A disposable microfluidic cassette for DNA amplification and detection
Jing Wanga, Zongyuan Chena, Paul L. A. M. Corstjensb, Michael G. Mauka and Haim H. Bau*a
aDepartment of Mechanical Engineering and Applied Mechanics, University of Pennsylvania, Philadelphia, Pennsylvania 19104-6315, USA. E-mail: bau@seas.upenn.edu
bDepartment of Molecular Cell Biology, Leiden University Medical Center, Wassenaarseweg 72, 2333 AL, Leiden, The Netherlands
First published on 5th December 2005
Abstract
A pneumatically driven, disposable, microfluidic cassette comprised of a polymerase chain reaction (PCR) thermal cycler, an incubation chamber to label PCR amplicons with up-converting phosphor (UPT) reporter particles, conduits, temperature-activated, normally closed hydrogel valves, and a lateral flow strip, was constructed and tested. The hydrogel valves, which were opened and closed with the aid of electrically controlled thermoelectric units, provided a simple means to seal the PCR reactor and suppress bubble formation. The hydrogel-based flow control was electronically addressable, leakage-free, and biocompatible. To test the device, a solution laden with genomic DNA isolated from B. cereus was introduced into the microfluidic cassette and a specific 305 bp fragment was amplified. The PCR amplicons were labelled with the phosphor (UPT) reporter particles, applied to the lateral flow strip, bound to pre-immobilized ligands, and detected with an IR laser that scanned the lateral flow strip and excited the phosphor (UPT) particles that, in turn, emitted light in the visible spectrum. The UPT particles do not bleach, they provide a permanent record, and they readily facilitate the filtering of background noise. The cassette described herein will be used for rapid testing at the point of care.
Introduction
In recent years, there has been a growing interest in developing microfluidic systems for Polymerase Chain Reaction (PCR) and nucleic acid detection.1–13 Microfluidic devices, which include miniaturized PCR thermal cyclers, conduits, chambers, and microvalves, have been fabricated in PDMS,1 silicon,3,4,12 glass,5,10,11 silicon-glass combinations,6,12 ceramics,7 and various plastic materials.2,8,9,13 Low-cost, disposable, plastic-based devices are of particular interest. A few of these devices include a means of detecting the nucleic acid amplicons directly integrated with the PCR thermal cycler reaction chamber. The detection of the nucleic acid material is either facilitated by capillary electrophoresis of the PCR products3,10,11 or by hybridization of the PCR products with a fluorescent dye.8,12,13 For example, Lagally et al.11 reported an amplification and electrophoresis device comprised of an integrated glass structure that achieved detection of 2 copies of a target DNA template. Yang et al.13 described a plastic-based microfluidic device integrating PCR and fluorescence detection in which DNA template, derived from samples of 10 E. coli cells, were detected.More recently, developments in nanotechnology have led to several novel, particle-based, quantitative, high sensitivity bioassays.14,15 One such technology utilizes “up-converting” phosphor particles (UPT) that exhibit anti-Stokes behaviour by converting low-energy, infrared (IR) excitation (∼980 nm) to high-energy phosphorescence emission in the visible spectrum.16 Several features of UPT distinguish it from other fluorescence techniques: Since up-conversion does not generally occur in natural biological materials, the use of UPT particles filters out background interference.17 Phosphor emissions can be of different colours, facilitating simultaneous multi-target detection.32 The UPT particles do not bleach, they facilitate relatively lengthy measurements, and they provide permanent records.21 UPT particle-based immunochromatographic assays have been reported to have up to 100 times higher sensitivity than conventional fluorescent assays.18,19,20,32,33 In our device, we used 400 nm diameter particles supplied by OraSure Technologies, Inc. (Bethlehem, PA).
Lateral flow (LF) assays provide a rapid, low cost technique for the identification of analytes and pathogens at the point of care and at home.22–26 Several LF assays have been developed for serodiagnosis of human leptospirosis,22 human fecal occult blood detection,23 HIV-1 diagnostics,24 virus diagnostics,25 and detection of drugs of abuse.26 LF assays were modelled in our previous works27,28 and are used here for bio-reaction and detection. In most cases, LF assays are used as stand-alone devices without any significant sample pre-processing. The capabilities of the LF assay can be greatly enhanced by integrating it with upstream microfluidics. One of the objectives of our work is to demonstrate the benefits of such integration.
In this work, for the first time, low cost, disposable, polycarbonate, microfluidic cassettes integrating nucleic acid amplification and UPT- lateral flow (LF) detection were constructed. Specifically, the DNA target was amplified, and the PCR amplicons were mixed and incubated with UPT particles. The DNA-UPT complexes were propelled through the LF strip, bound to the immobilized ligands in the test zone, and detected using the Uplink™ (OraSure Technologies, Inc., Bethlehem, PA) laser scanner and reader. The flow of the sample, buffers, and reagents was driven pneumatically with an external syringe pump. Temperature-sensitive hydrogel valves were constructed for both flow control and PCR chamber sealing. Thermoelectric units were used for the heating and cooling of the temperature-actuated hydrogel valves, incubation chambers, and PCR thermal cycler chamber. Ultimately, the cassette described here will serve as a component in a comprehensive system consisting of multiple analysis paths for the processing and identification of antibodies, antigens, and nucleic acids in oral fluids.29 Our cassette can also serve as a stand-alone device to detect free DNA in body fluids, such as saliva in this specific case, and as a device for the amplification and detection of DNA that was purified and isolated prior to its introduction into the cassette.
The paper is organized as follows. Section 2 describes the experimental setup, protocols, reagents and device fabrications. Section 3 describes the operation and characteristics of the hydrogel valves and the use of the valves in the microfluidic system for the sealing of the PCR chamber. The operation and performance of the PCR-UPT-Lateral flow integrated cartridge is also discussed. Section 4 concludes.
Experimental
Procedures
The protocol for the DNA-UPT-LF assay is depicted in Fig. 1. B. cereus genomic DNA isolated from bacterial cell cultures was diluted in saliva and provided the PCR amplification template. The primer pairs, targeting a 305-bp fragment (Fig. 1a), were tagged with digoxigenin (Dig) and biotin (Bio). PCR amplification resulted in the production of double-stranded DNA amplicons with 5′-Dig hapten on one strand and 5′-Bio hapten on the opposite strand (Fig. 1b). The tagged amplicons were then mixed and incubated in LF buffer containing avidin-conjugated UPT reporter particles. The mixture containing PCR amplicons bound to UPTavidin (biotin-avidin interaction, Fig. 1c) was then blotted to the LF strip. The nitrocellulose LF strip (45 mm, SRHF04000, Millipore) included a sample loading pad at its upstream end (20 mm, glass-fiber No. 33, Schleicher & Schuell), an absorbent pad at the downstream end (20 mm, paper No. 470, Schleicher & Schuell), a test zone and a flow-control zone. The test zone consisted of preloaded mouse anti-digoxigenin (MαDig) antibody in a Tris buffer (10 mM, pH 8.0, 1% (v/v) methanol) at a density of 20 ng mm−2. The control zone consisted of a biotin BSA conjugate, loaded at a density of 50 ng mm−2. During the lateral flow, the DNA-UPT hybrids were immobilized at the test zone by the Dig-MαDig (hapten-antibody) binding and the uncaptured UPTavidin reporters were immobilized at the control zone by biotin-avidin interaction (Fig. 1d). A standard UPT Reader (UPlink, Orasure Technologies, Inc., Bethlehem, PA 18015), adapted with a 980 nm infrared laser, excited the UPT particles and recorded the 550 nm emission, which corresponds to the green light spectrum of the particular UPT phosphor particles used here. The emission, through the 550 nm bandpass filter, was measured, amplified with a photo multiplier tube, and displayed in relative fluorescence units (RFU) as a function of position along the strip. The software filtered out interfering background signals and reported the peak position and the peak area.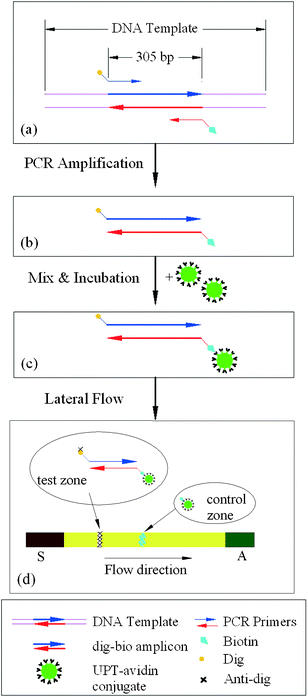 | ||
| Fig. 1 Protocol for DNA labelling and UPT lateral flow detection. (a) A primer pair with Dig and Bio hapten targets a 305 bp DNA fragment from the genomic DNA of B. cereus. (b) The resulting double-stranded PCR amplicons contain 5′-end Bio hapten on one strand and 5′-end Dig hapten on the opposite strand. (c) A Dig-DNA-Bio-avidin-UPT sandwich structure forms after mixing and incubating the amplicons with a UPT-avidin conjugate. (d) During the lateral flow, the DNA is captured by binding to the immobilized anti-Dig at the test line and the uncaptured UPT particles bind to the immobilized avidin at the control line. A signal is generated by exciting the UPT reporter particles. | ||
Reagents, chemicals and protocols
B. cereus (a Gram-positive bacterium) was used as the model organism for the pathogen detection assays. The bacterial cells were lysed, and the genomic DNA was isolated using QIAGEN DNeasy™ Tissue Kit (QIAGEN Inc., Valencia, CA 91355). The DNA concentrations were determined using fluorimetric assays in a Turner Biosystem (Sunnyvale, CA) TBS-380 fluorimeter with PicoGreen™ dye that selectively binds to double-stranded DNA. The B. cereus genomic DNA (5,224,283 bp30) was used as the PCR template. The PCR reagents were 50 mM Tris-HCl (pH 9.0), 20 mM (NH4)2SO4, 1.5–3.5 mM MgCl2, 200 µM dNTP, and 0.1 µg µL−1 BSA. The forward and reverse primers (0.3 µM) were, respectively, 5′-TCT CGC TTC ACT ATT CCC AAG T-3′ and 5′-AAG GTT CAA AAG ATG GTA TTC AGG-3′ (Operon Biotechnologies, Inc., Huntsville, AL). At the 5′-ends, the former and reverse primers were, respectively, tagged with Biotin (Bio) and Digoxinenin (Dig) hapten. The primers targeted a specific 305 bp gene fragment. The concentration of Taq DNA polymerase (Eppendorf AG, Hamburg, Germany) was 0.15 unit µL−1.The on-chip PCR protocol was initiated with a denaturation step at 95 °C for 120 s, followed by 25 amplification cycles (95 °C, 15 s; 55 °C, 25 s; 72 °C, 20 s), and terminated with an extension step at 72 °C for another 120 s. The total time for the on-chip PCR was 29 min. In parallel, control runs were carried out in a standard benchtop PCR thermocycler (TC-312, Techne Incorporated, Princeton NJ) with 10 µL sample-reagents volume. The benchtop PCR protocol used the same conditions for the initial denaturation and the final extension steps, but they used different cycling times due to the slower cooling rate inherent in the benchtop PCR system. The benchtop cycle times were: 95 °C, 20 s; 55 °C, 30 s; and 72 °C, 23 s. The initial series of PCR products were analyzed by agarose gel (1%) electrophoresis, stained with ethidium bromide.
150 ng UPT-avidin conjugate were suspended in 25 µL lyosphere solution matrix (100 mM Tris pH 9.0, 100 mM NaCl, 23.4 g L−1 EGTA, 1% (v/v) Tetronic 904, 1 g L−1 NaN3, 1 g L−1 sodium polyphosphate, 0.5 g L−1 Na-casein, 10 g L−1 α-cyclodextrin, 5 g L−1 dextran sulfate (MW 500 k), 20% (w/v) sucrose). The suspension was freeze-dried in vacuum to form a lyosphere and stored at 4 °C. Before its introduction into the cassette, the lyosphere was suspended in 90 µL of LF buffer (10 mM Hepes pH 7.2, 135 mM NaCl, 0.5% (v/v) Tween-20, 1% (w/v) BSA). The suspension was then mixed (in the cassette) with 8 µL of PCR products.
Fabrication of microfluidic devices
The microfluidic devices (Fig. 2a) were made of polycarbonate (Ensinger Ltd., Pontyclun, UK) and machined with a computer numerical control (CNC) milling system (Fadal VMC15XT, CA 91311 USA). The cassette consisted of two polycarbonate plates having a 58 mm × 15 mm footprint. The conduits, incubation chamber, and PCR chamber were machined in a 0.8 mm thick polycarbonate plate. The conduits had square cross-sections with widths and depths ranging from 250 µm to 500 µm. The PCR and incubation chamber volumes were, respectively, 8 µL and 100 µL. In order to reduce the roughness of the milled chambers' surfaces, the conduit and chambers' inner surfaces were polished after machining. Wells for the hydrogel valves and the upper part of the incubation chamber were machined in a second 1.5 mm-thick polycarbonate plate. After machining and cleaning with isopropyl alcohol, the hydrogel material was inserted into the wells, and the plates were bonded together in a hot press machine (Twelve Ton Press No. 3850, CARVER Inc., IN). The thermal bonding temperature was 140 °C, and the press force was 300 lbs. After 50 min, prior to releasing the press force, the hot plates were cooled to ambient temperature. Due to the high bonding temperature, the pre-loaded hydrogel dried out. A 200 µm diameter Type K thermocouple for temperature monitoring was inserted into the PCR chamber with its tip nearly flush with the sidewall. The thermocouple was then sealed with epoxy (Torr Seal Equivalent, LDS Vacuum Products, Inc. FL). The compatibility of the Type K thermocouple with PCR had been pre-tested in a benchtop system.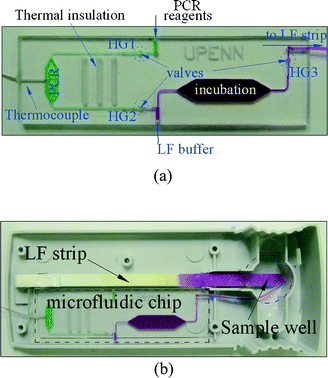 | ||
| Fig. 2 (a) A photograph of the microfluidic cassette comprising a PCR reactor, conduits, an incubation chamber and hydrogel valves. (b) A cartridge integrating the cassette (a) with the lateral flow strip. | ||
Instrumentation and data acquisition
The microfluidic cassette with the hydrogel microvalves was fitted into a 10 × 2.5 cm Uplink™ Reader Cartridge (Fig. 2b). A thermoelectric (T/E) module (Melcor Corp., NJ 08648-4587, USA) was set under the cassette for heating and cooling. A syringe pump KDS200 (KD Scientific Inc., Holliston MA) was connected to the cartridge through a manifold HGA010E1 (Humphrey Products Company, Kalamazoo, MI). The reagents were pre-loaded into the teflon tubes and pneumatically driven into the cassette. The T/E heaters, DC power supply (HP 6032A, Agilent Technologies, Inc., Palo Alto, CA), heavy duty relay module (ELK-912B, ELK Products, Inc., Hildebran, NC) and the syringe pump were controlled by a computer through an HP 3862A multi-channel multiplexer (Agilent Technologies, Inc., Palo Alto, CA). The pump motion, optical signal reading, and data acquisition were controlled by a custom-written LabVIEW™ program (National Instruments, Austin, TX) through a GPIB interface. A proportional-integral-differential (PID) algorithm was used for temperature control.Results and discussion
Properties of hydrogel valves
The hydrogel microvalves were composed of a flow conduit, a cavity, and a hydrogel plug. The top (left column) and side (right column) views of the hydrogel valve are depicted schematically in Fig. 3. The width, w and depth, h of the well that housed the hydrogel valve were larger than the corresponding dimensions of the conduit and varied from 500 µm to 750 µm. When the hydrogel was inserted into the well, it adhered to the floor of the well.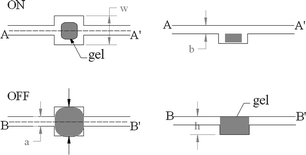 | ||
| Fig. 3 A cartoon depicting the hydrogel valve's volume changes as a function of the temperature. The top (A) and bottom (B) rows correspond, respectively, to temperatures above and below the critical (phase change) temperature. The left and right columns are, respectively, top and side views of the valve. | ||
Fig. 3 also depicts schematically the operation of the hydrogel valve. When the hydrogel valve was dry or as long as its temperature was above the critical (phase transition) temperature, Tc (32 °C), the hydrogel remained in the de-swelling (unexpanded) state, and the conduit was open to flow (top row in Fig. 3). When the temperature was decreased below the critical temperature, Tc in the presence of aqueous solution, the hydrogel swelled and blocked the flow (bottom row). For a more detailed description of the hydrogel valve fabrication, operation, and applications see Ref 31.
The hydrogel valves were tested prior to their use in the cassette. The water was supplied from an elevated reservoir, which was kept at constant pressure during the test. The pressure drop across the valve was controlled by adjusting the reservoir’s elevation. The water was discharged at atmospheric pressure. We subjected the valve to repetitive cycles of heating and cooling and measured the flow rate during each cycle. Fig. 4 depicts the valve's temperature (circles) and the flow rate (lines) through the valve as functions of time, when the conduit's dimensions are a = b = 500 µm and the valve chamber's dimensions are 1 mm × 1 mm × 1 mm. The valve's opening and closing times were, respectively, 6 s and 5 s. In the open state, a flow rate of 84 µL min−1 was measured by weighting the accumulated water collected when the pressure drop across the valve was 6 kPa. The closed valve did not exhibit any visible leakage up to a pressure of 200 kPa. We did not test the hydrogel at higher pressures due to the tendency of the polycarbonate plates that comprised the microfluidic chip to delaminate at higher pressures.
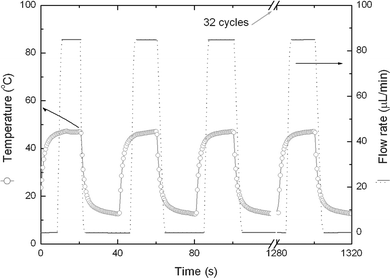 | ||
| Fig. 4 The flow rate (solid-dashed line) through and the temperature of (hollow circles) the valve are depicted as functions of time when the valve is subjected to alternating heating and cooling. | ||
PCR thermal cycling
Compared with silicon or glass, polycarbonate (PC) has a relatively low thermal conductivity. In order to achieve fast thermal cycling, the thickness of the PCR chamber wall that was in contact with the thermoelectric unit was 0.25 mm. A problem that is often experienced with stationary PCR micro-reactors is bubble formation. The bubbles may expel the reagents from the PCR chamber, adversely affect temperature uniformity and measurement, and reduce amplification efficiency. One means to suppress or eliminate the bubble formation is to pressurize the PCR chamber by sealing it. Here, we use the hydrogel valves for this purpose.The thermoelectric unit was used for both heating and cooling of the PCR chamber. We experimented with several heating and cooling protocols. Cooling from 95 °C to 55.5 °C through interaction with the ambient (∼25 °C) took about 30 s. The use of an external 14 W cooling fan during the cooling step reduced the cooling time to ∼20 s. The combined use of a cooling fan and a thermoelectric cooler reduced the cooling time to 15 s.
Fig. 5 depicts the PCR microreactor's temperature as a function of time. The temperature was measured at the inner wall of the reactor chamber. The temperature trace in the time interval from 600 s to 660 s is shown in Fig. 5b in enlarged form. In general, the heating time from room temperature to 95 °C was 14 s at a heating rate of 5 °C s−1. The cooling rate was about 2.6 °C s−1. Both the heating and cooling rates were faster than the ones of the benchtop thermal cycler (1 to 2 °C s−1). The time required for one complete cycle of denaturing, annealing, and extension was about 30 s without counting the dwelling time (7–15 s) at each temperature.
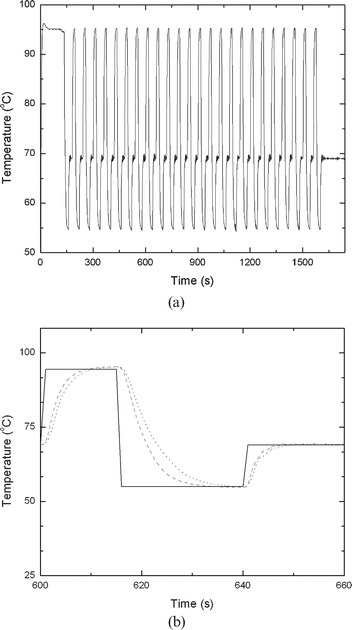 | ||
| Fig. 5 The PCR's temperature as a function of time (a). (b) is an enlargement of a segment of (a) spanning the time interval from 600 to 660 s. The solid, dotted, and dashed lines correspond, respectively, to the control temperature setting, the temperature variations when a cooling fan is used, and the temperature variations when both a fan and a thermoelectric unit are deployed. | ||
The actual PCR chamber volumes were ∼8 µL, slightly smaller than the design volume (10 µL) due to the deformation of the polycarbonate substrate during the thermal bonding. The PCR reagents as well as the target DNA were introduced into the PCR chamber while both the upstream (inlet, HG1 in Fig. 2) and downstream (outlet, HG2 in Fig. 2) hydrogel valves were open. During this process, the inlet valve was maintained well above the transition temperature, which allowed unhindered passage of the liquid and PCR mixture. The liquid filled the PCR chamber, displacing the air through the open exit hydrogel valve. At this stage, the exit valve was dry and allowed free passage of air. Once the liquid front arrived at the exit valve, the hydrogel swelled and blocked the passage of the liquid. Subsequently, the inlet hydrogel valve was allowed to cool to below the transition temperature, sealing the chamber. Since, at this instant, the sample was stationary, the precise closing time of the upstream valve was not critical, and the valve's actuation could therefore be programmed in an open-loop control mode. Once both the upstream and downstream valves were closed, the temperature of the PCR reactor was cycled according to the PCR protocol described earlier. Since the temperatures of the thermal cycling were much higher than the hydrogel valves' transition temperature, three slots were machined into the polycarbonate between the hydrogel valves and the PCR chamber to thermally isolate the valves from the PCR heating.
Although in principle the polycarbonate cassettes can be reused, to avoid possible cross-contamination, a virgin cassette was used for each investigation. Both positive and negative (no DNA) tests for the PCR reactor, as well as positive and negative benchtop PCR controls, were included in the experiments. Results of the gel electrophoresis of the PCR products (EtBr-stained 1% agarose gel) are shown in Fig. 6. The left lane is the marker. The 305 bp amplicon's position is indicated with an arrow. The lanes labelled “+” show the results for on-chip and benchtop control amplifications when the sample included 10 ng DNA template. The lanes marked “−” correspond to cases when the DNA template was absent. In the negative tests, the PCR mixture contained all the necessary ingredients except for the DNA templates. The lanes labelled “on-chip” correspond to the PCR products obtained from the cassette device, and the lanes labelled “benchtop” correspond to the commercial thermal cycler's products. The cassette device exhibited specific amplification of the 305 bp target.
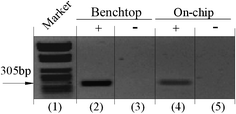 | ||
| Fig. 6 Agarose gel (1%) electrophoresis images of PCR products. (+) and (−) correspond, respectively, to actual tests and to negative controls. Lane 1 is a marker; lanes 2 and 3 are benchtop results; and lanes 4 and 5 are results of on-chip amplification. | ||
Flow control and integration
Once the individual components had been thoroughly tested, experiments were carried out with the integrated cassette. In describing the process steps, we refer to Fig. 2a. In order to improve image visibility, the chambers and the conduits in the figure were filled with dye.A sample of oral fluid spiked with DNA and mixed with the PCR reagents was introduced into the cassette. The mixture was pneumatically propelled into the PCR chamber. During this process, the hydrogel valve (HG1) upstream of the PCR chamber was maintained at an elevated temperature, and it allowed the passage of the mixture into the PCR reactor. The downstream valve (HG2) remained unheated. When the mixture arrived at the downstream valve (HG2), the valve swelled and blocked the passage. After a pre-determined time-interval, the heating of the upstream valve (HG1) was turned off, and the valve sealed the conduit. Since at this stage the liquid was stationary, accurate timing of the upstream valve was not necessary.
Subsequent to the completion of the PCR thermal cycling, the PCR chamber's heater was turned off, the PCR products cooled down to the ambient temperature, and both the inlet (HG1) and outlet (HG2) valves were opened by heating them to above their transition temperatures.
While the valves were heated and the PCR products were retained in the PCR chamber, the incubation-chamber was pre-cooled to −10 °C with a thermoelectric unit. Then, the PCR products were pneumatically propelled into the cold incubation chamber. Upon their arrival into the incubation chamber, the PCR products froze at the bottom of the chamber (Fig. 7a). The heating to the valve HG2 was terminated. Subsequently, the pneumatically-driven LF buffer, laden with the lyophilized UPT particle, was propelled into the cassette. When the liquid arrived at the location of the valve HG2, the valve swelled and blocked the passage. The LF-buffer then entered the upper part of the cooled incubation chamber (Fig. 7b). During this process, the downstream hydrogel valve (HG3) was at room temperature and allowed free passage of the displaced air. Once the solution's front arrived at the hydrogel valve's (HG3) location, the valve swelled and closed the passage. Next, the thermoelectric unit's polarity was switched from cooling to heating, and the unit increased the incubation chamber's temperature to 37 °C. The labelled DNA mixed and hybridized with the UPT-avidin conjugate. After 30 min of incubation, the hydrogel valve (HG3) downstream of the incubation chamber was heated and opened. The PCR amplicons with the bound UPT particles were propelled out of the incubation chamber and blotted onto the LF nitrocellulose strip. The loading pad at the upstream end of the LF strip absorbed the aqueous solution of the labeled amplicons. The mixture then progressed downstream along the LF strip by the action of capillary forces engendered by the pore structure of the nitrocellulose. The amplicons containing Dig-hapten were captured at the pre-printed ‘test line’ on the nitrocellulose strip where mouse-αDig antibodies were immobilized. The amount of bound avidin-UPT-Bio-DNA-Dig complex at the test line was indicative of the amount of PCR products. UPTavidin reporters that passed through the ‘test line’ were subsequently captured at the ‘control line’ by bio-avidin interaction.
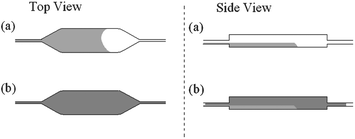 | ||
| Fig. 7 A cartoon of the freeze-based fusion of the PCR products with the lateral flow buffer in the incubation chamber. The first and second columns provide, respectively, top and side views. (a) The PCR products enter the pre-chilled incubation chamber and freeze. (b) The lateral flow buffer, laden with reporter particles, invades the volume above the frozen PCR products. | ||
The UPlink™ cartridge was then inserted into an UPlink™ reader equipped with a 980 nm IR laser exciter (1.2 W, Spectra Physics/Opt-Power Corp.). The IR light was guided with a fibre bundle through a focusing lens. The same fibre bundle collected the light that was emitted by the IR-excited, UPT particles. The emission signal was guided through a 550 nm band-pass filter appropriate for the emission wavelength of the UPT particles. The signal was then processed by a photomultiplier and was reported as RFU. To facilitate the scanning of the lateral flow strip, the reader was equipped with micro-stepper motors for position control. The UPlink™ system's software controlled the scanner movement, laser excitation, signal reception, and data collection, processing and display. The scanning was performed with 3 readings s−1 and the scan step was 0.2 mm. The strip was 45 mm long and scanning lasted 75 s.32,33
Fig. 8a depicts the detected fluorescence intensity in RFU as a function of the position along the strip when the sample contained 10 ng of DNA template. The signal resulting from the sample processed in the microfluidic cassette (Fig. 8c) is compared with a signal obtained with a benchtop PCR and incubation carried out in a water bath (Fig. 8b). The peaks' positions are identical. The peak area of the on-chip signal is slightly smaller than the benchtop result. We surmise that this lower on-chip PCR efficiency resulted from the presence of the epoxy-sealed thermocouple in the PCR chamber.
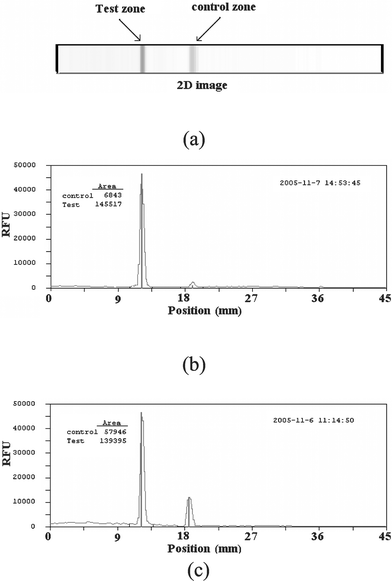 | ||
| Fig. 8 Illustration of the signal read from the lateral flow strip (a). The signal as a function of position along the strip when the sample was benchtop (b) and on-chip processed (c). The sample contained 10 ng of DNA. The signal is measured in relative fluorescent units (RFU). | ||
A second batch of twelve microfluidic devices without a thermocouple in the PCR chamber was fabricated and tested. The PCR thermal cycling was conducted in an open-loop control mode. Using the first batch of devices equipped with a thermocouple in the PCR chamber and operating in a closed-loop (feedback) control mode, we recorded the power supplied to the thermoelectric unit as a function of time. During the experiments with the second batch of devices, the power supply to the thermoelectric unit was pre-programmed using the recorded instruction sequence.
A series of samples containing 0, 1, 10, and 100 ng of B. cereus DNA template were PCR amplified on the cassette and the benchtop cycler. Fig. 9 charts the areas under the signal (in RFU) as functions of the initial target DNA amount in the sample for on-chip (hollow bars) and benchtop (shadowed bars) processing. Each bar represents the average of three independent measurements with three different cassettes. The scatter of the data is indicated with error bars. The cassette performance was nearly identical to that of the bench-top procedure. The magnitude of the signal decreased as the initial target DNA concentration decreased. Unfortunately, the negative test (0 ng DNA) resulted in a non-zero signal. This non-zero signal at 0 ng DNA is apparently caused by binding of PCR-artefacts (e.g. primer-dimers), and it may be reduced or eliminated by a better choice of primers. Thus, the test results indicate the presence of target analyte only when the signal is above a certain threshold.
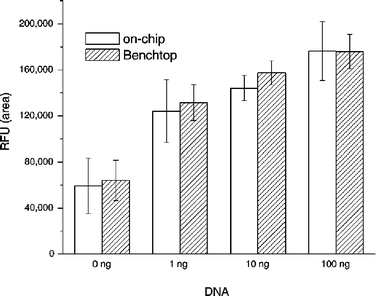 | ||
| Fig. 9 The detected signal's magnitude as a function of DNA concentration. Hollow and shaded bars correspond, respectively, to on-chip and benchtop experiments. The signal is measured in RFU. | ||
Fig. 10 depicts the peak areas at the test and control zones (in RFU) as functions of the initial amount of target DNA: 0, 1, 10, and 100 ng for the on-chip experiments. As the DNA concentration decreases, the test signal decreases and the control signal increases. This is not surprising since as the DNA concentration decreases, more reporter particles are available to migrate to the control zone.
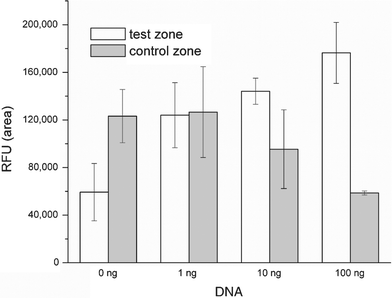 | ||
| Fig. 10 The detected on-chip signal magnitude as a function of DNA concentration. Hollow and filled bars correspond, respectively, to test zone and flow control zone. The signal is measured in RFU. | ||
Finally, Fig. 11 depicts the ratio of the test and the control signals. As the DNA concentration decreases so does the ratio. When the DNA concentration is 100 and 0 ng, the ratio is, respectively, 3 and 0.5. The bench-top data (not shown here) exhibited similar trends.
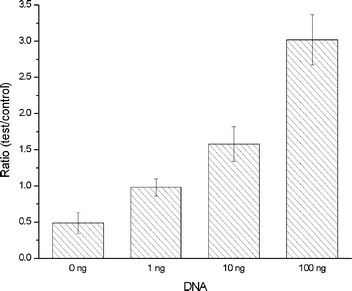 | ||
| Fig. 11 The ratio of the test and the control signals for on-chip experiments. The signal is measured in RFU. | ||
Conclusions
A disposable, inexpensive microfluidic cassette for nucleic acid amplification and integrated detection was constructed and tested. The PCR thermal cycling was realized with a thermoelectric module taking advantage of both the module's cooling and heating capabilities. With this strategy, the cooling speed from 95 °C to 55 °C was increased to 2.67 °C s−1 as compared to 1.6 °C s−1 in the absence of the thermoelectric unit. This cooling ramp improvement may increase PCR efficiency.34Flow control was realized with temperature-sensitive hydrogel valves. The hydrogel valves were also used to seal the PCR reactor during thermal cycling and to suppress bubble formation. The hydrogel valves operated with good reproducibility and without any visible deterioration in performance over many cycles. The valves could withstand pressures higher than 200 kPa without any noticeable leakage. The valve response time was less than 6 s, and the valves did not appear to significantly adsorb nucleic acids and enzymes.
The UPT reporter particles and lateral flow nitrocellulose strip were used to detect the PCR amplicons. The reporters were nano-crystalline phosphorescence particles, excited with infrared light and up-convert absorbed photon energy to emit visible light. The UPT particles do not bleach, and they provide a permanent record.
Experiments conducted with the microfluidic device demonstrated sensitivity comparable to results obtained in benchtop experiments. To further improve sensitivity, it would be necessary to reduce formation of PCR-artefacts. Utilizing benchtop UPT-LF format, detection down to 106 DNA amplicons has been demonstrated32 with UPT-conjugates and LF strips different than the ones that we used here. We hypothesize that a similar level of sensitivity can also be achieved in the microfluidic format with appropriate optimization.
Although most of the experiments consisted of samples of purified DNA diluted in sterile fluid, a few experiments in which the DNA was spiked into fresh saliva contributed by some of the authors were carried out. Significantly, the saliva did not inhibit PCR and it was possible to detect the presence of DNA in the saliva without a need for a purification step.
The microfluidic device reported here will be used as the downstream end of a more comprehensive, disposable, diagnostic system for non-invasive, rapid detection of pathogens at the point of care. The above platform will also include cell lysis and DNA isolation and purification. The microfluidic device described here can also be used as a standalone device for the detection of free DNA in body fluids. The work demonstrates the feasibility of enhancing the capabilities of lateral flow assays by integrating the lateral flow strip with upstream microfluidic sample processing.
Acknowledgements
The authors thank Dr. S. Yang (University of Pennsylvania), Dr. D. Malamud, Dr. W. R. Abrams, Ms. C. Davis, Mr. G. Tong, and Mr. F. Winslow (David B. Kriser Dental Center, New York University) for helpful discussions. OraSure Technologies Inc. supplied us with the UPlink reader and materials. This work was supported by NIH grant U01DE014964.References
- E. T. Lagally, J. R. Scherer, R. G. Blazej, N. M. Toriello, B. A. Diep, M. Ramchandani, G. F. Sensabaugh, L. W. Riley and R. A. Mathies, Anal. Chem., 2004, 76(11), 3162–3170 CrossRef CAS.
- R. A. Zangmeister and M. J. Tarlov, Anal. Chem., 2004, 76(13), 3655–3659 CrossRef CAS.
- M. A. Burns, B. N. Johnson, S. N. Brahmasandra, K. Handique, J. R. Webster, M. Krishnan, T. S. Sammarco, P. M. Man, D. Jones, D. Heldsinger, C. H. Mastrangelo and D. T. Burke, Science, 1998, 5388, 282, 484–487 Search PubMed.
- R. C. Anderson, X. Su, G. J. Bogdan and J. Fenton, Nucleic Acids Res., 2000, 28(12), E60–e60 CrossRef CAS.
- E. T. Lagally, C. A. Emrich and R. A. Mathies, Lab Chip, 2001, 1(2), 102–107 RSC.
- J. W. Hong, T. Fujii, M. Seki, T. Yamamoto and I. Endo, Electrophoresis, 2001, 22(2), 328–333 CrossRef CAS.
- D. J. Sadler, R. Changrani, P. Roberts, C. F. Chou and F. Zenhausern, IEEE Trans. Compon. Packag. Technol., 2003, 26(2), 309–316 CrossRef CAS.
- Y. J. Liu, C. B. Rauch, R. L. Stevens, R. Lenigk, J. N. Yang, D. B. Rhine and P. Grodzinski, Anal. Chem., 2002, 74(13), 3063–3070 CrossRef CAS.
- C. G. Koh, W. Tan, M. Q. Zhao, A. J. Ricco and Z. H. Fan, Anal. Chem., 2003, 75(17), 4591–4598 CrossRef CAS.
- J. Khandurina, T. E. McKnight, S. C. Jacobson, L. C. Waters, R. S. Foote and J. M. Ramsey, Anal. Chem., 2000, 72(13), 2995–3000 CrossRef CAS.
- E. T. Lagally, P. C. Simpson and R. A. Mathies, Sens. Actuators, B, 2000, 63(3), 138–146 CrossRef.
- D. Trau, T. M. H. Lee, A. I. K. Lao, R. Lenigk, I. M. Hsing, N. Y. Ip, M. C. Carles and N. J. Sucher, Anal. Chem., 2002, 74(13), 3168–3173 CrossRef CAS.
- J. N. Yang, Y. J. Liu, C. B. Rauch, R. L. Stevens, R. H. Liu, R. Lenigk and P. Grodzinski, Lab Chip, 2002, 2(4), 179–187 RSC.
- S. J. Park, T. A. Taton and C. A. Mirkin, Science, 2002, 5559, 295, 1503–1506 Search PubMed.
- W. C. W. Chan and S. M. Nie, Science, 1998, 5385, 281, 2016–2018 Search PubMed.
- D. A. Zarling, M.J. Rossi, N. A. Peppers, J. Kane, G. W. Faris and M. J. Dyer, US Patent, 5 674 698, 1997 Search PubMed.
- H. J. Zijlmans, J. Bonnet, J. Burton, K. Kardos, T. Vail and R. S. Niedbala, Anal. Biochem., 1999, 267(1), 30–36 CrossRef CAS.
- J. Hampl, M. Hall, N. A. Mufti, Y. M. M. Yao, D. B. MacQueen, W. H. Wright and D. E. Cooper, Anal. Biochem., 2001, 288(2), 176–187 CrossRef CAS.
- P. Corstjens, M. Zuiderwijk, A. Brink, S. Li, H. Feindt, R. S. Neidbala and H. Tanke, Clin. Chem., 2001, 47(10), 1885–1893 CAS.
- R. S. Niedbala, T. L. Vail, H. Feindt, S. Li and J. L. Burton, Proc. SPIE–Int. Soc. Opt. Eng., 2000, 3913, 193–203 CAS.
- F. Rijke, H. Zijlmans, S. Li, T. Vail, A. K. Raap, R. S. Niedbala and H. J. Tanke, Nat. Biotechnol., 2001, 19, 273–276 CrossRef.
- H. L. Smits, C. K. Eapen, S. Sugathan, M. Kuriakose, M. H. Gasem, C. Yersin, D. Sasaki, B. Pujianto, M. Vestering, T. H. Abdoel and G. C. Gussenhoven, Clin. Diagn. Lab. Immunol., 2001, 8(1), 166–169 CrossRef CAS.
- S. P. Johnston, M. M. Ballard, M. J. Beach, L. Causer and P. P. Wilkins, J. Clin. Microbiol., 2003, 41(2), 623–626 CrossRef.
- F. Ketema, C. Zeh, D. C. Edelman, R. Saville and N. T. Constantine, J. Acquired Immune Defic. Syndr., 2001, 27(1), 63–70 Search PubMed.
- A. Salomone and P. Roggero, J. Plant Pathol., 2002, 84(1), 65–68 Search PubMed.
- C. Barrett, C. Good and C. Moore, Forensic Sci. Int., 2001, 122(2–3), 163–166 CrossRef CAS.
- S. Z. Qian and H. H. Bau, Anal. Biochem., 2003, 322(1), 89–98 CrossRef CAS.
- S. Z. Qian and H. H. Bau, Anal. Biochem., 2004, 326(2), 211–224 CrossRef CAS.
- D. Malamud, H. Bau, S. Niedbala and P. Corstjens, Adv. Dent. Res., 2005, 18, 12–16 Search PubMed.
- D. A. Rasko, J. Ravel, O. A. Okstad, E. Helgason, R. Z. Cer, L. X. Jiang, K. A. Shores, D. E. Fouts, N. J. Tourasse, S. V. Angiuoli, J. Kolonay, W. C. Nelson, A. B. Kojsto, C. M. Fraser and T. D. Read, Nucleic Acids Res., 2004, 32(3), 977–988 CrossRef CAS.
- J. Wang, Z. Chen, M. Mauk, K. Hong, M. Li, S. Yang and H. H. Bau, Biomed. Microdevices, 2005, 7(4), 313–322.
- P. L. A. M. Corstjens, S. Li, M. Zuiderwijk, K. Kardos, W. R. Abrams, R. S. Niedbala and H. J. Tanke, IEE Proc. Nanobiotechnol., 2005, 152(2), 64–72 Search PubMed.
- R. S. Niedbala, H. Feindt, K. Kardos, T. Vail, J. Burton, B. Bielska, S. Li, D. Milunic, P. Bourdelle and R. Vallejo, Anal. Biochem., 2001, 293, 22–30 CrossRef CAS.
- J. Y. Lee, H. Lim, S. Yoo, B. Zhang and T. Park, Biochem. Eng. J., 2005 DOI:10.1016/j.bej.2005.02.023.
| This journal is © The Royal Society of Chemistry 2006 |