The mechanism of action of ramoplanin and enduracidin
Xiao Fangad, Kittichoat Tiyanontbd, Yi Zhanga, Jutta Wannerc, Dale Bogerc and Suzanne Walker*d
aDepartment of Chemistry and Chemical Biology, Harvard University, 12 Oxford St., Cambridge, MA 02128, USA. E-mail: fang@fas.harvard.edu.; Tel: 617-432-5498
bDepartment of Chemistry, Princeton University, Princeton, NJ 08544, USA
cDepartment of Chemistry and The Skaggs Institute for Chemical Biology, The Scripps Research Institute, La Jolla, CA 92037, USA
dDepartment of Microbiology and Molecular Genetics, Harvard Medical School, Harvard University, Boston, MA 02115, USA. E-mail: suzanne_walker@hms.harvard.edu
First published on 29th November 2005
Abstract
The lipoglycodepsipeptide antibiotic ramoplanin is proposed to inhibit bacterial cell wall biosynthesis by binding to intermediates along the pathway to mature peptidoglycan, which interferes with further enzymatic processing. Two sequential enzymatic steps can be blocked by ramoplanin, but there is no definitive information about whether one step is inhibited preferentially. Here we use inhibition kinetics and binding assays to assess whether ramoplanin and the related compound enduracidin have an intrinsic preference for one step over the other. Both ramoplanin and enduracidin preferentially inhibit the transglycosylation step of peptidoglycan biosynthesis compared with the MurG step. The basis for stronger inhibition is a greater affinity for the transglycosylase substrate Lipid II over the MurG substrate Lipid I. These results provide compelling evidence that ramoplanin's and enduracidin's primary cellular target is the transglycosylation step of peptidoglycan biosynthesis.
Introduction
Peptidoglycan is a crosslinked carbohydrate polymer that surrounds bacterial cells and prevents them from rupturing under high internal osmotic pressures. Because peptidoglycan is essential for survival and has no eukaryotic counterpart, peptidoglycan biosynthesis (Fig. 1) is the target of a large number of clinically used antibiotics, including the β-lactams, cephalosporins, and glycopeptide antibiotics.1 The emergence of resistance to all these classes of antibiotics represents a significant threat to public health and has stimulated efforts to develop structurally novel antibacterial agents that inhibit the peptidoglycan biosynthetic pathway. One molecule that has received considerable attention in recent years is ramoplanin (Fig. 2, 1), a lipoglycodepsipeptide antibiotic discovered in the 1980s in a screen for peptidoglycan synthesis inhibitors.2,3 Ramoplanin has good activity against a wide range of Gram-positive organisms and is regarded as a promising candidate for the treatment of many Gram-positive infections.4 It is currently in late stage clinical trials for two different indications.5,6 Due to hydrolytic instability and other issues, however, neither of these indications involves systemic administration of ramoplanin, and the full potential of this compound has yet to be realized.4 A better understanding of the mechanism of action of ramoplanin may enable the development of derivatives to treat systemic infections.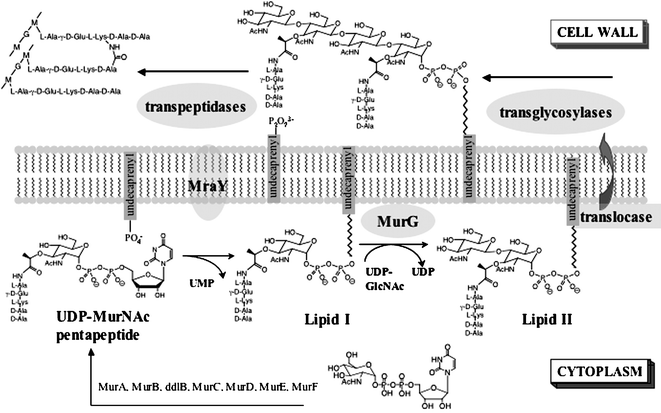 | ||
| Fig. 1 Peptidoglycan biosynthesis. | ||
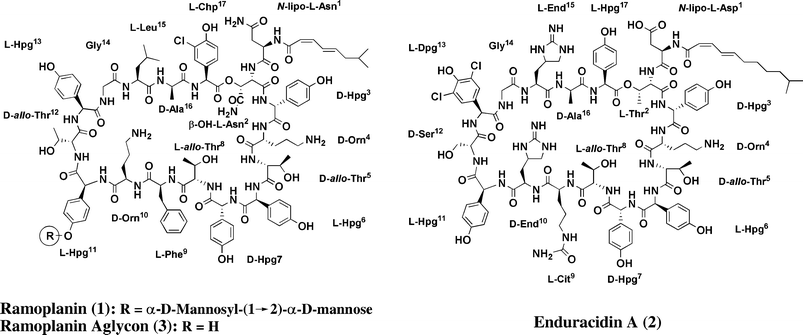 | ||
| Fig. 2 Structure of ramoplanin (1), enduracidin (2) and the ramoplanin aglycon (3). | ||
A mechanism of action for ramoplanin was first proposed in 1990 by Somner and Reynolds, who showed, using a cell-free, particulate membrane assay, that the antibiotic blocks the MurG-catalyzed conversion of Lipid I to Lipid II (Fig. 1) on the biosynthetic pathway to peptidoglycan.7 Although no direct evidence for an interaction with Lipid I was presented, these authors suggested that ramoplanin kills bacterial cells by binding to this substrate, rendering it inaccessible to MurG.7 A decade later, also using a cell-free, particulate membrane system, we showed that ramoplanin inhibits the transgly-cosylase-catalyzed coupling of Lipid II molecules to form the carbohydrate chains of peptidoglycan.8 We established that ramoplanin binds to synthetic Lipid II analogues, and so we proposed that ramoplanin acts primarily by binding to Lipid II and inhibiting the transglycosylation step of peptidoglycan biosynthesis.8,9
Our hypothesis that the primary mechanism of action of ramoplanin involves binding to Lipid II and inhibition of transglycosylation rather than binding to Lipid I and inhibition of MurG rested largely on the fact that Lipid II is translocated to the external surface of the bacterial membrane as soon as it is produced whereas Lipid I remains on the internal surface of the membrane.10 Ramoplanin is a large and highly water-soluble molecule, and in the absence of a dedicated transport mechanism, it seemed improbable that it could diffuse readily through bacterial membranes to reach an intracellular target. In fact, Somner and Reynolds made this point in their mechanistic papers on ramoplanin, but when they did their studies it was not known that Lipid I and MurG are intracellular.7,10
Comparative information on how well ramoplanin and various analogues inhibit MurG and the bacterial trans-glycosylases could provide more insight into the mechanism of action of the molecule. We have developed synthetic routes to Lipid I and Lipid II substrates and have developed assays to study E. coli MurG and E. coli PBP1b, the major bacterial transglycosylase in this organism.9,11,12 These tools enable us to carry out the studies required to assess the importance of the different proposed targets of ramoplanin and structurally related compounds. Below we report a comparative analysis of the inhibition kinetics of ramoplanin, the ramoplanin aglycon (Fig. 2, 3), and the related antibiotic enduracidin (Fig. 2, 2) with respect to MurG and PBP1b. We also present quantitative information on the binding affinities of ramoplanin for Lipid I and Lipid II. These studies support the hypothesis that the primary mechanism of action of ramoplanin involves binding to Lipid II and inhibiting the transglycosylation step of peptidoglycan biosynthesis. They also indicate that enduracidin operates by the same mechanism as ramoplanin.
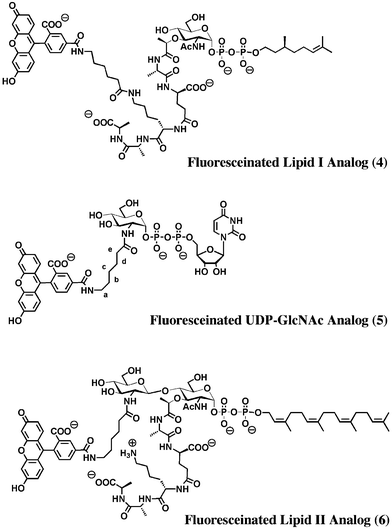 | ||
| Fig. 3 Structures of fluorescein-labeled peptidoglycan precursors. | ||
Experimental
Reagents
Ramoplanin was a gift from Oscient Pharmaceuticals. Enduracidin was purchased as the hydrochloride salt from Sigma-Aldrich (approx. 75% pure) and was further purified by HPLC (see below for conditions). The ramoplanin aglycon was prepared as previously reported.13 Alkaline phosphatase was purchased from Roche. UDP-[14C*]-GlcNAc (specific activity = 288 mCi mmol−1) was purchased from Perkin-Elmer Life Sciences, Inc. 6-(fluorescein-5-carboxamido)-hexanoic acid, succinimidyl ester was purchased from Invitrogen-Molecular Probes.Enduracidin purification
A solution of 5 mg enduracidin in 1.5 mL DMF was purified by reverse phase HPLC (VyDAC protein & peptide C18 column, Solvent A: H2O/0.1% trifluoroacetic acid; Solvent B: CH3CN/0.1% trifluoacetic acid) using a linear gradient from 0% B to 100% B over 145 min.Kinetic assay for enduracidin and ramoplanin aglycon in MurG reaction
MurG was overexpressed and purified as previously described.14 The reactions contained 0.01 mg mL−1 MurG (260 nM), reaction buffer (50 mM HEPES, pH 7.9), 5 mM MgCl2, 35µM UDP-[14C*]-GlcNAc, 10% methanol, varied concentrations of tetraprenyl Lipid I15 and inhibitor (see Fig. 4 for concentrations) in a total volume of 10µL in eppendorf tubes. After 2 min, the reactions were quenched with 1% SDS. The product was separated using paper chromatography (3 MM Whatman chromatography paper, 5 ∶ 3 isobutyric acid : 1N NH4OH), and quantified by scintillation counting. Kaleidagraph (Version 3.6.4, Synergy Software) was used to fit the data to an equation for substrate depletion.9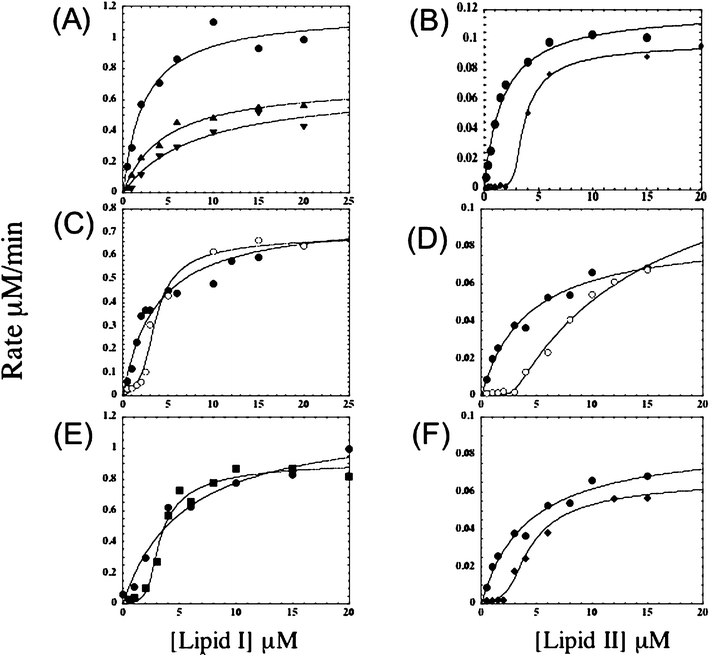 | ||
| Fig. 4 Representative velocity versus substrate concentration curves for MurG (A, C, E) and transglycosylase (B, D, F) reactions in the absence and presence of 1,2 and 3. (A) MurG reaction in the absence (●) and presence (▲ and ▼) of 1 (2 µM and 5 µM, respectively); (B) Transglycosylase reaction in the absence (●) and presence (◆) of 1 (6 µM); (C) MurG reaction in the absence (●) and presence (○) of 2 (6 µM); (D) Transglycosylase reaction in the absence (●) and presence (○) of 2 (6 µM); (E) MurG reaction in the absence (●) and presence (■) of 3 (5 µM); (F) Transglycosylase reaction in the absence (●) and presence (◆) of 3 (6 µM). | ||
Kinetic assays for enduracidin and ramoplanin aglycon in transglycoysylase reaction
E.coli PBP1b was expressed and purified as previously described.16 Assays were done in Eppendorf tubes containing transglycosylase buffer (50 mM HEPES at pH 7.5, 10 mM CaCl2, 1000 U mL−1 penicillin G, 0.2 mM decyl PEG and 11% DMSO) and varying concentrations of [14C*]-GlcNAc-labeled heptaprenyl Lipid II9 (specific activity = 288 mCi mmol−1), 6 µM of 2 or 3. The reactions were initiated by adding 1 µL PBP1b (from a solution freshly prepared by diluting a 50% glycerol stock of PBP1b 5-fold into 5 mM Tris buffer, pH 8.0, containing 8 mM decyl PEG) to the reaction mixtures for a final volume of 10 µL and an enzyme concentration of 30 nM. The reactions were stopped after 8 minutes by adding 10 µL of ice cold 10 mM Tris (pH 8.0) containing 0.2% Triton X-100. The products and starting material were separated using paper chromatography (3MM Whatman chromatography paper, 5∶3 isobutyric acid∶1 N NH4OH), and quantified by scintillation counting. Kaleidagraph (Version 3.6.4, Synergy Software) was used to fit the data to an equation for substrate depletion.9Synthesis of fluorescein-labeled Lipid I analog (4)
One mg (1 µmol) of citronellyl Lipid I (synthesized as previously described14) was dissolved in 120 µL saturated sodium bicarbonate solution. A solution of 2 mg (5 µmol) 6-(fluorescein-5-carboxamido)-hexanoic acid, succinimidyl ester in 120 µL dioxane was added, and the reaction was stirred for 1 h at room temperature. Another milligram (2.5 µmol) of 6-(fluorescein-5-carboxamido)-hexanoic acid, succinimidyl ester was added to the reaction mixture and stirred for an additional hour. The reaction mixture was then concentrated under vacuum, and purified by reverse phase HPLC using a linear gradient of H2O/CH3CN containing 0.1% ammonium bicarbonate. The product was eluted at approximately 75% H2O/CH3CN. The yield was 28%. Product 4 was analyzed by ESI-MS− (calculated: 1533.5, found 1532.5).Synthesis of fluorescein-labeled UDP-GlcNAc analog (5)
The fluoresceinated UDP-GlcNAc analog (5) was prepared by diluting UDP-glucosamine (11.4 µmoles) in a 1 ∶ 1 mixture of aqueous NaHCO3 (0.4 M) and dioxane. 6-(fluorescein-5-carboxamido) hexanoic acid, succinimidyl ester (20 mg, 34 µmoles) was added as a solution in dioxane. The reaction was run overnight at room temperature. To obtain the desired product 5, the reaction mixture was purified by reverse phase HPLC (Phenomenex Luna 5μ C18(2), 100A 250 × 21.2 mm; solvent A: H2O/0.1% ammonium bicarbonate; solvent B: CH3CN/0.1% ammonium bicarbonate). The following gradient was used for the purification: t = 0, %B = 0; t = 130, %B = 50; t = 145, %B = 100. 1H NMR (400 MHz, D2O): δ 8.08 (s, 1H, fluorescein), 7.87 (d, J = 6.4 Hz, 1H, fluorescein), 7.65 (d, J = 7.8 Hz, 1H, H6-uracyl), 7.19 (d, J = 6.0 Hz, 1H, fluorescein), 6.94 (m, 2H, fluorescein), 6.55 (m, br, 4H, fluorescein), 5.61 (d, J = 7.8 Hz, 1H, H5-uracyl), 5.56 (d, J = 4.8 Hz, 1H, H1-ribose), 5.33 (d, br, J = 4.0 Hz, 1H, H1-hexose), 4.16–3.18 (m, 13H), 2.22 (t, J = 7.2 Hz, 2H, He), 1.60–1.42 (m, br, 4H, Hb and Hd), 1.32–1.22 (m, 2H, Hc).Synthesis of fluorescein-labeled Lipid II analog (6)
0.5 µL of the fluoresceinated UDP-GlcNAc analog (5, 10 mM) and 0.5 µL of tetraprenyl C20 lipid I15 (10 mM) were incubated with 5 µL of 10 mg mL−1 MurG, 31.5 µL of alkaline phosphatase (3.2 U µL−1), 7.5 µL MeOH, and 5 µL reaction buffer (50 mM HEPES, pH 7.9 and 5 mM MgCl2) for 1 h. Ten of the above reactions were combined and purified using reverse phase HPLC at a liner gradient from 0% B to 100% B over 45 min (solvent A: H2O/0.1% trifluoroacetic acid; solvent B: CH3CN/0.1% trifluoroacetic acid). Product 6 was analyzed by ESI-MS− (calculated: 1827.7, found 1826.7).Fluorescence binding assays of ramoplanin against 4 and 6
Fluorescent measurements were done using an Aminco Bowman Series 2 Luminescence Spectrometer (λex = 492 nm, λem = 525 nm). Methanol stock solutions of 4 or 6 were diluted in buffer (25 mM potassium phosphate, 75 mM NaCl, pH 7.0) to yield a final concentration of 100 nM in 1 mL cuvettes. Aliquots of ramoplanin in DMSO were added and the mixtures were allowed to equilibrate 10 min to 1 h until the fluorescence intensity stopped changing. The titration data was fit using Prism (version 4.0a, GraphPad Software, Inc.) to the following equation describing binding of a ramoplanin dimer to 4 or 6 (i.e., R2F![[left over right harpoons]](https://www.rsc.org/images/entities/char_21cb.gif) R2 + F):
R2 + F):| I = I0 + [([R2] + [F] + Kd) − (([R2] + [F] + Kd)2 − 4[R2][F])1/2]*(I∝ − I0)/(2*[F]) | (1) |
Where I denotes fluorescence intensity (λex = 492 nm, λem = 525 nm); I0 denotes the fluorescence intensity in the absence of ramoplanin (λex = 492 nm, λem = 525 nm); I∝ denotes fluorescence intensity at the end of ramoplanin titration (λex = 492 nm, λem = 525 nm); [R2] represents the total concentration of ramoplanin dimer; and [F] represents the total concentration of 4 or 6.
Displacement assay of Lipid I and Lipid II against 4
Citronellyl Lipid I and Lipid II were prepared as previously reported.8,14,15 Fluorescent measurements were done using an Aminco Bowman Series 2 Luminescence Spectrometer (λex = 492 nm, λem = 525 nm). A methanol stock of 4 was diluted in buffer (25 mM potassium phosphate, 75 mM NaCl, pH 7.0) to a final concentration of 100 nM and the fluorescence intensity was monitored. Aliquots of a DMSO stock solution of 1 were added to 4 to a final concentration of 600 nM. Then aliquots of the unlabeled citronellyl Lipid I or Lipid II DMSO stock were added to the mixture, which was allowed to equilibrate until the fluorescence intensity stopped changing. The Lipid II titration was stopped when the fluorescence intensity returned to the level observed before 1 was added (7 × 10−5 M). At the same concentration, the fluorescence intensity was observed to increase only by 0.2 fluorescence units, corresponding to 10% displacement of 4 from the complex. Because of the limited amount of citronellyl Lipid I, the titration was not continued.Results
Previous results on the inhibition kinetics of ramoplanin with respect to MurG and PBP1b
Preliminary results on the inhibition of MurG and PBP1b by ramoplanin revealed significant differences in the mode of inhibition of these enzymes (Fig. 4A and 4B).9,17 Notably, the curves for inhibition of MurG indicated a non-competitive mode of inhibition whereas those for PBP1b were sigmoidal, which is consistent with a mechanism involving binding to Lipid II.9,17 The sigmoidal shape of the inhibition curves reflects the fact that the reaction rate is negligible at low concentrations because the substrate is bound to ramoplanin and unavailable for reaction; however, as soon as the substrate concentration exceeds a critical value, the rate jumps dramatically and inhibition is largely overcome. From the concentration at which the reaction rate was observed to jump (always one half the ramoplanin concentration) and structural studies, we concluded that ramoplanin binds to Lipid II as a dimer.9,18 A Job titration confirmed this stoichiometry.19 In contrast, the inhibition curves for MurG suggested a mode of inhibition involving a direct interaction between ramoplanin itself and the enzyme.17 Surprisingly, the inhibition was found not to depend on binding to Lipid I.17 Since ramoplanin had been shown to bind to Lipid I analogues in vitro,17 we were perplexed by these results. Therefore, we decided to investigate the behavior of a structurally related antibiotic, enduracidin, in the hope that this molecule would shed some more light on the behavior of ramoplanin.Inhibition kinetics of enduracidin with respect to MurG and PBP1b
Enduracidin (Fig. 2, 2) is a lipodepsipeptide that was isolated in the 1960s and found to be active against a wide range of Gram-positive bacteria,20–22 including methicillin-resistant Staphylococcus aureus (MRSA).22 Enduracidin was proposed to inhibit peptidoglycan biosynthesis because it causes the accumulation of UDP-MurNAc-pentapeptide (Fig. 1) in bacteria;23 however, the site of inhibition was never established.Like ramoplanin, enduracidin contains 17 amino acid residues, 16 of which form a macrocycle joined by a lactone linkage from the β-hydroxyl group on residue 2 (Thr2) to the carboxyl group of residue 17 (Hpg17). Enduracidin is identical to ramoplanin at residues 3 through 8, and at residues Gly14, Ala16, and Hpg17. In addition, it has the same chirality at all 17 amino acids and bears the same charge (+2) at physiological pH. Differences between enduracidin and ramoplanin occur at residue 9 (Phe in ramoplanin and Cit in enduracidin), at residue Hpg11 (which contains a dimannosyl moiety in ramoplanin but is unmodified in enduracidin), at residue 13 (Dpg in ramoplanin and Hpg in enduracidin) and at residues 10 and 15 (which are ornithine and leucine in ramoplanin but are both enduracididines in enduracidin).
A comparison of the solution structures of ramoplanin and enduracidin by NMR shows that they have quite similar backbone structures, as expected given the identical number and chirality of the residues in the macrocycles.24,25 The backbone structures feature two antiparallel β-sheets formed by residues 2–7 and 10–14, respectively.24,25 The side-chains of residues 3, 9 and 17, although located far apart in primary sequence, show strong NOEs, indicating that they are in close proximity and revealing a cup-shaped curvature in the molecule.24,25 Although minor structural differences may exist between ramoplanin and enduracidin,24,25 they are sufficiently similar that one might reasonably attribute any differences in behavior to specific differences in individual side chain residues.
The curves for inhibition of MurG and PBP1b by enduracidin are shown in Fig. 4C and 4D. The curve for inhibition of PBP1b is sigmoidal and inhibition is overcome at high concentrations of Lipid II, indicating that enduracidin inhibits this enzyme by binding to Lipid II (Fig. 4D). Furthermore, the reaction rate was negligible until the substrate concentration exceeded half the total enduracidin concentration, implying a 2 ∶ 1 enduracidin ∶ Lipid II binding. The kinetics shows that enduracidin is virtually identical to ramoplanin with respect to the mode of inhibition of this bacterial transglycosylase. However, the manner in which enduracidin inhibits MurG is different from ramoplanin. Whereas ramoplanin displays noncompetitive inhibition, enduracidin produces a sigmoidal inhibition curve that is consistent with substrate binding (Fig. 4A and 4C).
Based on the differences in the curves for the inhibition of MurG, we surmised that ramoplanin must contain some features, which enduracidin lacks, that explain its ability to interact directly with MurG. Although ramoplanin and enduracidin differ by several residues, the single largest structural difference between these two molecules is the presence of a dimannosyl moiety on residue Hpg11 of ramoplanin. This dimannosyl group on ramoplanin is not required for biological activity.26–28 To investigate whether it plays a role in MurG inhibition, we removed this glycosyl group by treatment with anhydrous HF to obtain the aglycon.13 The aglycon was tested for inhibition of both MurG and PBP1b, and the results are described in the following section.
Inhibition kinetics of ramoplanin aglycon with respect to MurG and PBP1b
Kinetic curves for MurG and PBP1b in the presence of the ramoplanin aglycon are shown in Fig. 4E and 4F. The curve for inhibition of PBP1b is sigmoidal, as it is for ramoplanin itself. The curve for MurG inhibition is also sigmoidal, in contrast to the behavior of ramoplanin. Therefore, the ramoplanin aglycon, like enduracidin, inhibits E. coli MurG by binding to Lipid I. We have concluded that the dimannosyl unit on ramoplanin entirely accounts for the noncompetitive inhibition pattern observed for the parent compound. This dimannosyl unit evidently promotes an interaction with E. coli MurG that is independent of Lipid I binding and that obscures the effects of binding to Lipid I.17 It remains to be established whether MurG homologues from other organisms also interact with ramoplanin directly, or whether this phenomenon is specific to the E. coli enzyme. However, even if ramoplanin directly interacts with other MurG homologues via the dimannosyl unit, the interaction is not likely to be biologically relevant because our studies with E. coli MurG suggest that it is not of sufficiently high affinity to compete with the much tighter binding observed to Lipid II.17Quantification of Lipid I and Lipid II binding using inhibition kinetics
Inhibition kinetics reveals a considerable amount about the interactions between a substrate-binding antibiotic and the target substrate. For example, the kinetics of transglycosylase inhibition has revealed that ramoplanin, the ramoplanin aglycon, and enduracidin all bind to Lipid II with a stoichiometry of 2 ∶ 1. Furthermore, the observation that there is virtually no reaction at low substrate concentrations in the presence of low micromolar concentrations of antibiotic implies that all of the substrate is sequestered at these concentrations, which indicates in turn that the Kd’s for binding are in the submicromolar range for all three compounds. By fitting the inhibition curves to an equation for inhibition via substrate sequestration, it is possible to estimate Kd’s for Lipid II binding of approximately 10 nM for the three compounds.Although we could estimate the Kd for Lipid II binding to ramoplanin via analysis of the curves for transglycosylase inhibition, we could not estimate the Kd for ramoplanin and Lipid I from the curves for MurG inhibition because the inhibition pattern was not consistent with substrate binding. However, we found that the ramoplanin aglycon inhibits MurG by binding to Lipid I. Because the dimannosyl unit does not play any role in the binding of ramoplanin to Lipid II (as determined by a comparison of ramoplanin and the ramoplanin aglycon inhibition patterns for PBP1b), it seems safe to assume that it does not play a role in binding to Lipid I. Therefore, we thought it would be reasonable to use the kinetic data obtained for inhibition of MurG by the ramoplanin aglycon to estimate the Kd for ramoplanin itself. Visual inspection of the kinetic curves for MurG and PBP1b inhibition by the ramoplanin aglycon reveals weaker binding to Lipid I, and fitting these data to the same equation used for PBP1b inhibition yields an estimated Kd for Lipid I binding that is approximately five-fold higher than the Kd for Lipid II binding (see Experimental). Nevertheless, because the kinetic experiments were done at high concentrations relative to the estimated binding constants, and thus are not as accurate as one might wish, we sought an alternative method that allows the direct measurement of binding constants at lower concentrations.
Quantification of Lipid I and Lipid II binding using fluorescent binding assays
Measuring the binding of ramoplanin to Lipid I and Lipid II analogues using standard biophysical methods has proven to be a considerable challenge because ramoplanin ∶ ligand complexes self-associate to form fibrils.8,29 Thus, prior to this work, the only quantitative data on binding reported for ramoplanin came from the laboratory of McCafferty and coworkers, who studied the binding of ramoplanin to analogues of Lipid I using NMR.29 In order to prevent aggregation of the ramoplanin complexes, these authors performed their experiments in DMSO–water mixtures.30 They reported a Kd of 180 ± 20 µM for UDP–MurNAc–pentapeptide (Fig. 1) and estimated the Kd for ramoplanin and Lipid I to be less than 200 µM.30 Because the inhibition kinetics for MurG and transglycosylase shows that the Kd’s of ramoplanin for both Lipid I and Lipid II are at least two orders of magnitude lower than previously reported estimates (i.e. < 1 µM), we sought a more appropriate method than NMR to measure the dissociation constants. Fluorescence-based methods are suitable for measuring binding constants in the nanomolar range. Because studies in our laboratory have suggested that the peptide chain on Lipid I plays a negligible role in binding to ramoplanin,31 we prepared an analogue of Lipid I containing a fluorescent label on the ε-amino group of the lysine in the pentapeptide chain (Fig. 3, 4). To evaluate the utility of this compound for measuring binding constants, we added increasing amounts of ramoplanin to a 100 nM solution of 4 and monitored the fluorescent emission of the sample at 525 nm following excitation at 492 nm. A decrease in fluorescence intensity was observed as ramoplanin was added. The change in fluorescence was observed to plateau by the time 2 µM of ramoplanin was in solution. Based on two sets of data, the dissociation constant for binding of the ramoplanin dimer to Lipid I was found to be 170 ± 30 nM (Fig. 5A).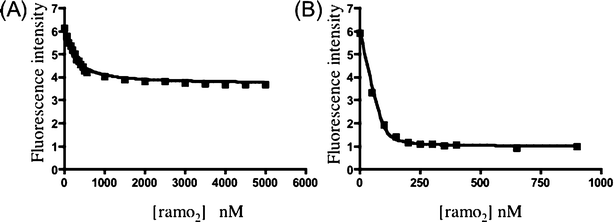 | ||
| Fig. 5 Fluorescent binding assays of ramoplanin with 4 and 6. (A) Fluorescence intensity of compound 4 as a function of ramoplanin concentration. (B) Fluorescence intensity of compound 6 as a function of ramoplanin concentration. | ||
We attempted to prepare the corresponding fluorescently labeled Lipid II analogue using MurG to form the β-(1,4) linkage to GlcNAc. Although we have previously shown that Lipid I analogues containing substituents on the lysine side chain are substrates for MurG,14 this particular compound did not turn over efficiently. However, we were able to prepare a Lipid II analogue (Fig. 3, 6) with a fluorophore on the C2-N-acyl position of the GlcNAc sugar using the corresponding fluorescently labeled UDP-GlcNAc donor 5. When ramoplanin was added to a 100 nM solution of compound 6, a dramatic decrease in fluorescence was observed, plateauing around 400 nM ramoplanin. A dissociation constant of 3 ± 2 nM for binding of the ramoplanin dimer to Lipid II was established from two sets of data (Fig. 5B).
Since the fluorophore on Lipid I analogue 4 and Lipid II analogues 6 was installed at different sites, we wanted to make certain that the preferential binding of ramoplanin to Lipid II was not related to the position of the fluorophore. Therefore, we sought to compare the concentrations of unlabeled Lipid I and Lipid II analogues required to displace the fluorescent probe 4 from ramoplanin. The displacement experiments were complicated by the fact that ramoplanin complexes self-associate; nevertheless, 10−5M concentrations of Lipid II displaced compound 4 fully; at the same concentrations of Lipid I, only 10% of 4 was displaced. The titration of Lipid I was not carried to completion due to compound limitations. The results, however, were consistent with all previous data indicating that Lipid II binds better to ramoplanin by a difference of one magnitude.
Discussion
The mechanism of action of ramoplanin has been the subject of extensive examination over the past fifteen years, with two possible cellular targets identified for the antibiotic.3,7–9 The first proposed target to be identified was MurG, the GlcNAc transferase that converts Lipid I to Lipid II on the pathway to peptidoglycan.7 The experiments pointing to MurG as a target were carefully performed but were not conclusive because they were conducted at a time when there were limited assays to identify the step at which peptidoglycan synthesis inhibitors take effect.7 Particulate membranes containing all of the enzymes involved in the membrane-linked steps of peptidoglycan biosynthesis were used, and the assays involved following the conversion of UDP–MurNAc–pentapeptide first to Lipid I and then to Lipid II and peptidoglycan polymer. Reynolds and coworkers showed that ramoplanin prevents the formation of Lipid II but were unable to investigate whether ramoplanin also inhibits subsequent steps.7 Ten years after Reynolds and coworkers proposed the MurG step as the target of ramoplanin, we adapted the particulate membrane assay to report on the transglycosylation step of peptidoglycan synthesis.8 We showed that ramoplanin also blocks this step.8 The identification of two possible cellular targets for ramoplanin raised the question of whether one target is more responsible for the antibiotic effects than the other. We argued that the primary mechanism of action of ramoplanin most likely involves binding to Lipid II/inhibition of transglycosylation rather than binding to Lipid I/inhibition of MurG because Lipid I is intracellular while ramoplanin does not have the physical properties characteristic of molecules that penetrate cell membranes (MW = 2554, aqueous solubility >100 mg mL−1).10While these arguments are reasonable, we had no direct, quantitative evidence that ramoplanin binds preferentially to Lipid II over Lipid I, or that it inhibits transglycosylases more strongly than it inhibits MurG. Furthermore, as we and others have discovered, the structural and energetic aspects of ramoplanin–substrate complexation have proven remarkably difficult to dissect.8,29,30 As soon as Lipid I and Lipid II are added to ramoplanin, the complexes self-assemble to form fibrils.8,29,30 This self-assembly process precludes efforts to characterize and quantify the binding interactions by most techniques. McCafferty and coworkers have been able, by using organic solvent–water mixtures and weakly binding ligands, to obtain some NMR data on 1 ∶ 1 complexes of ramoplanin with substrate analogues,29 but it is not clear that these data provide insight into the 2 ∶ 1 complexes that are involved in enzyme inhibition.
The inhibition kinetics described here for the ramoplanin aglycon and enduracidin show that both of these molecules bind with submicromolar affinity to Lipid I, more than 100-fold below the upper limit for the Kd established by McCafferty and coworkers using NMR. Therefore, we used fluorescent derivatives of Lipid I and Lipid II to evaluate binding to ramoplanin. Our studies show that there is a difference of about one order of magnitude in the Kds of ramoplanin for Lipid I and Lipid II, with Lipid II favored. The Kds estimated from the inhibition kinetics are consistent with fluorescence experiments showing that the ramoplanin aglycon binds Lipid II preferentially over Lipid I by a factor of approximately five. Preferential binding to Lipid II, combined with its greater accessibility on the outside surface of the bacterial cell membrane, strongly supports the hypothesis that the primary means by which ramoplanin kills bacterial cells involves binding to Lipid II and inhibiting transglycosylases. Because the inhibition kinetics shows that enduracidin binds better to Lipid II than to Lipid I, we propose that the primary mechanism of action of this molecule is the same as that of ramoplanin: it inhibits bacterial transglycosylases by binding to Lipid II. It is presumed that both ramoplanin and enduracidin also bind to the reducing end of the growing glycan chain, which contains the same disaccharide diphosphate moiety as Lipid II (see Fig. 1).
It is worth noting that the many residue differences between ramoplanin and enduracidin have no obvious effect on Lipid II binding. It has been suggested that the residues in these molecules that are most critical for interaction with peptidoglycan intermediates are located at positions 3–8, which are identical in ramoplanin and enduracidin.29 Therefore, it has been suggested that residues 3–8 comprise the “minimal pharmacophore” of ramoplanin.29 However, features other than the similarities in residues 3–8 may explain the observation that ramoplanin and enduracidin behave similarly with respect to ligand binding. For example, conformationally defined backbone interactions requiring the full structure may play the primary role in the recognition of Lipid I and Lipid II.32 Breukink and coworkers have reported that the lantibiotic nisin, which also binds to Lipid II, uses primarily backbone contacts in complexation, and they have identified a “pyrophosphate cage” as a key binding element.33 Structural studies of enduracidin and ramoplanin show that the backbones of these two molecules are very similar.24,25 The backbone conformations are determined by the number and chirality of the amino acids in the macrocycles rather than by the side chain identities, and it is quite possible that most of the side chains are individually unimportant for binding. Alanine scanning experiments on the ramoplanin structure should shed more light on the importance of each particular amino acid side chain in substrate binding and biological activity, and changes in chirality can provide insight into the importance of the backbone conformation in binding. Having established a convergent synthetic route to the ramoplanin aglycon analogues that enables such a detailed examination of the importance of each residue,28,32,34–36 and with suitable assays in place to probe Lipid II vs. Lipid I binding, transglycosylase vs. MurG inhibition, and biological activity, we should be able to determine the essential requirements for binding and biological activity, and we may be able to identify analogues with more desirable properties to use as antibiotics.
Conclusions
We compared the inhibitory behavior of ramoplanin and two structurally related compounds, enduracidin and the ramoplanin aglycon, with respect to two enzymes involved in the late steps of peptidoglycan biosynthesis. We have shown that the ramoplanin aglycon and enduracidin inhibit these two enzymes, MurG and the bacterial transglycosylase PBP1b, by binding to their substrates, Lipid I and Lipid II, respectively. The inhibition kinetics shows that both compounds bind more tightly to Lipid II than to Lipid I. We measured the Kds of ramoplanin for fluorescent analogues of Lipid I and Lipid II and found that Lipid II binds about ten-fold more tightly than Lipid I. Based on our studies, we conclude that the primary mechanism of action of both ramoplanin and enduracidin involves inhibition of the transglycosylation step of peptidoglycan biosynthesis.Acknowledgements
This work is supported by NIH grants AI50855 (to SW) and CA41101 (to DLB). We thank Oscient Pharmaceuticals for a generous gift of ramoplanin.References
- C. T. Walsh, Antibiotics: Actions,Origins,Resistance, ASM press, Washington, D. C., 2003.
- B. Cavalleri, H. Pagani, G. Volpe, E. Selva and F. Parenti, J. Antibiot. (Tokyo), 1984, 37, 309 CAS.
- S. Walker, L. Chen, Y. Hu, Y. Rew, D. Shin and D. L. Boger, Chem. Rev., 2005, 105, 449 CrossRef CAS.
- M. A. Montecalvo, J. Antimicrob. Chemother., 2003, 51, iii31 CAS.
- R. Patel, J. Antimicrob. Chemother., 2003, 51, iii13 CAS.
- N. Woodford, Expert Opin. Invest. Drugs, 2003, 12, 117 Search PubMed.
- P. E. Reynolds and E. A. Somner, Drugs Exp. Clin. Res., 1990, 16, 385 Search PubMed.
- M. C. Lo, H. Men, A. Branstrom, J. Helm, N. Yao, R. Goldman and S. Walker, J. Am. Chem. Soc., 2000, 122, 3540 CrossRef CAS.
- Y. Hu, J. S. Helm, L. Chen, X. Y. Ye and S. Walker, J. Am. Chem. Soc., 2003, 125, 8736 CrossRef CAS.
- K. Bupp and J. van Heijenoort, J. Bacteriol., 1993, 175, 1841 CAS.
- X. Y. Ye, M. C. Lo, L. Brunner, D. Walker, D. Kahne and S. Walker, J. Am. Chem. Soc., 2001, 123, 3155 CrossRef CAS.
- H. B. Men, P. Park, M. Ge and S. Walker, J. Am. Chem. Soc., 1998, 120, 2484 CrossRef CAS.
- J. Wanner, D. T. Tang, C. C. McComas, B. M. Crowley, W. L. Jiang, J. Moss and D. L. Boger, Bioorg. Med. Chem. Lett., 2003, 13, 1169 CrossRef.
- S. Ha, E. Chang, M. C. Lo, H. Men, P. Park, M. Ge and S. Walker, J. Am. Chem. Soc., 1999, 121, 8415 CrossRef CAS.
- L. Chen, H. Men, S. Ha, X. Y. Ye, L. Brunner, Y. Hu and S. Walker, Biochemistry, 2002, 41, 6824 CrossRef CAS.
- L. Chen, D. Walker, B. Sun, Y. Hu, S. Walker and D. Kahne, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 5658 CrossRef CAS.
- J. S. Helm, L. Chen and S. Walker, J. Am. Chem. Soc., 2002, 124, 13970 CrossRef CAS.
- M. C. Lo, J. S. Helm, G. Sarngadharan, I. Pelczer and S. Walker, J. Am. Chem. Soc., 2001, 123, 8640 CrossRef CAS.
- J. S. Helm, PhD thesis, Princeton University, 2004.
- E. Higashid, K. Hatano, M. Shibata and K. Nakazawa, J. Antibiot., 1968, 21, 126.
- E. Yourasso and R. Monsieur, Chemotherapy, 1972, 17, 182 Search PubMed.
- H. Komatsuzawa, J. Suzuki, M. Sugai, Y. Miyake and H. Suginaka, J. Antimicrob. Chemother., 1994, 33, 1155 CrossRef CAS.
- S. Tanayama, T. Fugono and T. Yamazaki, J. Antibiot., 1968, 21, 313 CAS.
- F. Castiglione, A. Marazzi, M. Meli and G. Colombo, Magn. Reson. Chem., 2005, 43, 603 CrossRef CAS.
- M. Kurz and W. Guba, Biochemistry, 1996, 35, 12570 CrossRef CAS.
- F. Espersen, Curr. Opin. Anti-infect. Invest. Drugs, 1999, 78 Search PubMed.
- R. Ciabatti, B. Cavalleri and B. I. S.p.A, US Patent, 5491128, 1990 Search PubMed.
- Y. Rew, D. Shin, I. Hwang and D. L. Boger, J. Am. Chem. Soc., 2004, 126, 1041 CrossRef CAS.
- P. Cudic, D. C. Behenna, J. K. Kranz, R. G. Kruger, A. J. Wand, Y. I. Veklich, J. W. Weisel and D. G. McCafferty, Chem. Biol., 2002, 9, 897 CrossRef CAS.
- P. Cudic, J. K. Kranz, D. C. Behenna, R. G. Kruger, H. Tadesse, A. J. Wand, Y. I. Veklich, J. W. Weisel and D. G. McCafferty, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 7384 CrossRef CAS.
- M. C. Lo, PhD thesis, Princeton University, 2000.
- W. Jiang, J. Wanner, R. J. Lee, P. Y. Bounaud and D. L. Boger, J. Am. Chem. Soc., 2003, 125, 1877 CrossRef CAS.
- E. Breukink, H. E. van Heusden, P. J. Vollmerhaus, E. Swiezewska, L. Brunner, S. Walker, A. J. Heck and B. de Kruijff, J. Biol. Chem., 2003, 278, 19898 CrossRef CAS.
- W. Jiang, J. Wanner, R. J. Lee, P. Y. Bounaud and D. L. Boger, J. Am. Chem. Soc., 2002, 124, 5288 CrossRef CAS.
- D. Shin, Y. Rew and D. L. Boger, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 11977 CrossRef CAS.
- L. Chen, Y. Yuan, J. S. Helm, Y. Hu, Y. Rew, D. Shin, D. L. Boger and S. Walker, J. Am. Chem. Soc., 2004, 126, 7462 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2006 |