Soft lithographic patterning of supported lipid bilayers onto a surface and inside microfluidic channels
Pilnam
Kim
a,
Sang Eun
Lee
b,
Ho Sup
Jung
b,
Hea Yeon
Lee
*bc,
Tomoji
Kawai
b and
Kahp Y.
Suh
*a
aSchool of Mechanical and Aerospace Engineering, Seoul National University, Seoul 151-742, Korea. E-mail: hylee@sanken.osaka-u.ac.jp; sky4u@snu.ac.kr
bThe Institute of Scientific and Industrial Research, Osaka University, Osaka 567-0047, Japan
cCore Research for Evolutional Science and Technology (CREST), Japan Science and Technology Corporation, Honcho, Kawanguchi, 332-0012 Saitama, Japan
First published on 24th November 2005
Abstract
We present simple soft lithographic methods for patterning supported lipid bilayer (SLB) membranes onto a surface and inside microfluidic channels. Micropatterns of polyethylene glycol (PEG)-based polymers were fabricated on glass substrates by microcontact printing or capillary moulding. The patterned PEG surfaces have shown 97 ± 0.5% reduction in lipid adsorption onto two dimensional surfaces and 95 ± 1.2% reduction inside microfluidic channels in comparison to glass control. Atomic force microscopy measurements indicated that the deposition of lipid vesicles led to the formation of SLB membranes by vesicle fusion due to hydrophilic interactions with the exposed substrate. Furthermore, the functionality of the patterned SLBs was tested by measuring the binding interactions between biotin (ligand)-labeled lipid bilayer and streptavidin (receptor). SLB arrays were fabricated with spatial resolution down to ∼500 nm on flat substrate and ∼1 µm inside microfluidic channels, respectively.
Introduction
Recently, supported lipid bilayer (SLB) membranes have attracted considerable attention as a biomimetic platform for various applications such as fundamental biological research of cell-membranes, lipid-assisted bioassays and biosensors.1,2 In particular, the micropatterning of SLBs has been used to study cell behavior on controlled surfaces, and to fabricate lipid-assisted biochips such as DNA-chips, and to study lipid activity kinetics.1,3,4 Various methods have been developed for the fabrication of lipid arrays such as microcontact printing,5–7 photolithography,8–11 polymer lift-off,12,13 modification of surface by UV exposure,14,15 electron beam lithography,16 and scanning probe lithography.17,18 Most of these methods utilized physical or chemical modification to the surface for restricting the diffusion of lipids (so called “self healing”) since the fluidity is an intrinsic nature of SLBs in cell membrane.19Micro/nanopatterning of SLBs within microfluidic devices is a prerequisite for the development of high-throughput biosensors and for performing chip-based studies of cellular interactions based on lipid bilayers. A number of strategies were demonstrated to pattern lipid bilayers inside fluidic channels such as microfluidic flow patterning20 and polymer lift-off.13 Microfluidic flow patterning, which utilizes laminar flowing streams to pattern within microfluidic channels, is a powerful method to obtain microarrays with varying composition but is limited to generating geometrical patterns in the shape of the laminarly flowing streams with pattern sizes on the order of a few tens of micrometres. Polymer lift-off is also an elegant way to fabricate micropatterned SLBs in a controlled fashion but has limitations due to the potential toxicity of the photoinitiator,21 the need for expensive equipment and the difficulty in patterning the surface without modifying the surface topography. Thus, the development of simple and economically viable method for patterning flat substrates and inside microfluidic channels with pattern size ranging from a few tens of micrometres to less than a micrometre is potentially of great benefit.
More recently, a simple technique, applicable to many soft lithographic methods, has been presented to create patterned microchannels with precise control over the spatial properties of the substrate.22,23 In this method, polyethylene glycol (PEG) copolymer microstructures were fabricated within microfluidic channels to deposit proteins or cells onto pre-defined locations. Due to the excellent non-biofouling properties of PEG copolymers, well-defined microarrays of proteins or cells were achieved with spatial resolution down to a few tens of micrometres. We found that this approach can be directly applied to patterning of SLBs inside microfluidic channels with slight modifications to the protocol. The overall process consists of three steps: generation of microstructures of PEG copolymer onto glass substrate, irreversible sealing of a microfluidic channel using oxygen plasma treatment with manual alignment, and continuous flow of a solution containing lipid vesicles followed by flow of targeting probes if necessary. The patterned PEG layers provided excellent resistance to non-specific adhesion of lipid vesicles as well as to the diffusion of adsorbed SLB membranes.
In this study, two soft lithographic methods were used to fabricate micropatterns: microcontact printing24 and capillary moulding.25 The microcontact printed patterns were formed by transferring the polymer from the PDMS mould to the substrate by direct contact. A thin layer of the PEG-based comb polymer was deposited on the plasma-cleaned PDMS mould and the pattern was subsequently transferred to the mould by slightly pressing the mould onto the substrate. In the capillary moulding method a thin film was prepared by drop dispensing onto the substrate, and a patterned PDMS mould was subsequently brought into conformal contact with the surface and left undisturbed until dried. The moulding occurred as a result of capillary depression within the void spaces (i.e. repulsion of the hydrophilic polymer solution from the PDMS mould) as well as the hydrodynamic forces at the contact regions.25,26 Therefore, a thin film remained at the contact regions while the void regions dewetted from the surface to expose the substrate. Both methods were shown to be effective in exposing the substrate surface with various heights and spatial resolutions as described below.
Methods and materials
Materials
Polydimethyl siloxane (PDMS) elastomer composed of prepolymer and curing agent was purchased from Dow Corning (Sylgard 184). For liposomes, 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC), 1,2-dimyristoyl-sn-glycero-3-phosphoethanolamine N-(Cap biotinyl)(biotinyl Cap-PE) were purchased from Avanti Polar Lipids Inc. (Alabaster, AL). DiI (1,1′-Dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate) was purchased from Aldrich Chem. Streptavidin, Alexa 488 conjugate was purchased from Molecular Probes. Two kinds of PEG-based copolymers were used: a PEG-based random copolymer, poly((3-trimethoxysilyl)propyl methacrylate-r-poly(ethylene glycol) methyl ether methacrylate) (poly(TMSMA-r-PEGMA))27 and a methacrylate-based comb polymer containing pendant oligoethylene glycol side chains.28 The PEG comb polymer was kindly supplied by Dr Jinho Hyun at Seoul National University.Liposome preparation and labeling
The lipid vesicles were prepared by an extrusion method.29,30 POPC, biotinyl Cap-PE, 1-octadecanethiol were dissolved in chloroform at a molar ratio of 10 ∶ 1 ∶ 0.1. The mixture was evaporated under nitrogen and dried in a desiccator under vacuum for 5 h. Multilamellar vesicles (MLVs) were obtained by resuspending the lipidic dried film in 100 mM phosphate-buffered saline solution (PBS, pH 7.4) at a 1mM final lipid concentration. Subsequently, five freeze–thaw cycles were applied and the lipid vesicles were repeatedly extruded through a polycarbonate film with 50 nm pores by use of an extruder device in order to produce lipid vesicles of uniform size. The lipid vesicle size was confirmed by the dynamic light scattering method (DLS-700 Ar; Otsuka Electronics Co., Ltd., Japan). After the formation, lipid vesicles were labeled with the fluorochrome DiI.PDMS mould fabrication and substrate pretreatment
To cure the PDMS prepolymer, a mixture of 10 ∶ 1 silicone elastomer and the curing agent was poured on the various etched silicon masters prepared by photolithography and subsequent dry etching and placed at 70 °C for 1 h. The masters used for microfluidics had protruding features with the impression of microfluidic channels (ranging from 400 µm to 600 µm in width and ∼60 µm in height). After curing, PDMS moulds were cleanly detached from the masters. Glass slides were prepared by washing in distilled water and cleaned by plasma for 1 min.Atomic force microscopy
Atomic Force Microscope, AFM measurements were performed using a commercial AFM (NanoScope IV MultiMode AFM, Veeco Metrology LLC, Santa Barbara, CA). The scan rate was 0.5 Hz and 256 lines were scanned per sample. Tapping mode tips, OMCL_AC240TM-B2 with spring constant 0.9–2.2 mN, were obtained from OLYMPUS (Japan). Data were processed using Nanoscope III 4.31r6 software (Veeco Instruments Inc.).Fabrication of the microstructures within microfluidic channels
To fabricate microstructures by capillary moulding, a few drops of 5% (w/v) PEG copolymer [poly(TMSMA-r-PEGMA)] solutions in water were placed on glass substrate. To make conformal contact, a patterned PDMS mould was carefully placed onto the surface and then the sample was stored overnight at room temperature to allow evaporation of water for complete evaporation of the solvent. To fabricate microstructures by microcontact printing, a patterned PDMS stamp was plasma cleaned for 1 min (60W, PDC-32G, Harrick Scientific Inc.) to ensure proper cleaning and to increase wettability. After pretreatment, the PDMS mould was inked with 1% (w/v) solution of the comb polymer in a 50 ∶ 50 (v/v) H2O/ethanol mixture and placed directly onto substrate. The stamp was left for 30 s and peeled off (Scheme 1). To complete the device fabrication both for capillary moulding and microcontact printing, a PDMS mould with the features of the microfluidic channel and a patterned glass slide were plasma cleaned for 45 s (60 W, PDC-32G) without disturbing the PDMS stamp used for patterning (i.e., in conformal contact with the substrate) (Scheme 1). After plasma treatment, the PDMS stamp was peeled off from the substrate and the microfluidic mould was aligned and brought in conformal contact with the substrate and firmly pressed to form an irreversible seal. Fluids were driven through the channels using a SP200i syringe pump (World Precision Instruments, Sarasota, FL) that was connected to the device using polyethylene tubing (BD, Franklin Lakes, NJ).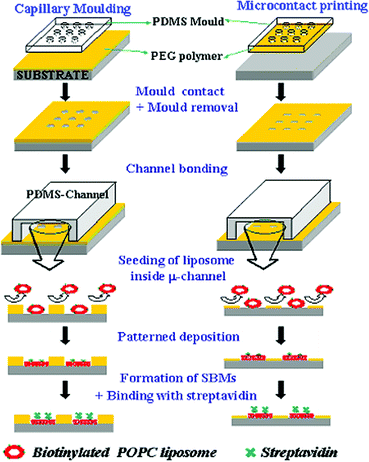 | ||
| Scheme 1 A schematic diagram for patterning supported bilayer membranes (SBMs) onto glass substrate and inside microfluidic channels either by using capillary moulding or microcontact printing with PEG-based polymers. | ||
Patterning of biotinylated lipid vesicles and biotin–streptavidin binding
A few drops of biotinylated liposome vesicles dissolved in PBS (pH = 7.4) at 100 mM were evenly distributed onto the patterned PEG substrates and incubated at room temperature for 40 min, and then the sample was rinsed thoroughly with PBS. After that, Alexa Fluor® 488 conjugated streptavidin dissolved in PBS (pH 7.4) at 50 µg mL−1 was stained onto the surface patterned lipid membrane at room temperature for 40 min and the sample was rinsed with PBS several times. To generate the lipid bilayer membrane micropatterns inside microfluidic channels, the solution of biotinylated lipid vesicles was seeded through the patterned microfluidic channel for 30 min at a flow rate of 5 µL min−1. For measuring biotin–streptavidin binding, a solution of streptavidin dissolved in PBS (pH 7.4) at 50 µg mL−1 was run through the channel for 45 min additively. All patterned surfaces were analyzed using an inverted fluorescent microscope (IX71, Olympus). All staining experiments were performed three to five times to ensure the reliability of the data. Fluorescent images were taken and quantified using Image-pro plus 5.1 (Olympus).Results and discussion
Patterning of lipids vesicles on glass substrate
To pattern lipid vesicles on glass substrate, PEG microstructures were fabricated either by using microcontact printing or capillary moulding methods (Scheme 1). For microcontact printing, a PDMS mould was inked with the methacrylate based comb polymer containing pendent oligoethylene glycol side chains. The microstructures formed by microcontact printing were reported to be very stable in water and could be exploited to spatially control adhesion and proliferation of biological species such as cells and proteins.28 For capillary moulding, a PDMS mould was placed onto a drop-dispensed solution of the PEG-based random copolymer [poly(TMSMA-r-PEGMA)]. The moulded PEG structures formed robust microstructures after the evaporation of the solvent. It was observed that the mobility of the two polymers was different on glass substrate; the comb polymer was not quite mobile while in contact with the substrate surface presumably due to the presence of methacrylate backbones that could be absorbed to the surface.31 As a result, the use of the comb polymer in capillary moulding was not successful. Similarly, the PEG copolymer was relatively mobile at the time of contact because of low viscosity such that the microcontact printing method was difficult to handle. For these reasons, the two polymers were used for different patterning methods.The three-dimensional and cross-sectional atomic force microscopy (AFM) images shown in Fig. 1(a–b) indicated that the substrate surface was completely exposed with good edge definition when patterned by contact printing. The height of the printed PEG layer was ∼13 nm which is higher than a few nanometres generally obtained for self-assembled monolayers due to high concentration and viscosity of the PEG comb polymer. The microstructures shown in Fig. 1(c–d) could also be generated with the clear surface exposure. The height of the microstructure was ∼294 nm, much higher than that for microcontact printing due to the fact that the capillary moulding involves a higher amount of the PEG copolymer.31 This increased height could act as a physical barrier to regulate the diffusion of adsorbed lipid bilayers as shown shortly.
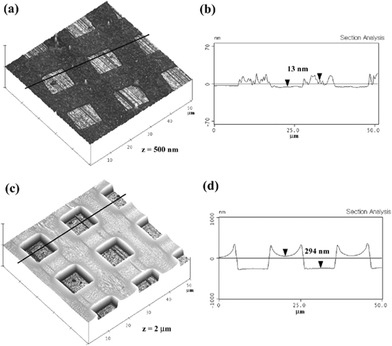 | ||
| Fig. 1 Three-dimensional and cross-sectional atomic force microscopy (AFM) images of the patterned PEG surfaces using microcontact printing (a), (b) and capillary moulding (c), (d). The box size was 10 µm. | ||
Next, we tested the ability of PEG microstructures to act as an adhesion-resistant layer and diffusion barrier of lipid vesicles. In previous research, PEG coatings have been used to minimize surface biofouling of extracellular matrix (ECM) proteins and to provide surfaces that are invisible to cells.32 This suggests that the adsorption of lipid vesicle would be significantly reduced on the PEG surfaces because lipid bilayer is a major component of cell membrane. Also, the adsorbed lipid vesicles would be converted to lipid bilayer membranes due to hydrophilic interactions with the exposed glass substrate.14 As shown in Fig. 2, lipid vesicles were selectively deposited onto the patterned surface by microcontact printing (Fig. 2(a)) and capillary moulding (Fig. 2(b)) with good fidelity. The box size was 10 µm. Although the selective deposition could be accomplished on a large area for all the PEG patterns tested, the lipid vesicles were sporadically adhered on the PEG layer in the case of microcontact printed PEG surfaces. There are two reasons for this. One is partial penetration of lipid vesicles into the thinner layer of the PEG comb polymer as seen from the cross-sectional AFM image in Fig. 1(b). As shown in the figure, the surface roughness was relatively high such that some regions of the substrate appeared to be nearly exposed. The other is diffusion of adsorbed lipid bilayers along the boundary between the glass and the PEG layer. As compared to microcontact printing, the moulded microstructures provided a clean interface between bare and glass substrate as shown in the inset of Fig. 2(b) and the adhesion on the PEG surface was strongly restricted. In Fig. 2, sub-micrometre patterns of SBMs were also presented along with fluorescent profiles using 500 nm lanes (c) and 500 nm grids (d). These patterns were neatly formed over a large area with partial interconnections and defects. The fluorescent intensities dropped to the background level on the exposed surface, suggesting that the current approach could offer the sub-micrometre patterning without modifying the experimental protocol.
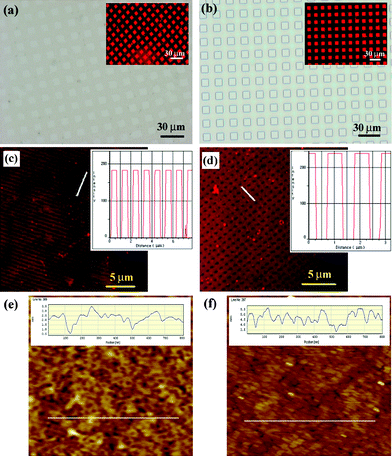 | ||
| Fig. 2 (a), (b) Optical images of the PEG patterns and fluorescent images of the biotinylated lipid layers (inset) using microcontact printing (a) and capillary moulding (b). The 10 µm box pattern was used for both methods. (c), (d) Fluorescent micrographs of the sub-micropatterned biotinylated SBMs along with intensity profiles using capillary moulding: (c) 500 nm lanes and (d) 500 nm grids. For grid pattern, lipid vesicles were adhered onto the matrix part since a negative PDMS mould was used. The inset shows the fluorescent intensity along the white line. (e), (f) AFM measurements of the roughness of glass substrate (e) before and (f) after liposome treatment. The scan area is 1 × 1 µm2. | ||
To verify the formation of lipid bilayer membranes, we measured the roughness of glass substrate before and after liposome treatment in Fig. 2(e),(f). The initial roughness of glass substrate was ∼2.3 nm, which was substantially reduced to ∼0.8 nm after liposome treatment. The normal size and height of an individual liposome aggregate were measured to be approximately 150 × 150 nm2 and 60 nm, respectively.16 The calculated volume is similar to that of the originally designed liposome used in this experiment. In Fig. 2(f), however, no such aggregates were found regardless of the slightly rough topography of glass substrate, suggesting that the lipid bilayer was formed instead of lipid vesicle.
We also measured the relative adsorption of lipid vesicles on bare glass and PEG surfaces by analyzing fluorescent intensities. The measurement showed that the surface covered with PEG copolymer provided 97 ± 0.5% reduction in lipid adsorption onto two dimensional surfaces and 95 ± 1.2% reduction inside microfluidic channels in comparison to glass control. The adsorbed amount was slighter higher for patterning inside microchannels probably due to limitations of mass transport in the washing step.
Two-dimensional micro/nano-patterning of SBMs is potentially useful in a number of bioassay devices.33,34 To test the functionality of patterned lipid bilayer membranes, lipid vesicles containing a biotinyl Cap-PE, were selectively adsorbed onto the patterned surface by capillary moulding. The presence of this biotin (ligand)-containing pattern was subsequently tested by studying their adhesion to Alexa 488-conjugated streptavidin (receptor). As shown in Fig. 3, using this approach micropatterns of various sizes and shapes, including 30 µm circles and 10 µm lanes, could be generated that were clearly visible under optical and fluorescence microscopes.
 | ||
| Fig. 3 (a), (d) Optical micrographs of the PEG patterned surfaces using 30 µm circles and 10 µm lanes, respectively. (b), (e) Fluorescent images of the patterned biotinylated SBMs after selective deposition onto the exposed regions. (c), (f) Fluorescent images of the same regions in (b), (e) after conjugation with Alexa Fluor® 488 streptavidin. | ||
Fabrication of microfluidic channels with patterned SLBs
Microfluidic devices with arrays of SLB membranes were created by irreversible sealing of a PDMS channel onto the pre-patterned glass substrate with proper alignment (Scheme 1). In this process, the covered mould during the moulding process was left for protecting the patterned surface during oxygen plasma treatment. After surface treatment by plasma, the covered PDMS mould was carefully detached from glass substrate, and the PDMS channel mould was subsequently bonded to the substrate in such a way that the patterned region was included in the channel.To demonstrate the ability of the microfluidic channels formed here to act as lipid based-bioassay and analytical tools, biotin–streptavidin bindings were analyzed using a fluorescence microscope. To analyze biotin–streptavidin binding within microfluidic devices, biotinylated lipid vesicles were labeled with the fluorochrome DiI, and Alexa 488-conjugated streptavidin was prepared as a receptor. As shown in Fig. 4(a)–(c), the lipid bilayer membrane was formed by fusion of patterned lipid vesicles onto pre-located regions of the substrate. Also, streptavidin was selectively deposited with the biotinylated lipid bilayer membrane (Fig. 4(d)–(f)), suggesting that the biotinylated lipid membrane could act as a platform for a wide range of applications such as bioassay-chips and biosensors using antigen–antibody interactions.
 | ||
| Fig. 4 (a)–(c) Fluorescent images of the patterned biotinylated SLBs using capillary moulding: (a) 70 µm wells, (b) 1 µm square strips, and (c) 5 µm boxes. (d)–(f) Fluorescent images of the same regions in (a)–(c) after conjugation with streptavidin. (g), (h) Fluorescent images of the patterned biotinylated SLBs using microcontact printing: (g) 10 µm lanes and (h) 70 µm wells. (i) A simple Y-shaped channel used in the experiment. | ||
The reason why the moulding process was mostly used to pattern inside microfluidic channels is to minimize the diffusion of adsorbed lipid layers. As shown in Fig. 4(g),(h), the adsorbed SLB membranes appeared to migrate across the boundary between the exposed surface and the adjacent microcontact printed PEG layer. In comparison, there was no significant diffusion of the lipid membrane when patterned by microcontact printing without flow, indicating that the destruction of the pattern was medicated by some flow-induced migration of the SLB membranes. On the contrary, the moulded microstructures were effective in restricting the diffusion of the lipid for ∼11 h in a continuous stream of streptavidin solution probably due to the increased height of the microstructures. After ∼11 h the lipid layer started to diffuse to the adjacent regions. The stability on a surface was almost the same as that within the microchannel, suggesting that the flow does not affect the stability of the SLB membranes substantially. In all the microfluidic experiments, a simple Y-shaped channel was used as shown in Fig. 4(i), which could be expanded other types of microchannels with proper handling.
Conclusions
We have presented two soft lithographic methods for patterning SLBs onto flat substrates and inside microfluidic channels. Microcontact printing and capillary moulding methods were used to create robust microstructures of the PEG-based polymers, which acted as resistant layers against non-specific adhesion of lipid vesicles. Both methods could be used to fabricate the patterned PEG surfaces with the substrate surface clearly exposed whereas the capillary moulding approach turned out to be more efficient in regulating the adhesion and migration of the lipid vesicles. It is hoped that this simple method provides an alternative platform for the fabrication of lipid-based immunoassay chips and a useful tool for research of lipid membrane within microfluidic devices applicable for high-throughput applications.Acknowledgements
This work was supported by the Micro Thermal System Research Center of Seoul National University and Core Research for Evolutional Science and Technology (CREST) of Japan Science and Technology Corporation. We thank Prof. Jinho Hyun for providing the PEG comb polymer and Dr Ali Khademhosseini for helpful discussions.References
- E. Sackmann, Science, 1996, 271, 43–48 CrossRef CAS.
- S. G. Boxer, Curr. Opin. Chem. Biol., 2000, 4, 704–709 CrossRef CAS.
- A. van Oudenaarden and S. G. Boxer, Science, 1999, 285, 1046–1048 CrossRef CAS.
- C. Xu, P. Taylor, P. D. I. Fletcher and V. N. Paunov, J. Mater. Chem., 2005, 15, 394–402 RSC.
- A. T. A. Jenkins, R. J. Bushby, N. Boden, S. D. Evans, P. F. Knowles, Q. Y. Liu, R. E. Miles and S. D. Ogier, Langmuir, 1998, 14, 4675–4678 CrossRef CAS.
- L. A. Kung, L. Kam, J. S. Hovis and S. G. Boxer, Langmuir, 2000, 16, 6773–6776 CrossRef CAS.
- M. P. Srinivasan, T. V. Ratto, P. Stroeve and M. L. Longo, Langmuir, 2001, 17, 7951–7954 CrossRef CAS.
- P. S. Cremer, J. T. Groves, L. A. Kung and S. G. Boxer, Langmuir, 1999, 15, 3893–3896 CrossRef CAS.
- R. N. Orth, J. Kameoka, W. R. Zipfel, B. Ilic, W. W. Webb, T. G. Clark and H. G. Craighead, Biophys. J., 2003, 85, 3066–3073 CrossRef CAS.
- L. A. Kung, J. T. Groves, N. Ulman and S. G. Boxer, Adv. Mater., 2000, 12, 731–734 CrossRef CAS.
- C. K. Yee, M. L. Amweg and A. N. Parikh, Adv. Mater., 2004, 16, 1184–1189 CrossRef CAS.
- R. N. Orth, M. Wu, D. A. Holowka, H. G. Craighead and B. A. Baird, Langmuir, 2003, 19, 1599–1605 CrossRef.
- J. M. Moran-Mirabal, J. B. Edel, G. D. Meyer, D. Throckmorton, A. K. Singh and H. G. Craighead, Biophys. J., 2005, 89, 296–305 CrossRef CAS.
- P. Lenz, C. M. Ajo-Franklin and S. G. Boxer, Langmuir, 2004, 20, 11092–11099 CrossRef CAS.
- V. I. Silin, H. Wieder, J. T. Woodward, G. Valincius, A. Offenhausser and A. L. Plant, J. Am. Chem. Soc., 2002, 124, 14676–14683 CrossRef CAS.
- J. M. Kim, H. S. Jung, J. W. Park, T. Yukimasa, H. Oka, H. Y. Lee and T. Kawai, J. Am. Chem. Soc., 2005, 127, 2358–2362 CrossRef CAS.
- J. W. Carlson, T. Bayburt and S. G. Sligar, Langmuir, 2000, 16, 3927–3931 CrossRef CAS.
- B. L. Jackson and J. T. Groves, J. Am. Chem. Soc., 2004, 126, 13878–13879 CrossRef CAS.
- J. T. Groves, N. Ulman and S. G. Boxer, Science, 1997, 275, 651–653 CrossRef.
- L. Kam and S. G. Boxer, J. Am. Chem. Soc., 2000, 122, 12901–12902 CrossRef CAS.
- V. A. Liu and S. N. Bhatia, Biomed. Microdev., 2002, 4, 257–266 Search PubMed.
- A. Khademhosseini, K. Y. Suh, S. Jon, G. Eng, J. Yeh, G. J. Chen and R. Langer, Anal. Chem., 2004, 76, 3675–3681 CrossRef CAS.
- A. Khademhosseini, J. Yeh, S. Jon, G. Eng, K. Y. Suh, J. A. Burdick and R. Langer, Lab Chip, 2004, 4, 425–430 RSC.
- Y. N. Xia, J. A. Rogers, K. E. Paul and G. M. Whitesides, Chem. Rev., 1999, 99, 1823–1848 CrossRef CAS.
- K. Y. Suh, A. Khademhosseini, J. M. Yang, G. Eng and R. Langer, Adv. Mater., 2004, 16, 584–588 CrossRef CAS.
- K. Y. Suh and R. Langer, Appl. Phys. Lett., 2003, 83, 1668–1670 CrossRef CAS.
- S. Y. Jon, J. H. Seong, A. Khademhosseini, T. N. T. Tran, P. E. Laibinis and R. Langer, Langmuir, 2003, 19, 9989–9993 CrossRef CAS.
- J. H. Hyun, H.W. Ma, Z. P. Zhang, T. P. Beebe and A. Chilkoti, Adv. Mater., 2003, 15, 576–579 CrossRef CAS.
- R. C. MacDonald, R. I. MacDonald, B. P. Menco, K. Takeshita, N. K. Subbarao and L. R. Hu, Biochim. Biophys. Acta, 1991, 1061, 297–303 CAS.
- R. R. C. New, Liposomes: A Practical Approach, Oxford University Press, Oxford, 1990 Search PubMed.
- H. W. Ma, J. Hyun, Z. P. Zhang, T. P. Beebe and A. Chilkoti, Adv. Funct. Mater., 2005, 15, 529–540 CrossRef CAS.
- G. M. Whitesides, E. Ostuni, S. Takayama, X. Y. Jiang and D. E. Ingber, Annu. Rev. Biomed. Eng., 2001, 3, 335–373 Search PubMed.
- K. S. Phillips and Q. Cheng, Anal. Chem., 2005, 77, 327–334 CrossRef CAS.
- G. Walter, K. Bussow, D. Cahill, A. Lueking and H. Lehrach, Curr. Opin. Microbiol., 2000, 3, 298–302 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2006 |