Particulate matter characterization at a coastal site in south-eastern Italy
Received
19th September 2005
, Accepted 22nd November 2005
First published on 6th December 2005
Abstract
Several samples of airborne particulate matter (PM), collected from 6th November to 6th December 2003 at a coastal site in the south-east of Italy, have been analyzed by different techniques to characterize elemental composition and morphological properties of the inorganic PM fraction and obtain preliminary results on anthropogenic contributions. Al, Cr, Cu, Fe, Mn, V, Pb, Ti, Ca and Zn mass concentrations, evaluated by an inductively coupled plasma atomic emission spectrometer, account for up to l% of the bulk PM mass in the investigated samples. According to geochemical calculations, Ca, Al, Fe and Mn are predominantly of crustal origin, while Cr, Cu, Pb, V, Ti and Zn heavy metals are of anthropogenic origin. Ion chromatography analyses have identified sulfate (SO42−) nitrate (NO3−), sodium (Na+), and ammonium (NH4+) as the main ionic components accounting for up to 38% of the total PM mass and up to 90% of the total ionic mass. Besides ion chromatography, X-ray energy dispersive (EDX) microanalyses have revealed the high variability of Cl: its weight concentration varies from about 24% to below the detection limit (≥0.5%) in the investigated samples. The marked anticorrelation between the excess of S and the Cl/Na ratio has allowed inferring that reactions between sea salt particles and acidic sulfates, which liberate HCl gas to the atmosphere leaving particles enriched in non-sea-salt sulfates, have significantly contributed to chloride depletion. Morphological analyses by scanning electron microscopy have shown that about 90% of the total sampled particles have a diameter ≤5 μm.
1. Introduction
Atmospheric particles are emitted from natural sources such as sea spray, crustal erosions, volcanic emissions, and dust outbreaks, and from anthropogenic processes such as fossil fuel combustion and industrial emissions.1 The detailed knowledge of the microphysical particle properties is becoming increasingly important since size and composition characterization of individual atmospheric particles is of relevance for modeling of atmospheric processes2 and for environmental control purposes.3 Atmospheric aerosols affect the Earth’s climate both directly through scattering and absorption of solar radiation and indirectly acting as active sites for chemical reactions occurring in the atmosphere. Moreover, epidemiological studies4 have demonstrated that atmospheric particles in urban areas have a clear correlation with human hospitalizations and deaths. The impacts that atmospheric particles have on public health are quite dependent on particles’ sizes besides their compositions. Size influences the site deposition in the human respiratory tract and the consequent degree of toxicity that may be experienced. Particle sizes also reflect the origin and formation of airborne particles: larger sized particles are often of crustal origin and smaller particles come from combustion processes or gas to particle conversion reactions in the atmosphere.5 Therefore, a detailed characterization of the individual atmospheric particles can provide much useful information about source, reactivity, transport, and removal of atmospheric chemical species.
The size and composition characterization of individual particles can be performed by a variety of analytical techniques including ion chromatography, inductively coupled plasma atomic emission spectrometry, scanning electron microscopy, transmission electron microscopy,3 and micro-PIXE analysis.6
In this study, several samples of airborne particulate matter have been collected from 6th November to 6th December 2003 at a coastal site in the south-east of Italy, to contribute to the characterization of aerosols over the Mediterranean basin, providing results on morphological and elemental particle composition and hence on anthropogenic contributions.7,8 The PM sampling was performed in the autumn to obtain samples less affected by the contribution of long-range transported emissions. The east Mediterranean basin and then south-east Italy are crossroads where aerosols from different sources may converge: urban/industrial aerosols and seasonal biomass burning from Central and Eastern Europe, maritime and long-range transported polluted air masses from the Atlantic Ocean,9 mineral dust from North Africa,10,11 and sea spray from the Mediterranean Sea itself. However, the aerosol loading significantly reduces in autumn–winter over the Mediterranean basin, as a consequence of the reduced weather stability and the more frequent wet removal processes of aerosols.
The experimental methods and results are reported in Section 2. The summary and conclusion are in Section 3.
2. Experimental methods and results
Particulate matter has been sampled at a coastal site in south-east Italy (40° 34′ N, 18° 01′ E) located at about 5 km away from the Adriatic Sea and 7 km away from an industrial area that includes a large petrochemical plant and a coal-burning power plant. The geographical location of the sampling site in the Mediterranean basin is represented in Fig. 1 by the letter I.
 |
| | Fig. 1 Geographical location in the Mediterranean basin of the sampling site of this paper (I), Barcelona (B), Crete (C), and Delos (D). | |
2.1. Sampling methodology
The FH 95 particulate sampler provided by ESM Andersen Instruments GmbH, has been used to collect atmospheric particles with a 2.3 m3 h−1 flow rate, on cellulose nitrate filters having a nominal pore size of 0.8 μm. The particulate sampler has operated on the roof of a building at a height of ∼10 m above ground level. The total suspended particulate (PTS) matter has mainly been sampled from the 6th of November to the 6th of December, rather few PM10 and PM2.5 samplings have been performed. Samplings with times of 24 and 12 hours have been performed to investigate if microphysical particle properties and concentrations vary from day to night. The gravimetric method has been used to evaluate particulate matter concentrations.
2.2. Results on PM concentrations
PTS, PM10 and PM2.5 concentrations referring to 24 hour sampling times are reported on Table 1 showing that PTS concentrations vary from 25 ± 1 to 55 ± 3 μg m−3, while PM10 concentrations vary from 22 ± 1 to 42 ± 1 μg m−3. PTS (full dots), PM10 (open dots), and PM2.5 (open box) concentrations, referring both to 24 and 12 hour samplings as available, are plotted in Fig. 2. It is worth observing from Fig. 2 that the PM concentrations retrieved by diurnal samplings differ by less than 25% than those retrieved on following nocturnal samplings. In addition, Fig. 2 reveals that samplings performed on the following days provide rather close values of PTS, PM10, and PM2.5 concentrations: the 24 hour samplings started on November the 22nd (10:00), 24th (10:00), and 25th (10:00) provide PM2.5, PTS and PM10 concentrations of 25 ± 1, 25 ± 1, and 26 ± 1 μg m−3, respectively. Latter results may suggest that particles with a diameter smaller than 2.5 μm dominated in the sampling area at least from 22nd November to 25th November.
 |
| | Fig. 2 Particulate matter concentrations of 12 and 24 hour samplings from 6th November to 6th December, 2003. | |
Table 1 Particulate matter concentrations of 24 hour samplings performed from 6th November to 6th December, 2003
| Day (Nov.–Dec. ’03) |
06 |
07 |
08 |
09 |
10 |
11 |
12 |
13 |
14 |
19 |
20 |
21 |
22 |
23 |
24 |
25 |
01 |
02 |
03 |
04 |
05 |
06 |
| PTS/μg m−3 |
26 ± 1 |
34 ± 1 |
|
|
55 ± 3 |
29 ± 2 |
26 ± 1 |
28 ± 2 |
50 ± 2 |
32 ± 1 |
|
40 ± 1 |
|
|
25 ± 1 |
|
|
48 ± 1 |
27 ± 2 |
|
|
|
| PM10/μg m−3 |
|
|
42 ± 1 |
|
|
|
|
|
|
|
22 ± 1 |
|
|
|
|
26 ± 1 |
|
|
|
35 ± 1 |
31 ± 1 |
|
| PM2.5/μg m−3 |
|
|
|
|
|
|
|
|
|
|
|
|
25 ± 1 |
|
|
|
|
|
|
|
|
31 ± 1 |
A field-emission scanning electron microscope SEM (Philips, Model XL-20), equipped with an energy dispersive X-ray system (EDX, DX-4) has been used to characterize morphological properties of the sampled particles and perform a semi-quantitative analysis of their elemental composition. The EDX system allows analysis of elements having an atomic number ≥6. Operating conditions for the SEM analysis were as follows: accelerating voltage: 15 kV; probe current: 53 μA; X-ray detector distance: 45 mm; working distance: 12 mm; accumulation time: 100 s; dead time: 10 to 25%.
2.4. Results of SEM-EDX analyses
SEM images of randomly chosen areas at magnifications of 1000 and/or 5000 have revealed in all samples that the shape of the particles, often clustered together, was quite irregular. In particular, particles with the equivalent diameter ranging from 0.8 to 20 μm have been observed in all samples. The particle’s equivalent diameter is the diameter of a circle with the same area.12 The lowest detected diameter values were determined by the SEM resolution under our operating conditions. Cumulative frequency distributions of the particle equivalent diameter obtained by analyzing 300 particles randomly selected on some PTS samples, have revealed that 90% of the particles have a diameter smaller than 5 μm, while 50% of the particles have a diameter smaller than 2.5 μm. One PM10 and 13 PTS samples have been examined by the SEM-EDX system. Single-particle elemental analyses have revealed the presence of particles containing two or more of the following elements: Na, Mg, Al, Si, S, Cl, K, Ca, and Fe. Even though C and O have been detected, they have not been considered in the data analysis because they are major components of cellulose nitrate filters. A typical EDX spectrum of cellulose nitrate filters is shown in Fig. 3a. Single particle EDX spectra have allowed inferring the presence of aluminosilicates, S-bearing particles (such as Na2SO4 and CaSO4), sea-salt particles, and oxides (such as SiO2 and Al2O3). All the compounds have been observed as single particles, with minor aluminosilicate content, and in mixtures with aluminosilicates, sulfates, and oxides. It is worth mentioning that EDX spectra do not allow the inferring of chemical species without ambiguity. However, we believe that it is possible to infer the presence of some chemical species when the chemical composition of particles is simple. Some single-particle EDX spectra are shown as example in Fig. 3b–f. We believe that the spectra of Fig. 3b and c can be considered to be representative of single particles of NaCl and CaSO4, respectively. The rather high intensity of the O line with respect to the C line of Fig. 3c supports the presence of a CaSO4 particle: O must also be ascribed to the sampled particle and not only to the cellulose nitrate filter, in accordance with Fig. 3a. A typical spectrum that can be ascribed to an aluminosilicate particle is shown in Fig. 3d. While the spectrum of Fig. 3e can be ascribed to a NaCl particle in a mixture with aluminosilicates and sulfates, the spectrum of Fig. 3f can be considered representative of a particle made of calcium and sodium sulfates. The intensities of the EDX spectrum lines have been converted to corresponding percentages of weight concentrations by a standardless ZAF correction method in which ideal flat samples have been assumed,3 to get semi-quantitative data on weight concentrations (%) of some selected elements (Na, Mg, Al, Si, S, Cl, K, Ca, and Fe). To this end, it is worth mentioning that SEM-EDX results are mainly representative of the “coarse” inorganic PM fraction since our operating conditions have allowed the detection of particles with the equivalent diameter ≥0.8 μm. The results referring to the analyzed samples are given in Fig. 4 and Table 2. Sampling day and time are also provided in Table 2 and it is worth noting that element weight concentrations (%) referring to diurnal and nocturnal samplings indicate that the particle chemical properties do not vary significantly from day to night. It is also worth observing from Fig. 4 and Table 2 that element weight concentrations (%) of the 24 hour PM10 sample of November 8 (sample #3) are rather similar to those of the 12 hour PTS sample of November 14 (sample #12). Latter results are in accordance with previous findings showing in the investigated samples that 90% of the particles have a diameter smaller than 5 μm. Table 2 reveals that predominant elements are Na and S: the weight concentration of both elements varies from 51 to 75% in the tested samples. While, Mg, Si, K, Ca, Fe, and Al are the minor elements: their concentration is 20% to 51% of total concentration. The high variability from sample to sample of the Cl concentration represents the most striking result of Table 2: the Cl weight concentration represents 24% of the total concentration of sample #1, while it is below the detection limit (≥0.5%) in samples #3, #10, and #12 where the Na concentration is more than 15%. Reactions between sea salt and acidic nitrate and sulfate that liberate HCl gas to the atmosphere leaving particles enriched in nitrate and non-sea-salt sulfate and depleted in chloride, are generally invoked to explain chloride depletion.13
 |
| | Fig. 3 Typical EDX elemental analysis of (a) cellulose nitrate filters, (b) NaCl particles, (c) CaSO4 particles, (d) aluminosilicate particles; (e) NaCl particle in a mixture with aluminosilicates and sulfates, and (f) particles made of calcium and sodium sulfates. | |
 |
| | Fig. 4 Elemental weight concentrations (%) in the investigated samples. | |
Table 2 Weight percentages of Na, Mg, Al, Si, S, Cl, K, Ca, Fe in the analyzed samples
| Sample |
Month/Day |
Sampling time |
Na |
Mg |
Al |
Si |
S |
Cl |
K |
Ca |
Fe |
| #1 |
11/06 |
10:00–10:00 |
39 |
5 |
2 |
5 |
17 |
24 |
2 |
6 |
0 |
| #2 |
11/07 |
10:00–10:00 |
29 |
4 |
5 |
8 |
22 |
10 |
4 |
15 |
4 |
| #3 (PM10) |
11/08 |
10:00–10:00 |
14 |
3 |
9 |
15 |
36 |
0 |
5 |
19 |
0 |
| #4 |
11/10 |
10:00–22:00 |
39 |
5 |
4 |
11 |
17 |
15 |
2 |
6 |
2 |
| #5 |
11/10 |
22:00–10:00 |
40 |
6 |
3 |
7 |
14 |
23 |
2 |
5 |
2 |
| #6 |
11/11 |
10:00–22:00 |
43 |
2 |
4 |
7 |
21 |
15 |
2 |
5 |
0 |
| #7 |
11/11 |
22:00–10:00 |
33 |
5 |
4 |
9 |
19 |
17 |
3 |
6 |
5 |
| #8 |
11/12 |
10:00–22:00 |
50 |
5 |
3 |
8 |
25 |
2 |
4 |
5 |
0 |
| #9 |
11/13 |
10:00–22:00 |
40 |
3 |
2 |
8 |
34 |
1 |
4 |
9 |
0 |
| #10 |
11/13 |
22:00–10:00 |
34 |
2 |
8 |
18 |
28 |
0 |
5 |
6 |
0 |
| #11 |
11/14 |
10:00–22:00 |
29 |
3 |
5 |
12 |
33 |
1 |
3 |
12 |
3 |
| #12 |
11/14 |
22:00–10:00 |
18 |
2 |
4 |
12 |
39 |
0 |
7 |
16 |
3 |
| #13 |
11/19 |
10:00–10:00 |
32 |
2 |
4 |
10 |
32 |
1 |
3 |
11 |
7 |
| #14 |
12/01 |
10:00–10:00 |
42 |
5 |
2 |
7 |
20 |
5 |
9 |
10 |
0 |
In order to identify the role of potential sources of natural and anthropogenic components, the enrichment factor (EF) technique has been used. Latter technique also allows distinguishing between elements associated with the two major natural sources of aerosols of the sampling area, i.e. those having a predominantly crustal and marine origin.14 Taylor’s model (1964)15 is used to calculate the EF for crustal rock with Al as reference element (EFcru), while values for seawater with Na as a reference element16 are used to calculate the EF of elements of marine origin (EFmar). The basic assumptions are that aluminium (sodium) is entirely of crustal (marine) origin17 and that the mean crustal (marine) composition represents the sampling area.18 According to the suggested classification criteria19 an EFcru (EFmar) value of <10 is taken as an indication that an element in an aerosol has a significant crustal (marine) source14 and it is termed a non-enriched element. While EF values of >50 are ascribed to an element of anthropogenic origin20 and it is termed enriched element. The statistical distribution of EFcru (EFmar) values is represented in Fig. 5a (Fig. 5b), where dots (boxes) represent mean values, and error bars define maximum and minimum values. EF values of Fig. 5 indicate that Ca, Mg and K have a mixed origin: marine–crustal, while Si and Fe appear to be of crustal origin. Fig. 5 also reveals the significant Cl depletion in comparison with seawater (Cl/Na = 1.8) and that S is little enriched in comparison with the marine model and very enriched in comparison with the crustal model. Then, we must assume that S, besides the marine and crustal origin, has an anthropogenic origin.21 The time evolution of the excess of S calculated by deducting marine and crustal contributions from the total S-weight percentage is shown in Fig. 6 (open boxes) with the time evolution of Cl/Na weight percentage ratios (full dots). The excess of S is calculated using the following equation:
| excess of S = S − (0.084 Na) − (0.0031 Al) |
where S, Na, and Al represent the weight percentage of sulfur, sodium and aluminium, respectively, 0.084 is taken as the ratio of S to Na in seawater, and 0.0031 is taken as the ratio of S to Al in crustal rock. Dashed lines in
Fig. 6 represent sampling times. It is worth nothing from
Fig. 6 the marked anticorrelation between the excess of S and the Cl/Na ratio: higher Cl/Na weight ratios correspond to lower values of excess of S. Moreover, the plot
versus S of ΔCl/Na, where ΔCl represents the Cl depletion in comparison with seawater, reveals that the total Cl-depletion (ΔCl/Na = 1.8) occurs in samples with S > 25% (
Fig. 7). Latter results (
Fig. 6 and 7) may indicate that Cl depletion also occurs when chlorides of marine aerosol salts react with
sulfuric acid liberating HCl gas to the atmosphere and leaving Na
2SO
4 particles.
13 The above conclusion is also supported by
EDX spectra that have allowed inferring the presence of
sodium sulfate particles as single particles or mixed with other chemical compounds (
Fig. 3f). Aden and Buseck
22 have observed significant quantities of Na
2SO
4 in emissions from a coal-burning power plant. Additionally, aerosol particle studies performed in summer by Ganor
et al.,
23 at Haifa Bay, on the eastern shore of the Mediterranean, which is surrounded by industrial sites including large petrochemical plants and an oil-fueled power plant, have revealed that sulfate and
nitrate concentrations were 5–10 times higher when a breeze was blowing from the land than when one was blowing from the sea, as a consequence of the emissions from local industries or the long-range transported pollution from countries of Eastern and Central Europe. Several studies have concluded that atmospheric
sulfates either can be emitted directly as primary particles or, more commonly, are the results of gas to particle conversion reactions in the atmosphere.
5,24 The time evolution of the relatively humidity (RH) provided by a ground meteorological station rather close to the monitoring site is also plotted in
Fig. 7 (grey line) and it is worth observing the strong correlation between relative humidity and excess of S: higher excesses of sulfate are observed when relative humidity takes values closer to 100% during the sampling time. Latter observation may indicate that the reactions between sea salt and acidic sulfate that contribute to both Cl depletion and excess of S are favored by high relative humidity in accordance to recent studies.
24
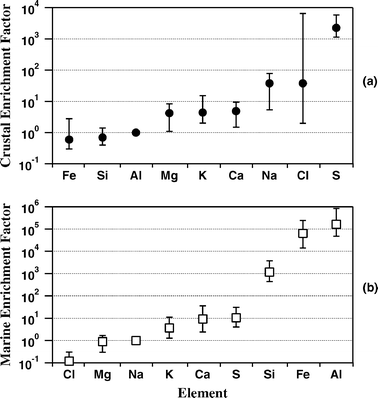 |
| | Fig. 5 (a) Crustal enrichment factors and (b) marine enrichment factors for the SEM analyzed samples: symbols represent mean values, and error bars define maximum and minimum values. | |
 |
| | Fig. 6 Time evolution of excess of S (open boxes), Cl/Na weight ratios (full dots), and ground relative humidity (grey line) during the sampling period. Dashed lines indicate sampling-time intervals. | |
 |
| | Fig. 7 Ratio of the Cl depletion in comparison with seawater (ΔCl) to Na versus the S weight percentage. | |
As it has been told, the Cl depletion can also be determined by reactions of nitric acid with NaCl that lead to the formation of NaNO3 particles. EDX microanalyses have not allowed N detection and then, the detection of NaNO3 particles. However, as we will show later on, ion chromatography analyses performed on rather few samples have revealed that sulfate and nitrate were the main ionic components of our sampling site.
Chloride depletion as a consequence of the formation of non-sea-salt sulfates has mainly been observed in spring–summer in several coastal areas of the Mediterranean basin. Analyses of PM10 and PM2.5 measurements performed in Barcelona (Spain) from June, 1999 to June, 2000, show that the time series of non-marine SO42− levels were characterized by high spring–summer and low autumn–winter concentrations: mean levels of 8 and 5 μg m−3, respectively.26
The higher summer oxidation rate of SO2 was considered responsible of the higher sulfate concentrations observed in summer.25 Bardouki et al.26 have also observed significant chloride depletion mainly in summer, by characterizing atmospheric aerosols collected at a coastal rural site on the island of Crete, in the eastern Mediterranean (Fig. 1) during July 2000 and January 2001. In particular, they have observed that the mean Cl/Na weight ratio that was 1.76 in winter reduced to 1.16 in summer. On the contrary, Chabas and Lefèvre21 in characterizing the atmospheric environment of Delos (Cyclades, Greece) on the south-east Mediterranean basin have found mean Cl/Na weight ratios of 0.52 and 0.81 in March and July 1995, respectively. Scanning electron microscopy has been used by Chabas and Lefèvre21 to characterize the collected aerosols.
According to Chabas and Lefèvre,21 the high excess sulfur concentration measured at Delos has an anthropic origin due to local and European pollution sources. The mean concentration in excess of sulfate at Delos approaches those measured in Corsica27 and in Malta,28 which are also ascribed to local and long-range transported anthropic pollution. In accordance to the above reported discussion, the quite high excess of sulfur revealed by Fig. 6 can also be ascribed to anthropic pollution sources. Sulfuric acid (H2SO4) that is the major oxidation products of atmospheric SO2,29 besides nitric acid (HNO3), represents the main species responsible of the formation of new aerosol particles. Sulfur dioxide is the predominant anthropogenic sulfur-containing air pollutant and it is emitted to the atmosphere mainly by industrial sources, particularly fossil fuel combustion. As it has been told, our sampling site is about 7 km away from an industrial area that includes a petrochemical and a coal-burning power plant and the samplings have been performed in the autumn, when the contribution of long range transported pollutants is less significant.30 Therefore, we believe that SO2 emissions by local industries have contributed to the presence in the atmosphere of acid chemical species responsible of the formation of new atmospheric particles.
Ion chromatography and inductively coupled plasma atomic emission spectrometry analyses have also been performed on some selected samples to better quantify anthropogenic contributions and further support SEM-EDX results. However, as a consequence of the limited instrumentation availability, analyses have only been performed on a few samples randomly chosen during the sampling period.
2.5. Ion chromatography analyses: methodology
A high performance ion chromatograph DIONEX DX-500 has been used to determine the mass concentration of the major anions (Cl−, NO3−, SO42−) and cations (Na+, NH4+, K+, Mg2+) in some selected samples. For anions analysis we used a self-regenerating suppressor ASRS®-ULTRA (4 mm) at 50 mA, a guard column IONPAC® AG4A-SC (4.50 mm) and an analytical column IONPAC® AS4A-SC (4.250 mm). All the anions were determined with isocratic elution at 2.0 mL min−1 of 1.8 mM Na2CO3/1.7 mM NaHCO3 eluent. For cations analysis we used a self-regenerating suppressor CSRS®-ULTRA (4 mm), a guard column IONPAC® CG12A (4.50 mm) at 100 mA and an analytical column IONPAC® CS12A (4.250 mm). All the cations were determined with isocratic elution at 1 mL min−1 of 20 mN H2SO4 eluent.
2.6. Results of ion chromatography analyses
Ion chromotography analyses have been performed on four samples randomly chosen during the sampling period. Table 3 reports mean (Mean), maximum (Max), and minimum (Min) values of the ion mass concentration retrieved in the four analyzed samples. Sulfate (SO42−) and nitrate (NO3−) have the highest mass concentration, followed by ammonium (NH4+) and sodium (Na+), accounting for up to 38% of the total PM mass and up to 90% of the total ionic mass. The sampled ion mass concentration accounts for up to 44% of the total PM mass, in the investigated samples. Table 3 also reports the mean variation of the non-sea-salt SO42− (nss-SO42−) that has been calculated by deducting the sea salt contribution to SO42−. The sea salt contribution to SO42− is estimated as measured [Na+] times 0.121, the SO42−/Na+ molar ratio in seawater,31 and accounts for less than 5% of the measured sulfate mass, in accordance to SEM-EDX results. It is worth mentioning that the predominance of nss-SO42− represents the striking feature of polluted marine boundary layers.2 The results of Table 3 indicate that the four analyzed samples are characterized by a distinctive positive ionic balance (ionic balance = sum of anions minus sum of cations) and Mihalopoulus et al.32 have found that polluted air masses from Europe have prevalently a positive ionic balance due to the excess of non-neutralized sulfate ions. Sulfuric acid produced via oxidation of SO2 is generally neutralized in the aerosol phase as ammonium bisulfate, or as ammonium sulfate.11 Free ammonia is also depleted by reaction with HNO3 to form NH4NO3.33 In our study NH4+/nss-SO42− molar ratios vary in the 0.3–0.5 range and as a consequence the measured ammonium is not enough to balance the negative charge of sulfate and nitrate. Therefore, reactions of nitric and sulfuric acid with coarse sea-salt and alkaline-soil particles may be favored in NH3 limited areas, just as SEM-EDX analyses have revealed. Finally, it is worth noting that we have observed that the Cl−/Na+ ratio varies in the 0.35–50 range in the four analyzed samples in satisfactory accordance with the SEM data of Fig. 6. The comparison of the ion mass concentration of Table 3 with those reported by several authors for different sites of the Mediterranean basin indicates that our monitoring site was characterized during the sampling period by ion concentrations typical of polluted rural coastal sites. Measurements performed at Barcelona, Spain (Fig. 1) have provided mean annual concentrations of 6.6, 5.7, and 2.7 μg m−3 for nss-SO42−, NO3−, and NH4+, respectively,34 while measurements performed at a coastal rural site on the island of Crete have provided mean winter concentrations of 2.4, 1.5, and 0.8 μg m−3 for nss-SO42−, NO3− and NH4+, respectively. Higher mean concentrations have been retrieved on summer times: 6.9, 2.7, and 2.4 μg m−3 for nss-SO42−, NO3−, and NH4+, respectively.26
Table 3 Mean, minimum (Min), and maximum (Max) mass concentrations (ng m−3) of the investigated species in some selected samples
| Species/ng m−3 |
Na+ |
NH4+ |
K+ |
Mg2+ |
Cl− |
NO3− |
SO42− |
nss-SO42− |
| Mean |
897 |
1444 |
320 |
567 |
509 |
4118 |
3884 |
3775 |
| Min |
675 |
1036 |
207 |
539 |
333 |
2201 |
2074 |
1953 |
| Max |
1077 |
1729 |
408 |
595 |
657 |
4886 |
4733 |
4602 |
2.7. Metal analyses: methodology
We have used an inductively coupled plasma atomic emission spectrometer ICP-AES Liberty 110 (Varian Inc., Palo Alto, USA) to determine the mass concentration of several metals. The instrument was equipped with a vertical torch inert to hydrofluoric acid and an ultrasonic nebulizer CETAC U-5000AT+ (Cetac Technologies Inc., Omaha, NE, USA). A microwave system MILESTONE MLS-1200 MEGA (Milestone, Bergamo, Italy) has been used to accomplish the dissolution of the samples. Details on sample preparation and analysis technique are reported elsewhere.35
2.8. Metal analysis results
Eight samples randomly chosen during the sampling period have been analyzed for determining the mass concentration of metals Al, Ca, Cr, Cu, Fe, Pb, Mn, Ti, V, Zn. Table 4 reports mean (Mean), maximum (Max), and minimum (Min) values of the mass concentration retrieved for each element in the analyzed samples. Uncertainties on metal mass concentrations are lower than 5% for Al, Fe, Ca, and Mn, while are lower than 8% for Cr, Cu, V, Ni, Pb, and Zn. We have observed that all analyzed metals account for up to 1% of the total PM mass in the investigated samples. Al, Fe and Ca are predominant elements, while Mn, Cr, Cu, V, Ti, Pb, and Zn account for up 7% of the total metal mass. The statistical distribution of EFs for crustal rock with Al as reference element is given in Fig. 8 where full dots represent mean values, and error bars define maximum and minimum values. Fig. 8 indicates that besides Ti and Mn, Fe and Ca have a significant crustal origin, in accordance to SEM-EDX analyses, while Cr, V, Cu, Pb, and Zn are mainly of anthropogenic origin. However, Table 4 reveals that heavy metal mass concentrations do not exceed guide and/limit values recommended by the World Health Organization.36 The comparison of metal mass concentrations of Table 4 with literature data20,34 also leads assuming that they are typical of rural sites of the Mediterranean basin.
 |
| | Fig. 8 Crustal enrichment factor for metals analyzed by an inductively coupled plasma atomic emission spectrometer: full dots represent mean values, and error bars define maximum and minimum values. | |
Table 4 Mean, minimum (Min), and maximum (Max) mass concentrations (ng m−3) of the investigated elements in some selected samples
| Element/ng m−3 |
Cr |
Cu |
Fe |
Mn |
Pb |
V |
Zn |
Ti |
Al |
Ca |
| Mean |
4.4 |
8 |
181 |
3.9 |
8.4 |
5.9 |
14.5 |
9.7 |
169 |
486 |
| Min |
1 |
3.1 |
79 |
2 |
3.9 |
2 |
6.8 |
3 |
55 |
297 |
| Max |
6 |
11.9 |
281 |
6 |
17 |
8.9 |
23.7 |
15.6 |
283 |
660 |
3. Summary and conclusions
Different complementary techniques have been used to characterize elemental composition and morphological properties of the inorganic fraction of airborne particles sampled at a coastal site of south-east Italy, and to get preliminary results on the effects of anthropogenic contributions. Ion chromatography and inductively coupled plasma atomic emission spectrometry analyses have been performed to quantify ion and metal contributions to the total collected PM. We have observed that the monitored ions (Cl−, NO3−, SO42−) and cations (Na+, NH4+, K+, Mg2+) account for up to 44% of the total PM mass concentration, while metals account for up to 1%. These analyses have been performed on rather few samples as a consequence of the laboratory instrumentation availability; nevertheless they have provided results in satisfactory accordance with SEM-EDX analyses performed on most of the collected samples and hence validate SEM-EDX results. Morphological SEM analyses of PTS samples and cumulative frequency distributions of the particle diameters have revealed that during the sampling time, ∼90% of the monitored particles were characterized by a diameter ≤5 μm. In fact, samplings performed on following days have provided rather close PTS and PM10 concentrations. X-ray energy dispersive microanalyses of selected samples have allowed getting semi-quantitative data on weight concentrations of Na, Mg, Al, Si, S, Cl, K, Ca, and Fe. It is shown that predominant elements were Na and S: the weight percentage of both elements varied from 51% to 75% in all tested samples. In contrast, Mg, Si, K, Ca, Fe, and Al were minor elements: their weight percentage varied from 20% to 51% in the tested samples. Enrichment factors have indicated that Ca, Mg and K have a mixed origin: marine–crustal, and that Si, Al, and Fe appear to be mainly of crustal origin. Chloride was the element characterized by the highest variability: its weight concentration varies from about 24% to below the detection limit (≥0.5%) in the investigated samples. The high excess of S and Cl depletion have partially been ascribed to reactions between sea salt and acidic sulfate that liberate HCl gas to the atmosphere leaving the particles enriched in non-sea-salt sulfate and depleted in chloride. Higher excess of sulfate and nearly total Cl depletion have been observed for ground relative humidity values of ∼100% during the sampling time. According to literature data, the latter results show that the atmospheric acid sulfuric reactions leading to the formation of new particles are favored by high relative humidities. It has been shown that the results of this paper are in satisfactory accordance with previous findings that have revealed significant chloride depletion as a consequence of the formation of non-sea-salt sulfates and nitrates, mainly in coastal sites of the Mediterranean basin close to industrial environments. However, it is worth noting that ion and metal mass concentrations indicate that the south-east Italy monitoring site was not significantly affected by anthropogenic emissions: the comparison with literature data shows that metal and ion mass concentration values of this paper are close to those of coastal rural sites.
To the best of our knowledge, this paper presents the first results on the airborne particulate matter characterization over coastal areas in south-east Italy and we believe that they contribute to the aerosol characterization over the Mediterranean basin. Work is in progress to better investigate the role of local and long-range transported anthropic emissions on our experimental findings. To this end, PM samplings will be performed during the year and at different sites in south-east Italy.
Acknowledgements
Work partially supported by ARPA-Agenzia Regione Puglia Ambiente (Italy) during the Project “Piano di Caratterizzazione del Sito Inquinato di Interesse Nazionale di Brindisi”. ARPA-Agenzia Regione Puglia Ambiente of Brindisi is also acknowledged for providing ground meteorological data. A. Turnone has carried out this work with the support of a grant provided by Università di Lecce.
References
-
G. A. D’Almeida, P. Koepke and E. P. Shettle, Atmospheric aerosols: Global climatology and radiative characteristics, A. Deepak Publishing, Hampton, VA, 1991 Search PubMed.
- F. Raes, R. Van Dingenen, E. Vignati, J. Wilson, J. P. Putaud, J. H. Seinfeld and P. Adams, Atmos. Environ., 2000, 34, 4215–4240 CrossRef CAS.
- L. Paoletti, B. De Berardis and M. Diociaiuti, Sci. Total Environ., 2002, 292(36), 265–275 CrossRef CAS.
-
K. Donaldson and W. MacNee, The mechanism of lung injury caused by PM10, in Air Pollution and Health, Issues in Environmental Science and Technology, ed. R. E. Hester and R. M. Harrison, Royal Society of Chemistry, Cambridge, 1998, vol. 10, pp. 21–32 Search PubMed.
- J. E. Post and P. R. Buseck, Environ. Sci. Technol., 1984, 18, 35–42 CAS.
- J. Smolík, V. Ždímal, M. Lazaridis, V. Havránek, K. Eleftheriadis, N. Mihalopoulos, C. Bryant and I. Colbeck, Atmos. Chem. Phys., 2003, 3, 2207–2216 CAS.
- S. Sobanska, C. Coeur, W. Maenhaut and F. Adams, J. Atmos. Chem., 2003, 44, 299–322 CrossRef CAS.
- J. Sciare, K. Oikonomou, H. Cachier, N. Mihalopoulos, M. O. Andreae, W. Maenhaut and R. Sarda-Estève, Atmos. Chem. Phys., 2005, 5, 2253–2265 CAS.
- F. De Tomasi and M. R. Perrone, J. Geophys. Res., 2003, 108, 4286–4297 CrossRef.
- A. Blanco, F. De Tomasi, E. Filippo, D. Manno, M. R. Perrone, A. Serra, A. M. Tafuro and A. Tepore, Atmos. Chem. Phys., 2003, 3, 2147–2159 CAS.
- D. Dordević, Z. Vukmirović, I. Tošić and M. Unkašević, Atmos. Environ., 2004, 38, 3637–3645 CrossRef CAS.
- S. Weinbruch, M. Wentzel, M. Kluckner, P. Hoffmann and H. M. Ortner, Mikrochim. Acta, 1997, 125, 137–141 CAS.
- L. M. McInnes, D. S. Covert, P. K. Quinn and M. S. Germani, J. Geophys. Res., 1994, 99(D4), 8257–8268 CAS.
- S. Guerzoni, E. Molinaroli and R. Chester, Deep-Sea Res., Part II, 1997, 44(3–4), 631–654 CrossRef CAS.
- S. R. Taylor, Geochim. Cosmochim. Acta, 1964, 28, 1237–1285.
-
CRC Handbook of Chemistry and Physics, ed. Robert C. Weast, CRC Press, Boca Raton, FL, 69th edn, 1988–1989 Search PubMed.
- D. S. Lee, J. A. Gardland and A. A. Fox, Atmos. Environ., 1994, 28(16), 2691–2713 CrossRef CAS.
- J. C. Chester, M. Nimmo, G. R. Fones, S. Keyse and Z. Zhang, Atmos. Environ., 2000, 34, 949–958 CrossRef CAS.
- S. R. Biegalski, S. Landsberger, M. Haik and E. L. Kothny, Atmos. Environ., 1984, 18(2), 409–416 CrossRef CAS.
- G. Lonati, M. Giugliano, P. Butelli, L. Romele and R. Tardivo, Atmos. Environ., 2005, 39, 1925–1934 CrossRef CAS.
- A. Chabas and R. A. Lefèvre, Atmos. Environ., 2000, 34, 225–238 CrossRef CAS.
-
G. D. Aden and P. R. Buseck, in Microbeam Analysis, ed. D. E. Newbury, San Francisco Press, Inc., San Francisco, CA, 1979, p.
254 Search PubMed.
- E. Ganor, Z. Levin and R. Van Grieken, Atmos. Environ., 1998, 32(no. 9), 1631–1642 CrossRef CAS.
- M. Kulmala, H. Vehkamäki, T. Petäyä, M. Dal Maso, A. Lauri, V. M. Kerminem, W. Birmili and P. H. McMurry, Aerosol Sci., 2004, 35, 143–176 Search PubMed.
- X. Querol, A. Alastuey, S. Rodríguez, F. Plana, C. R. Ruiz, N. Cots, G. Massagué and O. Puig, Atmos. Environ., 2001b, 35, 6407–6419 Search PubMed.
- H. Bardouki, H. Liakakou, C. Economou, J. Sciare, J. Smolík, V. Zdímal, K. Eleftheriadis, M. Lazaridis, C. Dye and N. Mihalopoulos, Atmos. Environ., 2003, 37, 195–208 CrossRef CAS.
-
G. Bergametti, PhD thesis, Paris VII University, 1987.
- K. Torfs and R. Van Grieken, Atmos. Environ., 1997, 31, 2179–2192 CrossRef CAS.
-
J. H. Seinfeld and S. N. Pandis, Atmospheric Chemistry and Physics: from Air Pollution to Climate Change, John Wiley, New York, 1998, p. 1360 Search PubMed.
- M. R. Perrone, M. Santese, A. M. Tafuro, B. Holben and A. Smirnov, Atmos. Res., 2005, 75, 111–133 CrossRef CAS.
- W. C. Keene, A. P. A. Pszenny, J. N. Galloway and M. E. Hawley, J. Geophys. Res., 1986, 91(N. D6), 6647–6658 CrossRef CAS.
- N. Mihalopoulos, E. Stephanou, S. Pilitsides, M. Kanakidou and P. Bousquet, Tellus, 1997, 49B, 314–326 Search PubMed.
- J. W. Erisman and M. Schaap, Environ. Pollut., 2004, 129(1), 159–163 CrossRef CAS.
- S. Rodríguez, X. Querol, A. Alastuey and F. Plana, J. Geophys. Res., 2002, 107(D24), 4777 CrossRef.
- A. Buccolieri, G. Buccolieri, N. Cardellicchio, A. Dell’Atti and E. T. Florio, Ann. Chim., 2005, 95, 15–25 CrossRef CAS.
-
World Health Organization, Air Quality Guidelines for Europe, WHO Regional Publications, Copenhagen, European Series, no. 91, 2nd edn, 2000 Search PubMed.
|
| This journal is © The Royal Society of Chemistry 2006 |
Click here to see how this site uses Cookies. View our privacy policy here.