Feasibility study of preparation and certification of reference materials for nitrogen dioxide and sulfur dioxide in diffusive samplers
Michel
Gerboles
*a,
Daniela
Buzica
a,
Luciano
Amantini
a,
Friedrich
Lagler
a and
Theo
Hafkenscheid
b
aEuropean Commission—Joint Research Centre, Institute for Environment and Sustainability, Emissions and Health Unit, 21020, Ispra, Italy. E-mail: michel.gerboles@jrc.it; Fax: +39-332-785652; Tel: +39-332-789364
bDutch Measurement Institute, Delft, Netherlands
First published on 10th November 2005
Abstract
This paper presents the results of a feasability study for the preparation and certification of reference materials (RMs) for nitrogen dioxide (NO2) and sulfur dioxide (SO2) in diffusive samplers. RMs for NO2 were prepared by exposure to gas mixtures in a chamber while the RMs for SO2 were prepared by liquid spiking. Certification of RMs for NO2 was found feasible with a certified uncertainty of 5.8% and a proposed shelf life of 5 years. The uncertainty was calculated with contribution from the homogeneity of preparation, stability and transport of the CRMs and from an external verification of the certified value. To reach 5.8% of uncertainty, the contribution of the differences between the results of analysis by ion chromatography and colorimetry must be eliminated. It is proposed to solve this by pre-extracting the samplers with water before analysis. The results of this study indicate that the samplers are stable for at least two years before and after exposure when stored in a refrigerator. By contrast, the certification of RMs for SO2 was found to not be feasible due to instability problems. This instability was attributed to reaction of sulfate on the walls of the samplers. Alternatively, the preparation of RMs by simultaneous exposure to SO2 and NO2 has been tested. Satisfying homogeneities has been reached both for NO2 and SO2.
Introduction
Among the general objectives of the European Directive 96/62/EC1 on assessment and management of ambient air quality, one is to monitor the ambient air in Europe on the basis of common methods and against limit values. The assessment and control of the measurement uncertainties is essential to meet this objective. It also requires measurement traceability to SI units and the implementation of quality assurance/quality control (QA/QC) programmes. For both requirements, the use of certified reference materials (CRM) is an indispensable tool.The 1st European Daughter Directive2 defines reference methods as well as indicative methods for monitoring nitrogen dioxide (NO2) and sulfur dioxide (SO2) in ambient air. Indicative methods can be used in a lowly-polluted area provided that their accuracy is within ±25%. A widely used indicative method for measuring NO2 and SO2 is the diffusive sampler technique,3 which provides an averaged measure of the pollutant over different exposure periods. The diffusive sampling method is being increasingly used because it is simple and inexpensive, it allows determination of air pollution levels in particular for screening the air quality in agglomerations or larger areas.4–6 However, CRMs are not available for NO2 and SO2 diffusive samplers. There is no track of producer of CRM for diffusive samplers in the database for index Code of Reference Materials (COMAR)7 which includes more than 200 producers of CRMs throughout the world. The catalogue of CRMs of the Institute for Reference Materials and Measurements (IRMM)8 presents CRMs for diffusive samplers but only for benzene in cartridges. In 2002, the National Institute for Standards and Technology (NIST)9 did not include this type of CRM in its catalogue.
The CERMATAIR project,10 partly funded by the European Community, studied the feasibility of producing and certifying reference materials appropriate to the concentration levels reflected by the limit values (LV) of the European Directives for ambient air monitoring.2,11 The gaseous pollutants SO2, NO2, carbon monoxide and benzene were considered. The study included CRMs in pressurized cylinders as well as CRMs for measurements by diffusive samplers. This paper presents the results of a feasability study carried out by the Joint Research Centre (JRC) for the preparation and certification of reference materials for NO2 and SO2 in diffusive samplers. The majority of results comes from the CERMATAIR project. The objective for these Reference Materials (RMs) was to reach an uncertainty of 5%. The study consisted of:
• the preparation of a batch of RMs at the LV of the Directive: 40 μg m−3 for NO2 and 20 μg m−3 for SO2
• the determination of their homogeneities
• small-scale external verifications performed by laboratories other than the preparation laboratory aimed at identifying discrepancies between concentration values from the preparation processes and verified values
• a two-year stability study of the prepared RMs
• an evaluation of uncertainty in view of the certification of the candidate CRM.
Experimental
Usually, RMs for diffusive samplers are prepared by field exposure12 or liquid spiking.13,14 However, field exposure does not allow the preparation of RMs at a target concentration while liquid spiking does not allow assessment of discrepancy of the chemical extraction prior to analysis. In this study it was intended to prepare RMs by exposure to standard gas mixtures.Coating and analysis of Palmes diffusive samplers
Several designs exist for diffusive samplers: Palmes diffusive samplers,15 radial samplers16 and badges.17–19 In order to focus on one of the most common diffusive samplers in Europe, the Palmes diffusive sampler15 supplied by Gradko Int. Ltd was chosen. In the year 2000, about 1200 sampling sites14,20 using Palmes diffusive samplers were implemented for monitoring ambient NO2 in the UK and 2000 sampling sites21 were implemented in France. This monitoring involves some 300 local authorities in the UK and 34 different laboratories in France.The Palmes diffusive sampler is capable of taking samples of gases from the atmosphere at a rate controlled by tube dimensions and gaseous diffusion, first through a porous material (a PTFE teflon membrane) and second through a static layer (see Fig. 1 left). Full details of the preparation of the diffusive samplers are given in Gerboles et al.22 Diffusive samplers are used to monitor the concentration of a pollutant in ambient air, C, according to eqn (1)23 where m is the mass of pollutant, U is the so-called uptake rate and t is the averaging time.
| C = m/Ut | (1) |
 | ||
| Fig. 1 (left) Details of preparation of the Palmes diffusive sampler with a membrane; (right) identified RM stored in a plastic container where the sampler is closed with a white plastic cap. | ||
Nitrite was analysed by colorimetry.28 Three ml of a mixed reagent (N-(1-naphtyl)-ethylendiamin dihydrochloride 0.007% v/v and sulfanilamide 2% v/v in orthophosphoric acid 5% v/v 29) were added in the Palmes diffusive samplers, which was then stirred up with a vortex for 1 min at 2000 turns min−1. The absorbance of the formed pink complex was measured at 540 nm after 45 min. In order to quantify nitrite, a 6-point calibration of nitrite, prepared in samplers, was plotted prior to each analysis.
Sulfate (and on occasions nitrite) was analysed by ion chromatography30 (IC). It was extracted adding 5 ml of MilliQ water directly into the Palmes diffusive sampler and then stirred up with a vortex for 2 min. The analysis was performed using an auto-sampler (Dionex AS 40) and 5 ml vials on a Dionex DX-100 with suppressed conductivity detection. The chromatographic system consisted of a IonPac AS 9 SC Analytical column with eluent flow rate of 2 ml min−1 and a loop of 1 ml. As for NO2, a 6-point calibration, in the range 0–0.3 μg ml−1 of sulfate, was plotted prior to each analysis in order to quantify sulfate. Sulfate and nitrite were identified by retention time and quantified by peak area.
It is necessary to optimise the method of measurement to reach a low uncertainty. Thorough cleaning of the Palmes samplers22 was required in order to limit the value and variability of blank. For NO2 analysis, the variability of the injected volume of reagent was controled to a relative standard deviation (RSD) of 0.1%. The uncertainty of the nitrite standards of the calibration line was also limited by systematically using micropipettes in a range of volumes for which the error is neglectable. For example, aliquots of 100 to 500 μl were pipetted from a 1000 mg l−1 standard solution of nitrite further diluted at 2 mg l−1.
The concentration of sulfate in the sampler was about 160 ng ml−1 (0.8 μg of sulfate extracted with 5 ml of MilliQ water), and thus a limit of quantification of 15 ng ml−1 is advised. At such a level, thorough cleaning of vials and their cap is compulsory. It is recommended that the analyst wears gloves. Since the DX-100 chromatograph suffers from a low signal/noise ratio, the volume of the loop must be as large as possible to avoid the fact that the variability of the baseline influences the integration of the peaks. The volume of the loop is limited by the size of the vial (5 ml) and by the need to flush the loop with a volume of sample coresponding to at least 3 times the loop. Therefore, a loop of 1 ml was found suitable.
Preparation of NO2 RM by exposure
The RMs for NO2 were prepared by exposure to a standard gas mixture using an exposure chamber. The exposure chamber was a ring tube system shaped “O” (size including support, 130 × 110 × 140 cm) made of borosilicate glass (see Fig. 2). The controlling system of the exposure chamber had the capacity to set target values and to control the concentration of the gas mixture, temperature, relative humidity and wind velocity inside the chamber.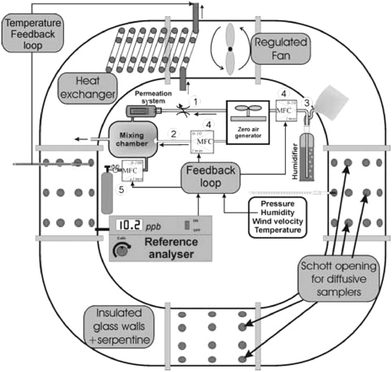 | ||
| Fig. 2 Scheme of the exposure chamber. 1—carrier flow with sonic orifice, 2—dry flow (complementary gas), 3—humid flow, 4—mass flow controller, 5—dynamic dilution of a highly concentrated NO2 cylinder. | ||
The wind velocity was created by a small fan (Papst series DV 6224) installed inside the chamber (see Fig. 2). Regulation of the fan speed allowed the control of the wind velocity between 0.4 and 4 m s−1. Gas mixtures were generated using a permeation system.31 The flow of the carrier gas (see Fig. 2-1) was controlled by a sonic orifice32 while the flow of complementary gas (called dry flow in Fig. 2-2) was controlled by thermal mass flow controllers33 (see Fig. 2-4). Relative humidity was controlled using the saturation method34 where dry air (called humid flow in Fig. 2-3) passes over the liquid and pushed air saturated with water vapour into the chamber. All flows were traceable to the reference standards of the laboratory which consisted of three mercury sealed chronovolumeters (Brooks SA).35 The balance between dry air and saturated air allowed the setting of relative humidity in the range of 0 to 85%. In the exposure chamber, the temperature was controlled using a heat exchanger inside the chamber and with a serpentine on the walls of the chamber. Two cryostats (Julabo FP 45) were used to supply cooling or warming liquid to the heat exchanger and to the serpentine. The temperature of the liquid was regulated using a Proportional-Integral-Derivative controller (PID). The walls of the exposure chamber were insulated to limit photochemical reactions and heat exchange with surroundings.
The Palmes diffusive samplers were introduced or removed through 72 Schott 32GL openings (see Fig. 2). In the exposure chamber, the NO2 concentration was monitored using chemiluminescence36,37 (analyser Environnement SA model AC 31M). The analyser was calibrated using transfer standards (gas cylinders) certified against the permeation method and cross-checked with the static volumetric method.38 Temperature, relative humidity, pressure and wind velocity were continuously recorded using a multifunction probe for temperature, humidity, wind velocity (Testo Term 0635.1540) and relative pressure sensor (Testo Term 0638.1445). The sensors and the analyser were connected to a data logger (ramlog 9000 abi data) for continuous monitoring of ambient conditions.
The target conditions of exposure were established according to the CEN validation protocol for diffusive samplers: 23 40 μg m−3 of NO2 concentration, 2 weeks of averaging time, temperature of 20 °C, wind speed of about 3.5 m s−1 and relative humidity of 50%. However, several exposures under the CEN conditions gave a higher nitrite variability than expected with RSD over 5%. According to Gerboles et al.,22 higher variability is observed for low concentration, long exposure period and high wind speed. The exposure conditions were thus modified in order to decrease the variability and were as following: wind speed of 1 m s−1, exposure period of 4 days and NO2 concentration of about 120 μg m−3 corresponding to the 40 μg m−3 over 2 weeks.
The exposure chamber was able to accommodate up to 72 samplers for each exposure. Half of each exposure set was used to check the homogeneity of the nitrite content. Four different exposures were performed in order to prepare 140 RMs for the characterisation, homogeneity and stability study. A fifth exposure was carried out to prepare RM for an external verification.
Preparation of SO2 RM by liquid spiking
The conditions of exposure defined in the CEN protocol23 were initially adopted: 20 μg m−3 of SO2, 2 weeks of averaging time, temperature of 20 °C, wind speed of about 3 m s−1 and relative humidity of 50%. Two experiments carried out in the exposure chamber under these conditions gave RSDs of 29% and 40% (n = 72) for the mass of sulfate.This variability in the preparation was attributed partly to high wind velocity and to absorption of SO2 in water that condensated in the chamber. Low wind velocity in the chamber favored the homogeneity of wind speed at all the positions where the samplers were installed. By contrast, higher wind speed created more turbulence in the chamber with different wind speed at different spots of the chamber. Several authors22,24 showed that the uptake rate changes with wind speed. Consequently, the high wind velocity produced higher variability of uptake rate and scattered response of the samplers. Furthermore, the temperature at the heat exchanger could go down to 12 °C to ensure a temperature of 20 °C in the chamber. This low temperature produced water condensation on the glass serpentine of the heat exchanger on which absorption of SO2 took place. This created inhomogeneity of the SO2 concentration in the chamber and therefore scattering of SO2 diffusive samplers responses (RSD of 40%). The vapor-phase water in the sampled air did not cause the variability, it was the result of the artificially created condensation. It was thus decided to prepare the RMs for SO2 by liquid spiking rather than by exposure to gas mixtures.
The spiking solution was prepared from a sulfate standard solution (1000 mg l−1) supplied by Merck (1.19813.0500). The sulfate standard was diluted using milliQ water so that 20 μl contained 0.8 μg of sulfate. This volume was injected in the sampler using a calibrated micropipette (Mettler-Toledo model VoluMate 20 μl). The micro-pipette was found to be more precise than a micro syringe which introduces readability and vaporization errors. Another 20 μl of a solution containing TEA in water 10% v/v and Brij 0.06% v/v (Brij 35 Merck 50067108) were previously coated on the meshes of each sampler. About 140 diffusive samplers were prepared by liquid spiking on September 24th and 25th 2002. They were identified using a unique number from 1 to 140.
Experimental results
NO2
The homogeneity of the nitrite content in the samplers was evaluated by analysing half of four 72-tube sets whose RSDs were 2.5, 1.5, 2.1 and 3.1% (see Table 1). All analysis pertaining to the same batch were carried out under conditions of repeatability.The stability of the nitrite content of the RMs was studied over a period of two years. After 3, 6, 12, 15 and 24 months of storage in a refrigerator and at room conditions, sets of 10 RMs were analysed by colorimetry by the JRC. The trends of nitrite in blank samplers and exposed RMs are given in Fig. 3. All values are in μg and the blank is already subtracted. The blank samplers were closed after preparation and imediately stored in a refrigerator during the whole stability testing exercise.
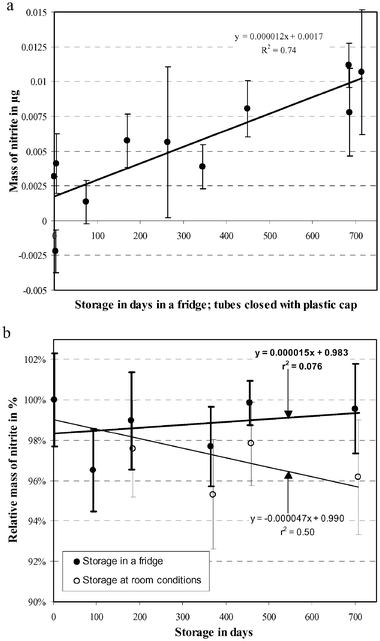 | ||
| Fig. 3 (a) Stability of blank NO2 RMs, (n = 3 to 8), (b) stability of NO2 RMs vs. storage duration for 2 different storages. The Y-error bars represent the relative standard deviations of homogeneity (n = 10 at least). | ||
A measurement of the nitrite content in the exposed RMs was carried out by 6 French network laboratories: AIRLOR, AIR LR, AIRPARIF, ATMO CHAMPAGNE-ARDENNES, ATMO PICARDIE, ATMO POITOU-CHARENTES, a national reference laboratory (Ecole des Mines de Douai, France) and the JRC-Institute for Environment and Sustainability (EC). 6 RMs prepared during the 5th exposure were sent to all participants. Samplers were transported by private courier. Upon receipt, the samplers had to be stored in a refrigerator in their containers and had to be protected from light. Before analysis the samples were exposed to ambient temperature for about 20 min. The results of the measurements are given in Table 2.
| Laboratories | Nitrite/μg | Analytical method |
|---|---|---|
| a The quoted values are standard deviations. | ||
| A | 0.817 ± 0.012 (n = 9) | Colorimetry30 |
| B | 0.909 ± 0.021 (n = 7) | Ion chromatography30 |
| C | 0.838 ± 0.016 (n = 6) | Colorimetry28 |
| E | 0.818 ± 0.012 (n = 6) | Colorimetry28 |
| F | 0.860 ± 0.012 (n = 6) | Ion chromatography39 |
| G | 0.835 ± 0.012 (n = 6) | Colorimetry28 |
| H | 0.815 ± 0.040 (n = 6) | Colorimetry28 |
| I | 0.692 ± 0.124 (n = 6) | Colorimetry28 |
| K | 0.735 ± 0.012 (n = 6) | Colorimetry28 |
SO2
The homogeneity of preparation was evaluated on four different batches of about 45 samplers. Their RSD was 2.3, 2.4, 1.4 and 1.9%, respectively. The method of the normalized deviation, described in ISO Guide 43-1,40 was used to evaluate whether the differences between gravimetry and IC remained within the uncertainty of the two methods of measurements. The normalised deviation is calculated according to eqn (2) where X0 is the gravimetric value with expanded uncertainty U(X0) and X is the IC value with expanded uncertainty U(X). As |En| is always ≤1 (see Table 3), it is confirmed that the difference remains within uncertainties.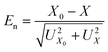 | (2) |
| 24/09/2002 | 25/09/2002 | |||||
|---|---|---|---|---|---|---|
| Gravimetry | IC | E n | Gravimetry | IC | E n | |
| a Gravimetry: U = 0.02 μg and IC: U = 0.032 (24/09) and 0.046 (25/09). | ||||||
| 0.780 | 0.795 | −0.4 | 0.784 | 0.804 | −0.4 | |
| 0.780 | 0.812 | −0.8 | 0.784 | 0.775 | 0.2 | |
| 0.780 | 0.787 | −0.2 | 0.784 | 0.816 | −0.6 | |
| 0.780 | 0.795 | −0.4 | 0.784 | 0.785 | 0.0 | |
| 0.780 | 0.797 | −0.5 | 0.784 | 0.811 | −0.5 | |
| 0.780 | 0.807 | −0.7 | 0.784 | 0.802 | −0.3 | |
| 0.780 | 0.778 | 0.1 | 0.784 | 0.802 | −0.4 | |
| 0.780 | 0.779 | 0.0 | 0.784 | 0.822 | −0.8 | |
| 0.780 | 0.789 | −0.2 | ||||
| Average | 0.780 | 0.793 | −0.5 | 0.784 | 0.802 | −0.6 |
| Initial values | Refrigerator | Room | |
|---|---|---|---|
| a The quoted values are standard deviations and n is the number of RMs. | |||
| 3 months | 0.784 ± 0.02 (n = 9) | 0.775 ± 0.186 (n = 10) | 0.645 ± 0.290 (n = 10) |
| 6 months | 0.784 ± 0.02 (n = 9) | 0.590 ± 0.236 (n = 10) | 0.681 ± 0.156 (n = 10) |
| 12 months | 0.784 ± 0.02 (n = 9) | 0.758 ± 0.302 (n = 10) | 0.672 ± 0.226 (n = 4) |
| 0.780 ± 0.02 (n = 9) | 0.792 ± 0.087 (n = 9) | 0.815 ± 0.130 (n = 10) | |
| 24 months | 0.784 ± 0.02 (n = 9) | 0.981 ± 0.342 (n = 17) | 0.871 ± 0.019 (n = 4) |
A limited external verification was also carried out. A set of 10 RMs was analysed by the National Physical Laboratory (NPL, UK), the Dutch Measurement Institute (Nmi, NL) and the Health and Safety Laboratory (HSL, UK). All the analysis were performed by IC. None of the laboratories reported analytical results that are useful for external verification. HSL found that the results obtained for sulfate after blank subtraction were not consistent due to analytical problems (method OSHA ID-18241) and ranged from 0 to 1 μg. NMi reported high blanks and highly variable sulfate levels upon analysis (0.65 ± 0.2 μg), NPL, giving no sulfate blank, reported a mean value of 0.6 with expanded uncertainty of ±0.3 μg for the 10 samples analyzed over 3 days.
Certification of RMs and uncertainty
NO2
Following the Guide to the Expression of Uncertainty in Measurement,42 the combined standard uncertainty for a CRM should include the uncertainty of the batch characterization (uchar) determined using a primary method of analysis, the uncertainty sources coming from the lack of homogeneity of preparation (uuh), from the instability during storage of the RM (ults) and from the transport to the customer (usts)43,44 as shown in eqn (3)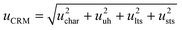 | (3) |
Batch characterization, traceability of preparation
The mass of nitrite in the RMs was determined according to eqn (4) where mNO2 is in μg, NO2− is the nitrite concentration in μg ml−1 found in the Palmes diffusive sampler after the addition of the reagent and V is the volume of the reagent (3 ml). In the Palmes diffusive sampler, the reagent is added using a dispenser (macro-transferpettor Brand-G) adjusted on balance and with RSD of repeatability of 0.1%. NO2− was determined using the direct comparison method (ISO 614345) considering the uncertainty of the nitrite standards and of the spectrophotometer responses. An optimising algorithm is then applied in order to fit the best regression line (eqn (5)) by minimising the error function weighted with the uncertainty of nitrite standards and of spectrophotometer responses. The slope b1 and intercept b0 of the linear curve are calculated together with their standard uncertainty and covariance. NO2− is calculated using eqn (5) where yc is the response of the spectrophotometer. The combined uncertainty of mNO2 (uchar) is obtained by applying the law of propagation of uncertainty (eqn (6)) which includes the correlation term between NO2− and V (r is assumed to be −1). The standard uncertainty for the volume of reagent u(V) includes contribution from the repeatability of the dispenser, from the used mass block and from the repeatability/linearity of the balance. The uncertainty of NO2− is calculated using eqn (7) where the standard uncertainty of yc is equal to the repeatability of the spectrophotometer. Applying eqn (6), the uncertainty uchar was found to be 1.0%.| mNO2 = NO2−V | (4) |
| NO2− = b0 + b1yc | (5) |
| uchar = (NO−2)2u2(V) + V2u2(NO2−) − 2VNO2−u(NO2−)u(V) | (6) |
| u2(NO−2) = b21u2(yc) + u2(b0) + y2cu2(b1) + 2yccov(b0,b1) | (7) |
The analytical method was calibrated using certified nitrite standard supplied by Merck-BdH and traceable to NIST with expanded uncertainty of 0.5% (nitrite 1000 ppm standard for IC, ARISTAR, Merck Eurolab, 458022S).
Homogeneity
Potential statistical outliers were not discarded from the homogeneity and stability testing in order to avoid influencing the final uncertainty by statistical treatment of data.The between RM standard uncertainty uexp (or the standard deviation of the sets tested for homogeneity) is the square root of the quadratic sum of standard deviations of measurement method under repeatability conditions (smeth) and uuh the between RMs variation according to eqn (8).46 The between RM variation comes from several parameters e.g.:
• reproducibility of cleaning, coating and membrane installation,
• variability of the sampler dimensions (both the diffusion path and cross-area) which results in variability of the uptake rate U of each sampler and hence in the nitrite content according to eqn (1),
• difference of the porosity of the sampler membrane,
• gradients of the experimental conditions (temperature, humidity, NO2 concentration or wind velocity22) in the exposure chamber modifying the uptake rate U of each sampler,
• position and orientation of the sampler in the chamber,
• reproducibility of extraction and analysis.
s meth also comes from several factors: the repeatability of the injection of reagents (u = 0.1%), the efficiency of the extraction of nitrite, the skill of the analyst in using the spectrophotometer, the repeatability of the spectrophotometer (u = 0.66%) and the daily drift of the spectrophotometer due to changes of conditions in the laboratory (in particular zero drift with u = 0.66%). To diminuish the effect of smeth, large batches (n = 33) were taken to estimate uuh. A sum of the relative variances of the repeatability of injection and of the repeatability/drift of the spectrophotometer gave smeth equal to 0.9%. Since the 4 batches of the homogeneity testing had a normal distribution, a mean of their standard deviation was used to estimate uexp (see Table 1). uuh was found to be 2.3% by subtracting of s2meth/n from u2exp and computing its square root according to eqn (8). The nitrite content of samplers was not affected by the orientation and position of the samplers when evaluated by analysis of variance.
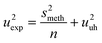 | (8) |
Stability testing and expiry date (shelf life)
The estimation of uncertainty instability is based on regression analysis of stability data with subsequent testing of the slope of the regression line for significance. The instability due to degradation during long term storage is then included in eqn (9)47 and is used in the determination of a shelf life of the CRM. The estimation of stability during storage was tested in the conditons relevant to storage in a refrigerator. Normalized results are given in Fig. 3b. A straight line can be fitted through the data with initial value C0, linear degradation rate b, time x and ε which denotes the random error component as shown in eqn (9). Using the results of Fig. 3b, the slope of the regression line corresponding to the degradation of long term storage is −1.5 × 10−5% day−1 with uncertainty ub equal to 7.7 × 10−6% day−1. The degradation rate does not show significant difference from 0 (tcritic of 2.02 for 45 degrees of freedom compared with a calculated value of 1.93) and therefore b is set to 0 in eqn (9). In order to avoid underestimation of the uncertainty of degradation of the CRMs, ults is set to xub where ub is the standard uncertainty of the degradation rate b.47 In this expression x is 5 years, the proposed shelf life of the CRM and ub is calculated using eqn (10). Ults is equal to 1.4% for a shelf life of 5 years.| C(C0,b,x) = C0 + bx + ε | (9) |
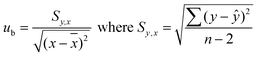 | (10) |
Degradation due to transport of RMs
In order to elucidate the possible degradation during transport of the CRM to the user, two types of experiments were carried out:• A stability testing with storage at room conditions (see Fig. 3b).
• A limited batch of RMs was sent to ATMO-Picardie and was analysed by the JRC before sending and after being received.
Even though the transport has a short duration, the stability testing was extended to two years in order to check whether refrigerator storage is necessary and to detect possible degradation. Applying the same treatment as for the long-term storage in a refrigerator to the storage at room conditions, the degradation rate b is −4.7 × 10−5% day−1 with uncertainty ub equal to 8.8 × 10−6% day−1. The degradation rate b is significantly different from 0 (t is 5.4 for a tcritic of 2.03 with 36 degrees of freedom). Therefore usts can be calculated using eqn (11)47 where C0 is the initial value (100%) and x is 60 days, a long-enough period for transport. Finally, the uncertainty of instability due to degradation during short term storage (transport to the user) usts is 0.3%.
| u2sts = (C0b)2u2(x) + x2u2(b) | (11) |
For the RMs that were sent by plane to ATMO-Picardie by private courier, 9 samplers were analysed before sending and gave a mass of nitrite of 0.81 ± 0.016. The ones analysed after being received by Atmo-Picardie gave a mass of nitrite of 0.80 ± 0.008 μg (n = 9). These two series of results showed a slight significant difference (t is 2.24 for tcritic of 2.15). The between-days variation of the analytical system may explain this difference rather than the degradation due to transport. Therefore this test did not appear to be fitted for purpose.
Certification and external verification
By applying eqn (3), the expanded uncertainty for the certification of RMs UCRM was found to be 5.8% using a factor of coverage k of 2.The following statistical tests were applied to the results of the external verification: Mandel k, Cochran’s test and Grubb’s tests.48 In this way, lab I was discarded because of its high scattered values. Laboratory I did not pass the Cochran’s test for homogeneity of variances and was requested to analyse another set of RMs after improvement of its analytical system. The new results of lab I (1.04 ± 0.02 μg) showed a better agreement with the simultaneous analysis by JRC (1.09 ± 0.02 with n = 6 21). After discarding lab I and applying the Grubb test on average values, lab K was found to present outliers. This outlier was due to a wrong analytical blank value.21
The normalised deviation En was computed for all pairs of results of the non-discarded participants. The normalised deviation is calculated according to eqn (2). In this test both U(X) and U(X0) are estimated with the sole uchar and Ults (2%). If |En| ≤ 1 then no significant differences are evidenced between two laboratory results and if |En| > 1 then the difference is significant. Table 5 shows that the En numbers for pairs consisting of one laboratory using colorimetry and one using IC or two using IC are always higher or equal to 1. On the contrary for pairs consisting of 2 laboratories using the colorimetric method, the En numbers are lower than 1.
JRC has further investigated this observation, analysing sets of samplers simultaneously exposed. The results confirmed the higher results for IC compared to colorimetric ones. For example, the analysis of a set of tubes gave 1.13 ± 0.03 μg using IC while colorimetry gave 1.03 ± 0.02 μg. The colorimetric method was then modified with pre-extraction with water before analysis, like for IC. By extracting with 1 ml of MilliQ water before analysis, the mass of nitrite determined by colorimetry became 1.11 μg ± 0.02 a value equivalent to the IC one. Several comparisons between the colorimetric analysis of exposed samplers, with and without previous water extraction, gave systematically higher results for the water extraction. On one set of analysis, JRC found 0.625 ± 0.008 μg for the extraction with water and 0.573 ± 0.008 μg for the extraction with the mixed reagent. On the same set of exposed samplers, AIR LR (F) found 0.611 ± 0.022 μg with extraction with MilliQ water and 0.541 ± 0.021 μg for the extraction with the mixed reagent. These findings confirm the increase of mass of nitrite when the samplers are extracted with water.
The mixed reagent has ortho-phosphoric acid because the Griess–Saltzmann reaction needs an acidic medium.49 In fact, the pH of the mixed reagent is 1.15. However, the efficiency of the extraction is limited by the pH as shown in Fig. 4. The plot was obtained by analysing samplers after extraction with 1 ml of MilliQ water at different pH prepared using ortho-phosphoric acid. The pH of the solution with mixed reagent and water was lower than 2 so that the Saltzman reaction itself was not limited in these tests.
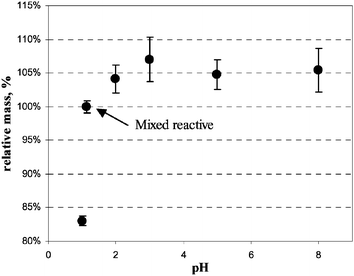 | ||
| Fig. 4 Effect of pH on the efficiency of extraction. | ||
The extraction with a solution containing 1.5% of TEA like that proposed in the OSHA method41 shall be rejected as well. Underestimation of 35% for NO2 and 40% for SO2 together with a RSD of 30% as compared to the results of the IC method have been observed by analysing sets of 10 samplers exposed to standard gas mixtures.
SO2
The characterization of the mass of sulfate in the samplers was found to be possible. The expanded uncertainty resulting from gravimetry was about 2.5% and was confirmed by comparison to IC analysis (see Table 3).The micro-pipette used for spiking was calibrated by gravimetry using a 10 μg readability balance and E2 mass blocks. The repeatability of the micro-pipette was evaluated daily (e.g. ±0.4% for one preparation). Solutions of sulfate were used for the liquid injection. The reference values of these solutions were derived from calibration using a certified standard of sulfate supplied by Merck-BdH and traceable to NIST with an expanded uncertainty of 0.5% (sulfate 1000 ppm standard for IC ARISTAR, Merck Eurolab, 458062D). At the time of preparation, the homogeneity as determined by IC was also satisfying.
However, the results of the instability testing and the external verification showed the impossiblity to certify RMs for SO2 prepared by spiking. The results do not indicate instability of the sulfate spiked onto the Palmes diffusive sampler. However, the spread found in the results is quite large (over 30%). Sometimes, sulfate contents near 0 were found for the external verification.
Discussion and conclusion
The preparation of RMs for NO2 by exposure to standard gas mixtures was found feasible. The RMs showed good homogeneity and long/short-term stability. The mass of nitrite in the samplers could be characterized. The target values for nitrogen dioxide, based on characterization, could be verified by external analyses. This indicated the feasibility of certification of the RMs from measurement data, when measurements are performed under conditions of full traceability to SI units. The relative expanded uncertainty of certification was 5.8%. As the objective was to reach less than 5%, it is proposed to use pairs of RMs whose average can be certified with expanded uncertainty of 4.2%. In this case, the contribution of homogeneity to the expanded uncertainty is reduced by a factor of square root of 2. The proposed shelf life of the CRM, used in the calculation, was 5 years. The biggest contribution to UCRM came from the homogeneity of preparation and the characterisation. The uncertainty estimation did not include a contribution of the differences between the results of analysis by IC and spectrophotometer. It is considered that the present implementation of the colorimetrc method underestimates the nitrite content. A solution is proposed to solve this by extracting with 1 ml of MilliQ water prior to the addition of the mixed reagent.The results of this study indicate that the nitrite content of Palmes diffusive samplers is stable for at least two years when stored in a refrigerator while a decrease at room temperature conditions is observed. Moreover, blanks tubes show a low increase in mass which does not interfere with normal use. This finding contradicts the recommendations generally given for preparation and analysis of NO2 Palmes diffusive samplers, namely that these should be analyzed within a few weeks after sampling.50
For SO2, it should be noted that during the external verification, all laboratories had difficulties in reproducing the preparation contents of sulfate with acceptable uncertainties. The apparent analytical problems underlying these findings—although unsatisfactory from the viewpoint of feasibility of preparing and certifying the RM for sulfur dioxide—emphasize the need for making available such a reference material.
In another study, using liquid spiking for the preparation of NO2 RMs, an important instability was also observed.21 This instability was attributed to the injection of nitrite on the walls of the samplers where the nitrite was then reacting. By including nitrite in the coating solution with the absorbent, good stability of the mass of nitrite over a month have been attained.21 This solution could be implemented for SO2 too by adding both nitrite and sulfate in the absorbing solution. The sulfate solution was injected on the walls of the samplers where it degenerated overtime, instead of being injected in the liquid absorbent where it could coupled with TEA. This caused the discrepancy. A correct injection is confirmed by the precise and exact analysis at the preparation time and further analysis with increasing bias and variability after 3, 6, 12, 24 months of the stability study (see Table 4).
For the preparation of RMs for SO2 by exposure, the condensation of water was the major cause of discrepancy. In order to avoid condensation, the temperature in the exposure chamber was increased so that the standard gas mixture would be far enough from its dew point. Simultaneous NO2 and SO2 exposure has been carried out under these conditions. Good homogeneity was achieved with RSD of 3% for nitrite and 6% for SO2 (compared with the RSD of 30% when water condensation took place in the chamber).
According to the present feasability study, the exposure chamber would allow the certification of only about 50 RMs. Certifying RMs needs a lot of work which may not be worthwhile for a set of only 50 CRMs. Therefore, the installation of the samplers in the 3 empty curves of the exposure chamber (see Fig. 2) has beeen tested. This would allow the exposure of 360 samplers at the same time. A verification both by spectrophotometry and IC of the homogeneity of 2 batches of 25 samplers out of 360 prepared by exposure at 25 °C confirmed previous results with RSDs of 2% for NO2 and 5% for SO2.
In conclusion, NO2 Palmes diffusive samplers should be prepared by exposure to gas mixtures rather than by liquid spiking since extraction of nitrite is problematic. Moreover, using the exposure method, SO2 can be added to the RMs for NO2 provided that condensation does not occur. Using this method, the RMs for SO2 showed a satisfying homogeneity. It is anticipated that the instability observed in the preparation of RMs for SO2 by liquid spiking could disappear in the preparation by exposure to gas mixtures. By modifying the method of installation of the samplers in the chamber sets of more than 300 RMs could be prepared and certified.
Acknowledgements
The authors wish to acknowledge the support of several laboratories for their analytical support: P. Quincey, National Physical Laboratory (NPL, UK), A. Baldan of the Dutch Measurement Institute (Nmi, NL) and M. Wright of the Health Safety Laboratory (HSL, UK), N. Marquis of Association pour la Surveillance et l’Etude de la Pollution Atmosphérique en Lorraine (AIRLOR, France), B. Dadolle of Air Languedoc-Roussillon (AIR LR, France), E. Le Bronnec of Surveillance de la Qualité de l’air en Ile-de-France (AIRPARIF, France), E. Chrétien of Association Régionale pour la Surveillance de la Qualité de l’Air en Champagne-Ardenne (ATMO Champagne Ardennes, France), B. Rocq et E. Roullet of Association pour la Surveillance de la Qualité de l’Air en Picardie (ATMO Picardie, France), S. Morenilla of Association Régionale pour la Mesure de la Qualité de l’Air en Poitou-Charentes (ATMO Poitou Charentes, France).This project was partly funded by the European Community under the ‘Competitive and Sustainable Growth’ Programme (1998–2002), contract No G6RD-CT-2001-00517, project No GRD1-2000-25015, acronym CERMATAIR title: Certified Reference Materials for the Measurement of Gaseous Compounds in Ambient Air.
References
- EEC Directive, L 296, 1996, No. 62.
- EEC Directive, L 163/41, 1999, No. 30.
- R. H. Brown, J. Environ. Monit., 2002, 2, 1–9 RSC.
- M. Gerboles, L. Amantini, F. Detimmermann and M. Paryssat, Studio della distribuzione del biossido di azoto a Bologna mediante campionatori passivi, EUR 18145 IT, Office for Official Publications of the European Communities, Luxembourg, 1998 Search PubMed.
- K. Stevenson, T. Bush and D. Mooney, Atmos. Environ., 2001, 35, 281–287 CrossRef CAS.
- M. Ferm and H. Rodhe, J. Atmos. Chem., 1997, 27, 17–29 CrossRef CAS.
- http://www.comar.bam.de.
- http://irmm.jrc.cec.eu.int.
- http://www.nist.gov.
- Th. L. Hafkenscheid, Certified Reference Materials for the measurement of gaseous pollutants in ambient air, Contract G6RD-CT-2001-00517, European Commission, www.cermatair.com Search PubMed.
- EEC Directive, L 313/12, 2000, No. 69.
- M. Hangartner, M. Kirchner and H. Werner, Analyst, 1996, 121, 1269–1272 RSC.
- P. Stacey, Proficiency testing for benzene, toluene, m-xylene and nitrogen dioxide in the air: experience in the WASP quality assurance scheme, EUR 20242 EN, Office for Official Publications of the European Communities, Luxembourg, 2002 Search PubMed.
- K. Stevenson, T. Bush and D. Mooney, Atmos. Environ., 2001, 35, 281–287 CrossRef CAS.
- E. D. Palmes, A. F. Gunnison and C. Tomczik, Am. Ind. Hyg. Assoc. J., 1976, 37, 570–577 CAS.
- V. Cocheo, C. Boaretto and P. Saccho, Am. Ind. Hyg. Assoc. J., 1996, 57, 897–904 CrossRef CAS.
- G. R. Carmichael, M. Ferm, N. Thongboonchoo, J.-H. Woo and L. Y. Chan, Atmos. Environ., 2003, 37, 1293–1308 CrossRef CAS.
- F. De Santis, F. Dogeroglu, T. Fino, A. Menichelli, C. Vazzana and I. Allegrini, Anal. Bioanal. Chem., 2002, 373, 901–907 CrossRef CAS.
- Ogawa Company, NO, NO2, NOx and SO2, Sampling Protocol using the Ogawa sampler, Pompano Beach, Florida, USA, http://www.ogawausa.com Search PubMed.
- T. Bush, K. J. Stevenson, S. Moorcroft and S. Smith, Validation of nitrogen dioxide diffusion tube methodogy, Stanger Science and Environment/National Environmental Technology Centre, Culham, UK, 1998 Search PubMed.
- M. Gerboles, D. Buzica, L. Amantini, H. Plaisance and H. Marfaing, Pollut. Atmos., 2004, 183, 345–359 Search PubMed.
- M. Gerboles, D. Buzica and L. Amantini, Atmos. Environ., 2005, 39, 2595–2608.
- European Committee for Standardization, Ambient air quality—Diffusive samplers for the determination of concentrations of gases and vapours—Requirements and test methods—Part 2: Specific requirements and test methods, EN 13528-2, Brussels, 2002 Search PubMed.
- Y. Yanagisawa, C. P. Hemphill, J. D. Spengler and P. B. Ryan, 86-37.7, in Proceedings of the 79th Annual Meeting of the Air Pollution Control Association, Minneapolis MN June 22–27, Air Pollution Control Association, Pittsburgh, PA, 1986 Search PubMed.
- B. A. Scheeren, F. De Santis, I. Allegrini and P. Heeres, Int. J. Environ. Anal. Chem., 1994, 56, 73–85 CrossRef CAS.
- D. Krochmal and A. Kalina, Atmos. Environ., 1997, 31, 3473–3479 CrossRef.
- J. E. Sickles, P. M. Grohse, L. L. Hodson, C. A. Salmons, K. W. Cox, A. R. Turner and E. D. Estes, Anal. Chem., 1990, 62, 338–346 CrossRef CAS.
- Passive Samplers for Nitrogen Dioxide, Ref 4414, Agence de l’Environnement et de la Maîtrise de l’Energie, ADEME Edition, Paris, 2002 Search PubMed.
- D. H. F. Atkins, C. Healy and J. B. Tarrant, Report, AERE R 9184, United Kingdom Atomic Energy Authority, Harwell Laboratory, Oxfordshire, UK, 1978.
- D. Buzica and M. Gerboles, Evaluation of the Palmes Tube Sampler with Membrane for the Simultaneous Determination of Nitrogen Dioxide and Sulfur Dioxide. Measurement Campaign in the Industrial Area of Martigue (F), Technical Note No. I.02.110, Office for Official Publications of the European Communities, Luxembourg, 2002 Search PubMed.
- International Organisation for Standardisation, Gas analysis—Preparation of calibration gas mixtures using dynamic volumetric methods—Part 10: Permeation method, ISO 6145-10, Geneva, 2002 Search PubMed.
- International Organisation for Standardisation, Gas analysis—Preparation of calibration gas mixtures—Dynamic volumetric methods—Part 6: Sonic orifices, ISO 6145-6, Geneva, 2003 Search PubMed.
- International Organisation for Standardisation, Gas analysis—Preparation of calibration gas mixtures using dynamic volumetric methods—Part 7: Thermal mass-flow controllers, ISO 6145-7, Geneva, 2001 Search PubMed.
- International Organisation for Standardisation, Gas analysis—Preparation of calibration gas mixtures using dynamic volumetric methods—Part 9: Saturation method, ISO 6145-9, Geneva, 2001 Search PubMed.
- French Standards Association, Qualité de l’air—Air ambient, Manuel d’instruction sur le calibrage des analyseurs et des échantillonneurs de polluants atmosphériqu—Echantillonnage—Station de mesurage et debits des gaz, NF XP X 43-053, Paris, 1997 Search PubMed.
- International Organisation for Standardisation, Ambient air—Determination of the mass concentration of nitrogen oxides—Chemiluminescence method, ISO 7996, Geneva, 1985 Search PubMed.
- European Committee for Standardization, Ambient air quality—Measurement method for the determination of the concentration of nitrogen dioxide and nitrogen monoxide by chemiluminescence, EN 14211, Brussels, 2005 Search PubMed.
- International Organisation for Standardisation, Gas analysis—Preparation of calibration gas mixtures—Static volumetric method, ISO 6144, Geneva, 2002 Search PubMed.
- H. Plaisance, I. Sagnier, J. Y. Saison, J.-C. Galloo and R. Guillermo, Environ. Monit. Assess., 2002, 79, 301–315 CrossRef CAS.
- International Organisation for Standardisation, Proficiency testing by interlaboratory comparisons—Part 1: Development and operation of proficiency testing schemes, ISO Guide 43-1, Geneva, 1997 Search PubMed.
- Occupational Safety & Health Administration, Nitrogen dioxide in workplace atmospheres (Ion Chromatography), OSHA–ID 182, U.S. Department of Labor, http://www.osha-slc.gov Search PubMed.
- International Organisation for Standardisation, Guide to the expression of uncertainty in measurement, ISBN 92-67-10188-9, Geneva, 1995 Search PubMed.
- T. P. J. Linsinger, J. Pauwells, A. M. H. Van der Veen, H. Schimmel and A. Lamberty, Accredit. Qual. Assur., 2001, 6, 20–25 Search PubMed.
- International Organisation for Standardisation, General requirements for the competence of reference material producers, Guide 34, Geneva, 2000 Search PubMed.
- International Organisation for Standardisation, Gas analysis—Comparison methods for determining and checking the composition of calibration gas mixtures, ISO 6143, Geneva, 2001 Search PubMed.
- J. Pauwells, A. Lamberty and H. Schimmel, Accredit. Qual. Assur., 1998, 3, 51–55 CrossRef.
- T. Linsinger, J. Pauwels, A. Lamberty, H. Schimmel, A. Van der veen and L. Siekman, Fresenius’ J. Anal. Chem., 2001, 370, 183–188 CrossRef CAS.
- International Organisation for Standardisation, Accuracy (trueness and precision) of measurement methods and results—Part 2: Basic method for the determination of repeatability and reproducibility of a standard measurement method, ISO 5725-4, Geneva, 1994 Search PubMed.
- French Standards Association, Teneur de l’air atmosphérique en dioxide d’azote. Methode de dosage par piégeage sur filtre imprégné de triéthanolamine, NF-X 015, AFNOR, Paris, 1976 Search PubMed.
- C. Kirby, M. Fox and J. Waterhouse, J. Environ. Monit., 2000, 2, 307–312 RSC.
| This journal is © The Royal Society of Chemistry 2006 |