovering high-affinity binding proteins using molecular display. First, displayed peptides with affinity for the target are selected from a large naïve library. Then, more-focused, non-stochastic libraries of homologous substitutions are created for affinity maturation of the receptor–ligand interaction. In addition to the homolog shotgun scanning method illustrated here, look-through mutagenesis and other techniques can explore optimization by small changes to an initial lead isolated from the naïve library. | ||
Two additional examples of peptide affinity maturation also illustrate the utility of phage display for discovering lead peptides, and then applying a second phage-displayed library to enhance affinity. Landon, Deutscher, and co-workers required small, high-affinity anti-Thomsen-Friedenreich (TF) antigen molecules for use as tumor cell targeting agents.45 A large P3-displayed library initially identified a consensus ligand to TF (HGRFILP-WWYAF-SPS). To improve the affinity and solubility properties of the lead peptide, the researchers then created a second library with the middle region of the peptide fixed and randomized flanking residues. This process yielded a final peptide that was more soluble, exhibited a fivefold affinity increase, and had possible clinical applications as anti-adhesive and anti-metastatic agents.45–47 Fleming, Sexton, and co-workers used similar methodology to identify high-affinity, high-selectivity peptides that bound B lymphocyte stimulator (BLyS).48 An initial library produced low µM Kd ligands to BLyS, but a second selectant-focused library produced short peptides with Kd values as low as 26 nM. An affinity-matured peptide could disrupt a high-affinity cytokine–receptor interaction. In addition to the potential use for treating autoimmune diseases, this example illustrates the effectiveness of small peptides for disrupting protein–protein interactions.
The T-cell receptor (TCR), an antigen recognition molecule with a naturally diverse repertoire, recognizes peptide–human leukocyte antigen complexes (pHLA).49 Artificial TCRs could potentially find applications in anti-cancer therapeutics or as reagents to block autoimmune T-cell activation. Li, Boulter, and co-workers developed a robust phage display method for displaying and evolving TCRs on the surface of phage. Their method displays a stable TCR on P3 as a disulfide-linked heterodimer, and generates libraries at the CDRs (complementary determining region) with the NNK codon, which encodes 12 amino acids.50 High-affinity receptors were selected by decreasing the concentration of the target pHLA during each round of selection, an effective thermodynamic control selection strategy. TCRs with affinities as low as 26 pM were discovered, an increase of ≈106-fold over naturally occurring TCRs.22
Like TCRs, antibodies provide a stable scaffold amenable to amino acid substitutions for the development of high-affinity receptors to a specific ligand. The subject of numerous affinity maturation studies, monoclonal antibodies (mAbs) can play a significant role in the treatment and imaging of cancer.51–53 Single-chain Fv (scFv) and Fab fragments can have the same affinity and specificity for antigens as full-size mAbs, and are preferred for molecular display due to their much smaller size.1,54–56 Also, scFvs and Fabs can be used directly as therapeutics with potentially better tumor-penetrating ability and faster clearance than full-size mAbs. In addition, scFvs and Fabs can often be converted to mAbs if necessary.57 The tremendous interest in developing specific antibodies for therapeutic applications and the highly modifiable nature of the antibody scaffold has inspired numerous examples of affinity maturation of specific scFvs with typically P3-displayed Ab fragments.
For example, using a thermodynamic control selection, Schier, Marks, and co-workers selected a scFv with a 1230-fold increase in affinity from the wild-type scFv for binding to tumor antigen c-erbB-2. In this study, the combination of mutations from four VH CDR3 libraries yielded a final scFv with picomolar affinity. The resultant high affinity scFv receptors had a predicted half-life (t1/2) on the tumor cell surface of 24 hours, compared to the wild-type t1/2 of only 13 minutes. This result illustrates the power of thermodynamic control selections to improve a kinetic parameter, in this case tumor retention.57,58 In the first example of homolog shotgun scanning, Vajdos, Sidhu and co-workers mapped the functional epitope of a different anti-c-erbB-2 antibody, revealing a small set of amino acids involved in precise contact with the antigen.41 Further efforts to affinity mature scFvs using phage display have targeted VEGF,59,60 CD22,61 anthrax toxin,62 and scorpion toxin Cn2.63,64 Display of Fabs on phage has also resulted in minimally immunogenic high-affinity antibody variants65 and anti-HIV-1 gp120 variants.66
Many outstanding examples of enhancing the specificity of antibody fragments binding to small molecules have been demonstrated using phage display.67–71 For example, Saviranta, Lövgren, and co-workers employed random mutagenesis and a simple selection strategy to enhance the steroid specificity of a Fab. Motivated to improve antibody specificity for clinical applications, the researchers aimed to decrease the testosterone-binding ability of an estradiol-binding Fab. A large excess of free testosterone was added as a competitor ligand, during selections of a phage-displayed (P3-fused) Fab library binding to estradiol. This strategy resulted in 20-fold improvements to Fab specificity.71 In a similar example, Dubreuil, Ducancel, and co-workers applied a strategy of saturation mutagenesis and error-prone PCR together with competition selections to enhance discrimination between 5α- and 5β-dihydroprogesterone. From these experiments, two scFvs retained picomoler binding affinity, but emerged with 23-fold and 15-fold improvements to binding specificity.67 The results illustrate the tremendous accuracy of binding possible through clever selection conditions, even for complex scaffolds such as antibodies.
Bacterial display
The development of a molecular display method on the surface of E. coli provided an effective alternative to phage display.13,14 Bacterial display retains many useful features of phage display, such as the high transformation efficiency, ease of manipulation, and familiarity of handling E. coli. Cell-surface display methodology such as bacterial display and yeast display (described below) present thousands of copies of the displayed protein on the cell surface. The most important advance made possible by cell-based molecular display methods is the use of FACS for quantitative screening and potentially high enrichment ratios of proteins bound to fluorescently labeled ligands under solution equilibrium conditions. FACS eliminates the uncertainty that elution successfully recovers the most tightly bound clones and the potential for nonspecific binding to the solid support used to immobilize the target molecule. In addition, different display levels of receptor variants can be normalized by quantifying display levels with a second fluorescently-labeled probe.15Kd values in agreement with measurements made with non-displayed proteins can also be obtained using flow cytometry.72Though bacterial display was developed in part for production of live bacterial vaccines,73 bacterial display of scFvs has been used successfully for affinity maturation. For example, Daugherty, Georgiou, and co-workers developed a quantitative system using FACS for selecting affinity-matured bacterial-displayed scFv mutants bound to fluorescently-labeled haptens.74,75 After library construction, the first round of selection applied thermodynamic control conditions, and the successive two rounds applied kinetic control conditions. The relative dissociation constants obtained using flow cytometry were verified by SPR, yielding Kd values of eight clones ranging from 0.9 to 2.4 nM. This method of rapidly isolating rare, high-affinity clones was next applied to measure the effect of mutation frequency on the effectiveness of affinity scFv maturation.76 Libraries with various mutation frequencies (from 1.7 mutations for a low rate to 22.5 for a high rate of mutations per scFv gene) were generated by error-prone PCR. Each library produced affinity matured clones, but the libraries with a moderate and high number of mutations yielded the strongest binders. Also, the mutations in the highest affinity binders included substitutions often outside the antigen binding site. Thus, unpredictable second sphere mutations led to the greatest enhancement of binding. The researchers also found that in vitro scanning saturation mutagenesis could be applied to obtain affinity-matured variants.77
The most diverse peptide libraries of any cell-surface display methods have been produced by bacterial display. Due to the high transformation efficiency of bacteria, Bessette, Daugherty, and co-workers could construct a bacterial display library consisting of 5 × 1010 different polypeptides.78 Using this diverse library, high-affinity binders to five unrelated proteins were isolated. The fine affinity discriminating abilities of FACS-based selections allowed determination of lengthy consensus motifs from a series of high-affinity binders to each protein. In conjunction with previously mentioned affinity maturation techniques and library construction methods such as homolog shotgun scanning, the technique could conceivably be used for affinity maturation of proteins.
Yeast display
Yeast display benefits from the advantage of FACS-based selections,79 yet has the additional feature of including eukaryotic protein folding pathways and codon usage very similar to mammalian cells.80 As a result, yeast display is perhaps the most ideally suited system for the surface display of mammalian cell-surface and secreted proteins such as antibodies and T-cell receptors. Since phage and bacterial display both require bacterial hosts, such libraries could potentially contain variants with improper folding or decreased representation due to mismatched human/bacterial codon bias.58In an early demonstration of affinity maturation by yeast display, Kieke, Wittrup, and co-workers isolated scFv mutants with improved affinity for T-cell receptors.81 Mutant scFvs, created with an E. coli mutator strain, were selected using a powerful combination of FACS and thermodynamic control selections. Although the library size was only 3 × 105 different scFvs, several mutants with affinities higher than the starting scFv were discovered. Recommendations for cell-sorting stringency to result in optimal screening of surface-displayed libraries, using a statistics algorithm were developed with yeast display.82 As a test system, two yeast-displayed scFvs were mixed at a ratio of 1000 ∶ 1 with the 2-fold higher affinity scFv at the lower concentration. Using a thermodynamic control selection, an approximately 125-fold enrichment level for the higher affinity mutant was achieved in a single pass, demonstrating that yeast display can recover rare, slightly improved mutants from a large population.83
As previously described, the potential for diagnostic and therapeutic applications using TCRs motivates affinity maturation for specific pHLA complexes. A TCR displayed on yeast with a wild-type Kd of 1.5 µM was affinity-matured 100-fold over wild-type with a library of only 105 mutants in five amino acid positions. When the wild-type and affinity-matured TCRs were tested for binding to a pHLA complex on the surface of T-cells, the mutant demonstrated detectable levels of binding unlike the wild-type TCR.84 In another example, a TCR was affinity matured for binding to the superantigen staphylococcal enterotoxin C3 (SEC3) to uncover the details of binding between TCR and SEC3.85 After a total of two rounds of mutagenesis (in the CDR3 region of the TCR) and six FACS selections, a TCR mutant with a Kd of 7 nM was isolated, representing a ≈1000-fold improvement in binding affinity. The improved variant was an effective inhibitor of T-cell activity mediated by SEC3, unlike the low affinity TCR variants. This approach to TCR antagonists against various superantigens could prove useful for treatment of diseases. More recently, it was demonstrated that the CDR1 and CDR2, in addition to the CDR3,85,86 regions of TCRs could be mutated to generate high-affinity TCR mutants and contribute to antigen specificity.87
The cytokine IL-2 is used for the treatment of metastatic renal cell carcinoma and melanoma. However, IL-2 is rapidly cleared from the body with a t1/2 of around an hour.88 Rao, Wittrup, and co-workers demonstrated yeast-displayed affinity maturation of IL-2 for the receptor IL-2Rα with the goal of engineering an IL-2 variant with improved therapeutic value.89,90 By subjecting the IL-2 gene to error-prone PCR and using a thermodynamic selection against soluble IL-2Rα at concentrations as low as 0.2 nM, an IL-2 mutant (Kd ≈ 1.1 nM) with a 25-fold improvement over the original IL-2 Kd of 28 nM was obtained.90 A pulsed bioassay, which approximated clearance of IL-2 from the body, indicated that the higher affinity mutant demonstrated increased activity for T-cell proliferation.
In one of the largest yeast displayed libraries to date, Fab antibody fragments were displayed on the surface of yeast.31,91,92 To construct the 5 × 109 diversity library, separate VH and VL libraries were created in two different yeast strains, and then combined into one library through highly efficient yeast mating. This technology was also applied to the construction of a large heterodimeric immune Fab library with diversity of 3 × 109 different members. This innovative approach should prove very useful for the field of affinity maturation by accessing larger yeast display libraries.91
Using the 4-4-20 anti-fluorescein antibody model system, a monovalent scFv mutant with a Kd of 48 fM and an off-rate >5 days was found through four rounds of error-prone PCR and kinetic control selections. Error-prone PCR was applied to the entire scFv rather than focusing on the CDR loops. This strategy produced a collection of very high affinity mutants through combinations of small improvements.93,94 A similar method was successfully applied to improve the retention time of a scFv against carcino-embryonic antigen (CEA) in tumor tissue.95 Starting with hMFE (a humanized antibody already known to bind CEA), Graff, Wittrup, and co-workers obtained hMFE variants with a large range of improved off-rates (10 to 1000-fold). The tightest binding hMFE variant had a t1/2 of nearly 4 days, an improvement over the 10 min t1/2 for wild-type hMFE. Unfortunately, the hMFE variants were not stable at 37 °C; therefore, the mutants were next matured for enhanced stability by selecting for improved display levels on yeast. This resulted in the highest affinity-matured hMFE variant retaining 80% binding activity after incubation at 37 °C for 9 days. These techniques could prove valuable to enhancing the affinities and stabilities of current scFvs with short off-rates.
In an example with a new approach to focused mutagenesis, look-through mutagenesis (LTM) was used to affinity mature an anti-TNFα scFv previously optimized to a Kd of 1 nM. LTM, like alanine and homolog shotgun scanning, uses a binomial set of amino acids in targeted positions. In LTM, targeted positions are substituted with either the wild-type residue or one of nine amino acids representing the major sidechain chemistries—small, nucleophilic, hydrophobic, aromatic, amide, or basic. The nine libraries required for complete coverage of this chemical diversity could also map functional group requirements for antibody binding. An initial thermodynamic control selection yielded variants with affinities increased two-fold. Final mutants were selected from a kinetic control selection, and had dissociation constants improved by an impressive 870-fold (Kd ≈ 1.1 pM).96
Intrabodies, a disulfide-free form of antibodies, show potential for controlling intracellular protein function. Using yeast display, Colby, Wittrup, and co-workers generated single domain antibodies to inhibit huntingtin (htt) aggregation by binding to the first 20 amino acids of the Huntington's disease-associated htt. Next, the researchers engineered disulfide-free variants to create an intrabody capable of functioning outside or inside the reducing environment of the cell.97,98 Having replaced the disulfide with hydrophobic residues, the intrabody was next affinity matured to regain the massive affinity loss following disulfide replacement. The resultant disulfide-free intrabody could rescue toxicity in a neuronal model of Huntington's Disease. Affinity-matured intrabodies have potential therapeutic value for the treatment of other diseases caused by protein aggregation.
Ribosome and mRNA display
Both ribosome and mRNA display apply an in vitro transcription/translation (IVTT) system that circumvents growth in and transformation of cells (Fig. 3).18 As a result, IVTT-based methods can potentially create much larger libraries (routinely >1012) than cell-based systems and potentially avoid expression bias.99 Libraries are selected using methods similar to phage display, because the nucleic acid–protein complexes are too small for detection by FACS. Another advantage of the IVTT methods is the potential for affinity maturation through recursive mutagenesis, in which selectants are further mutated after each round of selection. One example of ribosome display applied recursive evolution using standard Taq DNA polymerase (≈1 mutation every 20,000 bases) to amplify the library after each round of selection.100 This recursive mutagenesis and evolution with ribosome display was first applied to the affinity maturation of a scFv binding to a monomeric variant of the transcription factor GCN4; the best mutant had a 65-fold higher affinity than the starting scFv.101 More extensive diversification beyond the capabilities of Taq can be performed by employing methods such as error-prone PCR,102 mutagenic dNTP analogs,103 or DNA shuffling.104 | ||
| Fig. 3 Schematic of ribosome and mRNA display. A) Ribosome display uses IVTT to yield a ribosome–mRNA–protein complex for selection against an immobilized target. In step 1, library-encoding DNA is transcribed with RNA polymerase. The resultant mRNA (step 2) is translated using bacterial cell extracts, and the displayed protein bound to the ribosome–mRNA complex is trapped due to the lack of a stop codon in the transcript. Following selection and elution conditions (steps 3 and 4, respectively), selectants are reverse transcribed (step 5), before the process is repeated. (B) mRNA display creates a covalent protein–mRNA complex by means of a DNA–puromycin linker. After transcription (step 1), the transcript is attached to a DNA–puromycin handle (step 2). During translation, the ribosome stalls at the mRNA–DNA junction (step 3), which allows puromycin to form a covalent bond with the nascent protein. The resultant mRNA–protein hybrid can be used for selection and elution steps (steps 4 and 5), and the cycle repeated, after reverse transcription (step 6). | ||
A ribosome-displayed library of synthetic scFvs yielded many scFv variants with 40-fold higher affinities and dissociation constants as low as 82 pM to bovine insulin.105 In another example, a scFv antibody fragment with a starting affinity of 1.1 nM was improved 30-fold for binding to fluorescein. In the same report, ribosome display and DNA shuffling were used to mature the stability of an antibody under reducing conditions.4 A peptide-binding antibody fragment with a Kd of 5 pM, a 500-fold improvement over its precursor, was generated by ribosome display and then characterized by X-ray crystallography.106 The crystal structure revealed that none of the mutations made direct contact with the peptide ligand, demonstrating again that affinity maturation can occur through subtle backbone changes or through networks of sidechain–sidechain interactions.
Ankyrin repeats (AR), disulfide-free, easily-expressed, versatile binding proteins, are well suited for generating diversity, since repeated structural motifs can be varied while retaining a constant hydrophobic core. Furthermore, non-antibody scaffolds could have superior properties for specific therapeutic uses.107 As an alternative to antibody libraries, Binz, Plückthun, and co-workers evolved ARs for binding to maltose binding protein (MBP) and two kinases.108 Selections were performed on microtiter plates with biotinylated antigen, and ligands to each of the three target proteins were affinity-matured to the low nM range. The results demonstrate the high specificity and applicability of AR proteins as scaffolds for a wide range of applications.107,108
mRNA display, independently invented by two groups, is functionally very similar to ribosome display with a protein linked to its encoding mRNA.19,109 However, in ribosome display, the ternary complex is quite stable, but non-covalent; in mRNA display, the covalent mRNA–protein complex can allow for more stringent selection methods such as elevated temperature. With mRNA display, the mRNA is attached to puromycin, which can form a covalent bond with the protein after translation (Fig. 3). As an IVTT method, mRNA display has the advantage of harnessing easily constructed, often massive libraries.110 In one example, Wilson, Szostak, and co-workers used an mRNA display library of nearly 1013 members to identify high-affinity (with Kd as low as 5 nM), streptavidin-binding linear peptides lacking disulfides.111 Reduced from 88 residues, one 38-residue peptide could be used as an affinity tag for protein purification.
mRNA display has also been used to select for new protein motifs that bind to ATP. With 80-mer completely random peptides lacking structural constraints, a large starting library of 6 × 1012 was necessary to identify new ATP-binding motifs.112 Receptors were obtained after eight rounds of selection, and the library was further mutagenized using error-prone PCR to affinity mature the remaining clones. After a total of 18 rounds of selections, new ATP-binding motifs made up 34% of the library. The strongest binding mutant had a Kd of 100 nM and was not homologous to any known ATP-binding motifs. This attractive method to discover new protein motifs also demonstrates the requirement for affinity maturation of selectants from even the most diverse molecular display libraries reported to date.
Diagnostic protein microarrays generally require high-affinity, high-specificity proteins that can function properly when immobilized on a solid surface. Xu, Lipovsek, and co-workers designed the small-sized 10Fn3 protein as an antibody mimic, and engineered higher affinity and specificity than the starting protein using mRNA display.11310Fn3 contains three solvent-accessible loops, similar to the three CDRs in the VH portion of an antibody. A >1012 diversity library was constructed at the three 10Fn3 loops and selected for binding to TNFα. After ten rounds of enrichment for functional variants, a thermodynamic control selection at elevated temperature was performed to obtain variants with subnanomolar dissociation constants. Comparison of selectant binding to TNFα versus two other cytokines demonstrated that the selected binders exhibited high specificity. A 10Fn3 antibody-mimic also retained high affinity and specificity when spotted on a microarray plate.
Conclusion
A tremendous number of engineered proteins with enhanced affinity and specificity have been created using a variety of different molecular display techniques. Harnessing large libraries and powerful selection methods, new proteins can be discovered and affinity matured for almost any target. A general strategy for the affinity maturation of proteins can be derived from the papers reviewed here. First, moving from thermodynamic control (affinity-based) to kinetic control (off-rate-based) selections, or using only kinetic control selections, generally produced the greatest affinity enhancements. Second, selecting proteins from libraries constructed by focused mutagenesis methods, such as LTM or homolog shotgun scanning, after lead selections from an initial naïve library, can produce affinity-enhanced variants in addition to providing structure–activity data. These efforts will eventually expand applications of proteins used in biomedical applications. Consequently, molecular display will continue to impact proteomics, diagnostics, and therapeutics.References
- P. S. Chowdhury and H. Wu, Methods, 2005, 36, 11 Search PubMed.
- W. J. Dower and L. C. Mattheakis, Curr. Opin. Chem. Biol., 2002, 6, 390 CrossRef CAS.
- J. Foote and H. N. Eisen, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 10679 CrossRef CAS.
- L. Jermutus, A. Honegger, F. Schwesinger, J. Hanes and A. Plückthun, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 75 CrossRef CAS.
- A. L. Kurtzman, S. Govindarajan, K. Vahle, J. T. Jones, V. Heinrichs and P. A. Patten, Curr. Opin. Chem. Biol., 2001, 12, 361 CAS.
- R. C. Ladner, A. K. Sato, J. Gorzelany and M. de Souza, Drug Discovery Today, 2004, 9, 525 CrossRef CAS.
- D. Lipovsek and A. Pluckthun, J. Immunol. Methods, 2004, 290, 51 CrossRef CAS.
- E. V. Shusta, J. VanAntwerp and K. D. Wittrup, Curr. Opin. Biotechnol., 1999, 10, 117 CrossRef CAS.
- K. D. Wittrup, Curr. Opin. Biotechnol., 2001, 12, 395 CrossRef CAS.
- P. Amstutz, P. Forrer, C. Zahnd and A. Plückthun, Curr. Opin. Biotechnol., 2001, 12, 400 CrossRef CAS.
- G. P. Smith, Science, 1985, 228, 1315 CrossRef CAS.
- J. K. Scott and G. P. Smith, Science, 1990, 249, 386 CrossRef CAS.
- J. A. Francisco, R. Campbell, B. L. Iverson and G. Georgiou, Proc. Natl. Acad. Sci. U. S. A., 1993, 90, 10444 CAS.
- J. A. Francisco, C. F. Earhart and G. Georgiou, Proc. Natl. Acad. Sci. U. S. A., 1992, 89, 2713 CAS.
- G. Georgiou, C. Stathopoulos, P. S. Daugherty, A. R. Nayak, B. L. Iverson and R. Curtiss, 3rd, Nat. Biotechnol., 1997, 15, 29 CrossRef CAS.
- M. Little, P. Fuchs, F. Breitling and S. Dubel, Trends Biotechnol., 1993, 11, 3 CrossRef CAS.
- E. T. Boder and K. D. Wittrup, Nat. Biotechnol., 1997, 15, 553 CAS.
- L. C. Mattheakis, R. R. Bhatt and W. J. Dower, Proc. Natl. Acad. Sci. U. S. A., 1994, 91, 9022 CAS.
- R. W. Roberts and J. W. Szostak, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 12297 CrossRef CAS.
- N. Doi and H. Yanagawa, FEBS Lett., 1999, 457, 227 CrossRef CAS.
- M. Kurz, K. Gu, A. Al-Gawari and P. A. Lohse, ChemBioChem, 2001, 2, 666 CrossRef CAS.
- R. E. Hawkins, S. J. Russell and G. Winter, J. Mol. Biol., 1992, 226, 889 CrossRef CAS.
- H. B. Lowman and J. A. Wells, J. Mol. Biol., 1993, 234, 564 CrossRef CAS.
- H. B. Lowman, S. H. Bass, N. Simpson and J. A. Wells, Biochemistry, 1991, 30, 10832 CrossRef CAS.
- K. H. Pearce, Jr., B. C. Cunningham, G. Fuh, T. Teeri and J. A. Wells, Biochemistry, 1999, 38, 81 CrossRef CAS.
- K. H. Pearce, Jr., M. H. Ultsch, R. F. Kelley, A. M. de Vos and J. A. Wells, Biochemistry, 1996, 35, 10300 CrossRef CAS.
- W. D. Fairlie, A. D. Uboldi, J. E. McCoubrie, C. C. Wang, E. F. Lee, S. Yao, D. P. De Souza, S. Mifsud, D. Metcalf, N. A. Nicola, R. S. Norton and M. Baca, J. Biol. Chem., 2004, 279, 2125 CAS.
- M. Isalan, A. Klug and Y. Choo, Nat. Biotechnol., 2001, 19, 656 CrossRef CAS.
- K. Sato, M. D. Simon, A. M. Levin, K. M. Shokat and G. A. Weiss, Chem. Biol., 2004, 11, 1017 CrossRef CAS.
- J. A. Wells, Biochemistry, 1990, 29, 8509 CrossRef CAS.
- L. Blaise, A. Wehnert, M. P. Steukers, T. van den Beucken, H. R. Hoogenboom and S. E. Hufton, Gene, 2004, 342, 211 CrossRef CAS.
- B. L. Roberts, W. Markland, A. C. Ley, R. B. Kent, D. W. White, S. K. Guterman and R. C. Ladner, Proc. Natl. Acad. Sci. U. S. A., 1992, 89, 2429 CAS.
- S. S. Sidhu, H. B. Lowman, B. C. Cunningham and J. A. Wells, Methods Enzymol., 2000, 328, 333 CrossRef CAS.
- J. A. Wells and H. B. Lowman, Curr. Opin. Biotechnol., 1992, 3, 355 CrossRef CAS.
- H. B. Lowman, S. H. Bass, N. Simpson and J. A. Wells, Biochemistry, 1991, 30, 10832 CrossRef CAS.
- Y. Choo, I. Sanchez-Garcia and A. Klug, Nature, 1994, 372, 642 CrossRef CAS.
- H. A. Greisman and C. O. Pabo, Science, 1997, 275, 657 CrossRef CAS.
- H. Wu, W. P. Yang and C. F. Barbas, 3rd, Proc. Natl. Acad. Sci. U. S. A., 1995, 92, 344 CAS.
- R. P. Laura, A. S. Witt, H. A. Held, R. Gerstner, K. Deshayes, M. F. Koehler, K. S. Kosik, S. S. Sidhu and L. A. Lasky, J. Biol. Chem., 2002, 277, 12906 CrossRef CAS.
- G. Fuh, M. T. Pisabarro, Y. Li, C. Quan, L. A. Lasky and S. S. Sidhu, J. Biol. Chem., 2000, 275, 21486 CAS.
- F. F. Vajdos, C. W. Adams, T. N. Breece, L. G. Presta, A. M. de Vos and S. S. Sidhu, J. Mol. Biol., 2002, 320, 415 CrossRef CAS.
- K. Murase, K. L. Morrison, P. Y. Tam, R. L. Stafford, F. Jurnak and G. A. Weiss, Chem. Biol., 2003, 10, 161 CrossRef CAS.
- T. Clackson and J. A. Wells, Trends Biotechnol., 1994, 12, 173 CrossRef CAS.
- N. J. Deacon, A. Tsykin, A. Solomon, K. Smith, M. Ludford-Menting, D. J. Hooker, D. A. McPhee, A. L. Greenway, A. Ellett, C. Chatfield, V. A. Lawson, S. Crowe, A. Maerz, S. Sonza, J. Learmont, J. S. Sullivan, A. Cunningham, D. Dwyer, D. Dowton and J. Mills, Science, 1995, 270, 988 CrossRef CAS.
- L. A. Landon, E. N. Peletskaya, V. V. Glinsky, N. Karasseva, T. P. Quinn and S. L. Deutscher, J. Protein Chem., 2003, 22, 193 CrossRef CAS.
- E. N. Peletskaya, G. Glinsky, S. L. Deutscher and T. P. Quinn, Mol. Diversity, 1996, 2, 13 Search PubMed.
- E. N. Peletskaya, V. V. Glinsky, G. V. Glinsky, S. L. Deutscher and T. P. Quinn, J. Mol. Biol., 1997, 270, 374 CrossRef CAS.
- T. J. Fleming, M. Sachdeva, M. Delic, J. Beltzer, C. R. Wescott, M. Devlin, R. C. Lander, A. E. Nixon, V. Roschke, D. M. Hilbert and D. J. Sexton, J. Mol. Recognit., 2005, 18, 94 CrossRef CAS.
- J. M. Boulter, M. Glick, P. T. Todorov, E. Baston, M. Sami, P. Rizkallah and B. K. Jakobsen, Protein Eng., 2003, 16, 707 CrossRef CAS.
- Y. Li, R. Moysey, P. E. Molloy, A. L. Vuidepot, T. Mahon, E. Baston, S. Dunn, N. Liddy, J. Jacob, B. K. Jakobsen and J. M. Boulter, Nat. Biotechnol., 2005, 23, 349 CrossRef CAS.
- http://www.accessdata.fda.gov/scripts/cder/drugsatfda/index.cfm. .
- http://www.rxlist.com/. .
- Z. Qu, G. L. Griffiths, W. A. Wegener, C. H. Chang, S. V. Govindan, I. D. Horak, H. J. Hansen and D. M. Goldenberg, Methods, 2005, 36, 84 Search PubMed.
- R. E. Bird, K. D. Hardman, J. W. Jacobson, S. Johnson, B. M. Kaufman, S. M. Lee, T. Lee, S. H. Pope, G. S. Riordan and M. Whitlow, Science, 1988, 242, 423 CrossRef CAS.
- H. R. Hoogenboom, A. D. Griffiths, K. S. Johnson, D. J. Chiswell, P. Hudson and G. Winter, Nucleic Acids Res., 1991, 19, 4133 CrossRef CAS.
- J. McCafferty, A. D. Griffiths, G. Winter and D. J. Chiswell, Nature, 1990, 348, 552 CrossRef CAS.
- R. Schier, A. McCall, G. P. Adams, K. W. Marshall, H. Merritt, M. Yim, R. S. Crawford, L. M. Weiner, C. Marks and J. D. Marks, J. Mol. Biol., 1996, 263, 551 CrossRef CAS.
- R. Schier, J. Bye, G. Apell, A. McCall, G. P. Adams, M. Malmqvist, L. M. Weiner and J. D. Marks, J. Mol. Biol., 1996, 255, 28 CrossRef CAS.
- D. Lu, J. Shen, M. D. Vil, H. Zhang, X. Jimenez, P. Bohlen, L. Witte and Z. Zhu, J. Biol. Chem., 2003, 278, 43496 CrossRef CAS.
- C. V. Lee, W. C. Liang, M. S. Dennis, C. Eigenbrot, S. S. Sidhu and G. Fuh, J. Mol. Biol., 2004, 340, 1073 CrossRef CAS.
- M. Ho, R. J. Kreitman, M. Onda and I. Pastan, J. Biol. Chem., 2005, 280, 607 CAS.
- J. A. Maynard, C. B. Maassen, S. H. Leppla, K. Brasky, J. L. Patterson, B. L. Iverson and G. Georgiou, Nat. Biotechnol., 2002, 20, 597 CrossRef CAS.
- V. R. Juarez-Gonzalez, L. Riano-Umbarila, V. Quintero-Hernandez, T. Olamendi-Portugal, M. Ortiz-Leon, E. Ortiz, L. D. Possani and B. Becerril, J. Mol. Biol., 2005, 346, 1287 CrossRef CAS.
- L. Riano-Umbarila, V. R. Juarez-Gonzalez, T. Olamendi-Portugal, M. Ortiz-Leon, L. D. Possani and B. Becerril, FEBS J., 2005, 272, 2591 Search PubMed.
- R. De Pascalis, N. R. Gonzales, E. A. Padlan, P. Schuck, S. K. Batra, J. Schlom and S. V. Kashmiri, Clin. Cancer Res., 2003, 9, 5521.
- K. Koefoed, L. Farnaes, M. Wang, A. Svejgaard, D. R. Burton and H. J. Ditzel, J. Immunol. Methods, 2005, 297, 187 CrossRef CAS.
- O. Dubreuil, M. Bossus, M. Graille, M. Bilous, A. Savatier, M. Jolivet, A. Menez, E. Stura and F. Ducancel, J. Biol. Chem., 2005, 280, 24880 CrossRef CAS.
- Y. Iba, N. Hayashi, J. Sawada, K. Titani and Y. Kurosawa, Protein Eng., 1998, 11, 361 CrossRef CAS.
- U. Lamminmaki, S. Pauperio, A. Westerlund-Karlsson, J. Karvinen, P. L. Virtanen, T. Lövgren and P. Saviranta, J. Mol. Biol., 1999, 291, 589 CrossRef CAS.
- C. Miyazaki, Y. Iba, Y. Yamada, H. Takahashi, J. Sawada and Y. Kurosawa, Protein Eng., 1999, 12, 407 CrossRef CAS.
- P. Saviranta, M. Pajunen, P. Jauria, M. Karp, K. Pettersson, P. Mantsala and T. Lövgren, Protein Eng., 1998, 11, 143 CrossRef CAS.
- E. A. Burks, G. Chen, G. Georgiou and B. L. Iverson, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 412 CrossRef CAS.
- G. Georgiou, H. L. Poetschke, C. Stathopoulos and J. A. Francisco, Trends Biotechnol., 1993, 11, 6 CrossRef CAS.
- P. S. Daugherty, G. Chen, M. J. Olsen, B. L. Iverson and G. Georgiou, Protein Eng., 1998, 11, 825 CrossRef CAS.
- P. S. Daugherty, M. J. Olsen, B. L. Iverson and G. Georgiou, Protein Eng., 1999, 12, 613 CrossRef CAS.
- P. S. Daugherty, G. Chen, B. L. Iverson and G. Georgiou, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 2029 CrossRef CAS.
- G. Chen, I. Dubrawsky, P. Mendez, G. Georgiou and B. L. Iverson, Protein Eng., 1999, 12, 349 CrossRef CAS.
- P. H. Bessette, J. J. Rice and P. S. Daugherty, Protein Eng., Des. Sel., 2004, 17, 731 Search PubMed.
- M. J. Feldhaus, R. W. Siegel, L. K. Opresko, J. R. Coleman, J. M. Feldhaus, Y. A. Yeung, J. R. Cochran, P. Heinzelman, D. Colby, J. Swers, C. Graff, H. S. Wiley and K. D. Wittrup, Nat. Biotechnol., 2003, 21, 163 CrossRef CAS.
- B. K. Cho, M. C. Kieke, E. T. Boder, K. D. Wittrup and D. M. Kranz, J. Immunol. Methods, 1998, 220, 179 CrossRef CAS.
- M. C. Kieke, B. K. Cho, E. T. Boder, D. M. Kranz and K. D. Wittrup, Protein Eng., 1997, 10, 1303 CrossRef CAS.
- E. T. Boder and K. D. Wittrup, Biotechnol. Prog., 1998, 14, 55 CrossRef CAS.
- J. J. VanAntwerp and K. D. Wittrup, Biotechnol. Prog., 2000, 16, 31 CrossRef CAS.
- P. D. Holler, P. O. Holman, E. V. Shusta, S. O'Herrin, K. D. Wittrup and D. M. Kranz, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 5387 CrossRef CAS.
- M. C. Kieke, E. Sundberg, E. V. Shusta, R. A. Mariuzza, K. D. Wittrup and D. M. Kranz, J. Mol. Biol., 2001, 307, 1305 CrossRef CAS.
- P. D. Holler, L. K. Chlewicki and D. M. Kranz, Nat. Immunol., 2003, 4, 55 CrossRef CAS.
- L. K. Chlewicki, P. D. Holler, B. C. Monti, M. R. Clutter and D. M. Kranz, J. Mol. Biol., 2005, 346, 223 CrossRef CAS.
- M. W. Konrad, G. Hemstreet, E. M. Hersh, P. W. Mansell, R. Mertelsmann, J. E. Kolitz and E. C. Bradley, Cancer Res., 1990, 50, 2009 CAS.
- B. M. Rao, I. Driver, D. A. Lauffenburger and K. D. Wittrup, Mol. Pharmacol., 2004, 66, 864 CAS.
- B. M. Rao, A. T. Girvin, T. Ciardelli, D. A. Lauffenburger and K. D. Wittrup, Protein Eng., 2003, 16, 1081 CrossRef CAS.
- J. M. Weaver-Feldhaus, J. Lou, J. R. Coleman, R. W. Siegel, J. D. Marks and M. J. Feldhaus, FEBS Lett., 2004, 564, 24 CrossRef CAS.
- T. van den Beucken, H. Pieters, M. Steukers, M. van der Vaart, R. C. Ladner, H. R. Hoogenboom and S. E. Hufton, FEBS Lett., 2003, 546, 288 CrossRef CAS.
- E. T. Boder, K. S. Midelfort and K. D. Wittrup, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 10701 CrossRef CAS.
- K. S. Midelfort, H. H. Hernandez, S. M. Lippow, B. Tidor, C. L. Drennan and K. D. Wittrup, J. Mol. Biol., 2004, 343, 685 CrossRef CAS.
- C. P. Graff, K. Chester, R. Begent and K. D. Wittrup, Protein Eng. Des. Sel., 2004, 17, 293 Search PubMed.
- A. Rajpal, N. Beyaz, L. Haber, G. Cappuccilli, H. Yee, R. R. Bhatt, T. Takeuchi, R. A. Lerner and R. Crea, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 8466 CrossRef CAS.
- D. W. Colby, Y. Chu, J. P. Cassady, M. Duennwald, H. Zazulak, J. M. Webster, A. Messer, S. Lindquist, V. M. Ingram and K. D. Wittrup, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 17616 CrossRef CAS.
- D. W. Colby, P. Garg, T. Holden, G. Chao, J. M. Webster, A. Messer, V. M. Ingram and K. D. Wittrup, J. Mol. Biol., 2004, 342, 901 CrossRef CAS.
- C. Schaffitzel, J. Hanes, L. Jermutus and A. Plückthun, J. Immunol. Methods, 1999, 231, 119 CrossRef CAS.
- J. Hanes and A. Plückthun, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 4937 CrossRef CAS.
- J. Hanes, L. Jermutus, S. Weber-Bornhauser, H. R. Bosshard and A. Plückthun, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 14130 CrossRef CAS.
- J. L. Lin-Goerke, D. J. Robbins and J. D. Burczak, Biotechniques, 1997, 23, 409 CAS.
- M. Zaccolo, D. M. Williams, D. M. Brown and E. Gherardi, J. Mol. Biol., 1996, 255, 589 CrossRef CAS.
- W. P. Stemmer, Nature, 1994, 370, 389 CrossRef CAS.
- J. Hanes, C. Schaffitzel, A. Knappik and A. Plückthun, Nat. Biotechnol., 2000, 18, 1287 CrossRef CAS.
- C. Zahnd, S. Spinelli, B. Luginbuhl, P. Amstutz, C. Cambillau and A. Plückthun, J. Biol. Chem., 2004, 279, 18870 CrossRef CAS.
- H. K. Binz and A. Plückthun, Curr. Opin. Biotechnol., 2005, 16, 459 CrossRef CAS.
- H. K. Binz, P. Amstutz, A. Kohl, M. T. Stumpp, C. Briand, P. Forrer, M. G. Grutter and A. Plückthun, Nat. Biotechnol., 2004, 22, 575 CrossRef CAS.
- N. Nemoto, E. Miyamoto-Sato, Y. Husimi and H. Yanagawa, FEBS Lett., 1997, 414, 405 CrossRef CAS.
- T. T. Takahashi, R. J. Austin and R. W. Roberts, Trends Biochem. Sci., 2003, 28, 159 CrossRef CAS.
- D. S. Wilson, A. D. Keefe and J. W. Szostak, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 3750 CrossRef CAS.
- A. D. Keefe and J. W. Szostak, Nature, 2001, 410, 715 CrossRef CAS.
- L. Xu, P. Aha, K. Gu, R. G. Kuimelis, M. Kurz, T. Lam, A. C. Lim, H. Liu, P. A. Lohse, L. Sun, S. Weng, R. W. Wagner and D. Lipovsek, Chem. Biol., 2002, 9, 933 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2006 |