Simultaneous determination of atmospheric sulfur and nitrogen oxides using a battery-operated portable filter pack sampler
Mirai
Watanabe
a,
Takejiro
Takamatsu
b,
Masami K.
Koshikawa
*b,
Kazunori
Sakamoto
c and
Kazuyuki
Inubushi
c
aGraduate School of Science and Technology, Chiba University, 648 Matsudo, Chiba, 271-8510, Japan
bSoil Science Section, National Institute for Environmental Studies, 16-2 Onogawa, Tsukuba, Ibaraki 305-8506, Japan. E-mail: mkanao@nies.go.jp; Fax: +81-29-850-2576; Tel: +81-29-850-2440
cFaculty of Horticulture, Chiba University, 648 Matsudo, Chiba, 271-8510, Japan
First published on 25th November 2005
Abstract
We developed a method to analyze atmospheric SOx (particulate SO42− + gaseous SO2) and NOx (NO + NO2) simultaneously using a battery-operated portable filter pack sampler. NOx determination using a filter pack method is new. SOx and NOx were collected on a Na2CO3 filter and PTIO (2-phenyl-4,4,5,5-tetramethylimidazoline-3-oxide-1-oxyl) + TEA (triethanolamine) filters (6 piled sheets), respectively. Aqueous solutions were then used to extract pollutants trapped by the filters and the resulting extracts were pre-cleaned (e.g. elimination of PTIO) and analyzed for sulfate and nitrite by ion chromatography. Recoveries of SO2 and NOx from standard pollutant gases and consistency of the field data with those from other instrumental methods were examined to evaluate our method. SOx and NOx could be analyzed accurately with determination limits of 0.2 ppbv and 1.0 ppbv (as daily average concentrations), respectively. The sampler can determine SOx and NOx concentrations at mountainous or remote sites without needing an electric power supply.
Introduction
Increases in atmospheric SOx and NOx cause acid depositions that have adverse effects on the forest ecosystem, as do ozone and water stress.1–4 Measuring changes in SOx and NOx concentrations is very important for evaluating the effects of atmospheric pollutants on vegetation. However, in mountainous and remote areas it is usually difficult to measure these pollutants because of the lack of an electric power supply. Passive samplers, which are inexpensive non-electric devices, are exclusively used for extensive monitoring at such sites,2,3,5 but they are not useful for short-term sampling because their slow collection rate is controlled by diffusion, and local meteorological conditions often affected the results.3,6,7Active samplers such as filter pack (FP) methods are widely used for sampling atmospheric pollutants.8–10 FPs have been employed in acid deposition monitoring networks in various countries, such as in EANET,11 CASTNet,12 and EMEP.13 Although FPs provide sensitive measurements and are relatively inexpensive and simple, sampling artifacts often occur in distinguishing particulate (SO42−, NO3− and NH4+) and gaseous (SO2, HNO3 and NH3) forms.14–16 To overcome these problems, annular denuder17 and impactor/honeycomb denuder/filter pack18 systems were introduced. Although these denuder systems are sophisticated and precise, they are relatively expensive, complex, and more labor-intensive than the more usual FPs.10
Active samplers are usually operated using a fixed power supply, and field-oriented samplers operated by batteries are limited;19–21 although many personal samplers to monitor occupational atmospheric environments have been developed and are usefully employed.22–27
We developed a battery-operated portable FP sampler to simultaneously collect atmospheric SOx and NOx at mountainous sites without the need for a power supply, and to obtain daily (or hourly if concentrations are relatively high) average concentrations. In mountainous areas, NOx concentrations can rapidly fluctuate depending on mobile sources (automobiles), local meteorological (e.g. formation of a temperature inversion layer) and weather conditions.1,2,28 They may also fluctuate as a result of NO, which is naturally emitted from the forest soils.29,30 On the other hand, SOx concentrations often change daily or hourly under the influence of volcanic gases (a specific condition in Japan). Forest health may be more seriously affected by short-term high concentrations of the pollutants than by long-term averages. Therefore, our FP sampler may provide a new tool for information gathering, one that would be available to monitor and diagnose forest health.
We analyzed SOx and NOx as total concentrations without distinguishing particulate SO42− and gaseous SO2, or NO and NO2 due to the following reasons: (1) SOx and NOx deposited to vegetation are converted rapidly to SO42− and NO3−, respectively; (2) The analytical precision for SOx is decreased by speciation, because of the very low SOx concentrations in Japan and unexpected artifacts (partial adsorption of SO2 on a pre-filter to collect particulate pollutants14,31); (3) NOx concentrations may be a useful index of air pollution, because most of NOx from anthropogenic sources is emitted initially as NO, and then converted to NO2. In Japan, NOx emissions (especially from automobiles) have been controlled under effluent standards (e.g. NOx and PM Act); and (4) All acidic pollutants including NO should be removed and so facilitate clean exhaust emission from our sampler to a small field chamber32 (details are to be published elsewhere).
The novelty of our method is highlighted by the following points: (1) Determination of NOx using a FP sampler is new, although such samplers (and personal samplers) have been applied for NO2.15,27,33; (2) Our FP sampler is a field-oriented type, which can operate continuously for periods of several days at mountainous sites; and (3) The system is completely air-tight from inlet to outlet; thus, clean exhaust at a constant flow rate can be supplied to a small field chamber, for example, for studying the material leaching from tree leaves.
The pollutants collected on the filters were analyzed by ion chromatography after extraction into aqueous solutions. The best sampling and analytical conditions to obtain the maximal and reproducible recoveries were investigated using standard pollutant gases. Then, field data obtained by our sampler were compared with those from other instrumental measuring methods to confirm performance.
Experimental
Sampler design
We developed battery-operated portable samplers (Fig. 1) and used them experimentally. SOx and NOx were collected on a Na2CO3 filter (1 sheet, 25 mm id, filter holder: Advantec PP-25) and PTIO (2-phenyl-4,4,5,5-tetramethylimidazoline-3-oxide-1-oxyl) + TEA (triethanolamine) filters (6 sheets in a pile, 47 mm id, filter holder: Advantec PP-47), respectively, by passing air sequentially through the filters. When sampling in the field, a Teflon net (127 mesh inch−1, Sefar) was attached to the air intake to avoid trapping insects, and a polypropylene shade was attached as a rain shelter. A carbon vane pump (G12/01-4EB, Thomas) and an integrating mass flow meter (CMS20, Yamatake) were operated using two parallel rechargeable 12 V DC batteries (NP7-12, Yuasa). Although some of the other pumps (e.g., the PCXR-8, SKC Inc.), used in personal samplers (e.g., the IOM aerosol sampler24), may be applicable to our sampler after slight modification, the pump used here was good in respect to its air-tightness, cleanliness (because it is oil-less), and torque (a constant flow rate was maintained after passing through 7 sheets of filters). Also, the flow meter was not affected by temperature or pressure; it recorded values at 20 °C and 1 atm. Silicone tubing (3 mm in inner diameter, 6 mm in outer diameter) was used to connect components. All the components were put in a plastic container (340 L, 230 W, 160 H mm), with the sampler weighing about 6 kg. The rapid decrease in the flow rate when the air intake or outtake was clogged verified that there was no air leakage. The sampler can be operated continuously for more than 24 h at a flow rate of ca. 1.0 L min−1 (the decrease in the flow rate was <10%).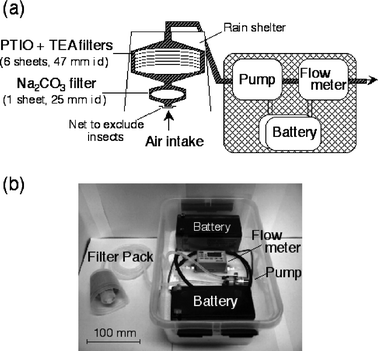 | ||
| Fig. 1 Scheme (a) and picture (b) of a portable filter pack sampler for SOx and NOx. | ||
Preparation of filters
Na2CO3 filter: Quartz fiber filters (Pallflex 2500QAST, 25 mm id) were washed with 0.1 M Na2CO3 by sonication for 30 min (28–100 kHz, Honda Electronics), and rinsed 3 times with deionized water. The filters were then soaked in 0.5 M Na2CO3 for 30 min (the solution was replaced with a fresh one during the soaking). After draining excess solution, the filters were dried in a vacuum desiccator, in which NaOH was used as the desiccant to prevent excessive drying.PTIO + TEA filter: Cellulose filters (Advantec No. 5C, 47 mm id) were washed with deionized water by sonication for 30 min, and rinsed 3 times with deionized water. The filters were then dried for more than 12 h at 60 °C in an electric oven. 0.5 ml of 0.1 M PTIO and 0.5 M TEA (in acetone) was impregnated homogeneously into the filter. The filters were then shaken gently to evaporate the acetone, stored in an opaque desiccator containing NaOH (or silica gel) as the desiccant, and used within five days (the filters can be stored for more than 1 month in a refrigerator34).
Post-sampling treatment of filters and analysis
After sampling, 30 ml of 0.3% H2O2 solution was used as an extractant on the Na2CO3 filter to oxidize the SOx collected and to extract it by dissolving it as SO42−. The extract was filtered (pore size 0.45 μm, GL Science), and subjected to SO42− analysis (and NO2− and NO3− analysis, if necessary). 30 ml of deionized water was applied to PTIO + TEA filters for more than 30 min with occasional shaking. Then 10 ml of chloroform (pre-washed with deionized water) was added to the extract, and the mixture shaken vigorously for 30 sec by hand. After centrifuging (800 G, 10 min), the filters were removed, and the mixture was shaken and centrifuged again. By this treatment, PTIO was removed from the extract into chloroform. Then, the extract (aqueous phase) was passed through a C18-cartridge (Sep-Pak, Waters) to remove the remaining traces of chloroform and PTIO. The resulting solution was subjected to NO2− analysis (and NO3− analysis, if necessary). Anions (SO42−, NO2− and NO3−) were analyzed by an ion chromatograph with a conductivity detector (DX-100, Dionex). The analytical conditions are summarized in Table 1. Filter blanks were also analyzed using unexposed filters, if necessary.| a Dionex. b Hitachi. | |
|---|---|
| Analyzer: | DX-100a |
| Column: | IonPac AS12A/AG12A (4 mm id)a |
| Eluent: | 2.7 mM Na2CO3 and 0.3 mM NaHCO3 |
| Flow rate: | 1.5 ml min−1 |
| Suppressor: | ASRS-ULTRA or ASRS-ULTRA IIa |
| Autosuppression recycle mode | |
| Sample injection volume: | 250 μl |
| Detector: | Conductivity detectora |
| Integrator: | D-2500 chromato-integratorb |
All chemicals were obtained from Wako Pure Chemical Industries, except for PTIO (Tokyo Kasei Kogyo). Deionized water was obtained from a Milli-Q system (Millipore).
Recovery tests
SO2, NO and/or NO2 in artificially polluted air samples (described below) were collected on various filters at flow rates of ca. 1.0 L min−1. Although recovery tests are usually conducted using dilute standard gases,33 relatively concentrated gases prepared in Tedlar bags were used because we wanted to accurately understand the loadings, and to quickly and inexpensively carry out the tests. Recoveries of pollutants were calculated using the following equation: recovery (%) = 100 × [SO42− or NO2− (+NO3−) found/SO2 or NOx loaded]. Since temperature was not controlled, it was assumed to be 20 °C (average room temperature), and deviations in recovery due to possible temperature changes were expected to be <3%. Because they were much lower than the loadings, corrections for filter blanks were not performed in this experiment.Preparation of artificially polluted air samples
Artificially polluted air samples were prepared in 1 L or 10 L Tedlar bags (GL Science) (thoroughly pre-cleaned with N2) with small Teflon balls (10 balls, 3 mm id, GL Science). The bag was filled with N2 (or air), and then appropriate small amounts of certified standard gases (SO2: 4178 ppmv at 20 °C, N2 balance, Takachiho Chemical; NO: 8150 ppmv at 20 °C, N2 balance, Takachiho Chemical; NO2: 2018 ppmv at 20 °C, N2 balance, Sumitomo Seika) were injected into the bag using a gastight syringe (1050TLL, Hamilton). The bag was then shaken vigorously to homogenize the gases. The amounts of pollutants in the bags corresponded approximately to those for 48 h sampling periods at polluted urban areas, i.e. SO2 was ca. 100 ppmv (in a 1 L bag) or 10 ppmv (in a 10 L bag), NO was 400 ppmv (1 L bag) or 40 ppmv (10 L bag), and NO2 was 200 ppmv (1 L bag) or 20 ppmv (10 L bag). The sampling duration for each test was ca. 1 min (for the 1 L bags) or 10 min (10 L bags). To prevent reaction and adsorption of gases in the bags, air samples were used just after preparation.Comparison of our data with that obtained by other instrumental methods
Daily average concentrations of SOx and NOx in suburban air were analyzed by this method at and around (within 10 m of) the Air Monitoring Station of the National Institute for Environmental Studies (NIES) (in Tsukuba, Ibaraki, Japan). The results were compared with those obtained by other instrumental methods;35i.e., concentrations of SO2 obtained by UV fluorescence (GFS-32, DKK) and those of NOx obtained by chemiluminescence (GLN-254, DKK). The comparisons were conducted for 56 sets of data obtained at a frequency of 1–3 times per month from January 2004 to February 2005.Statistical analysis
Statistical calculations (linear regression, correlation, and the Student t-test using paired data) were performed with StatView 4.5j (Abacus Concepts).Results and discussion
Recovery of SO2 from various alkaline filters
SO2, from the artificially polluted air samples containing SO2 alone or SO2 and NOx, was collected on various alkaline filters including NaOH, Na2CO3, K2CO3, tetrabutylammonium hydroxide, and on PTIO + TEA filters (Table 2). All filters were prepared by a method similar to that described above. Collection by an aqueous absorbent36 was also carried out, as an indication of the quantitative recovery.| Alkaline solutions impregnated into filters | Recovery of SO2 (%) | |||
|---|---|---|---|---|
| Alkali | Solvent | Volume/ml | From SO2 aloneb | From SO2 + NOxc |
| Values: means (standard deviations), replicates. The cellulose filters (Advantec No. 5C, 47 mm id) were used.a Tetrabutylammonium hydroxide.b 100 ppmv SO2 in 1 L N2.c 100 ppmv SO2 + 400 ppmv NO + 200 ppmv NO2 in 1 L N2.d Not tested. | ||||
| 1 M Na2CO3 | Water | 0.3 | 98.7 (0.3), 3 | 25.1 (3.5), 3 |
| 1 M Na2CO3·1.5H2O2 | Water | 0.3 | —d | 56.7 (6.4), 3 |
| 1 M NaOH | Water | 0.3 | 98.8 (5.5), 3 | 16.7 (2.1), 3 |
| 1 M NaOH + 0.2%Ca(OH)2 | Water | 0.3 | 98.2 (0.9), 3 | 12.7 (0.7), 3 |
| 6% K2CO3 + 2% glycerol | Water | 0.3 | 101.8 (2.1), 3 | 52.0 (1.9), 3 |
| 0.5 M TEA + 0.1 M PTIO | Acetone | 0.5 | 87.5 (13.8), 13 | 48.7 (2.4), 5 |
| 10% TBAHa | Methanol | 0.5 | 100.4 (2.5), 3 | 67.2 (3.7), 3 |
| 10% TBAH + 0.5 M TEA + 0.1 M PTIO | Methanol | 0.5 | — | 44.9 (2.3), 3 |
| Aqueous absorbend (0.1 M NaOH) | 0.6% H2O2 | 10.2 | 96.6 (3.0), 13 | 103.1 (0.7), 3 |
The recoveries of SO2 from air samples with SO2 alone (without NOx) were quantitative, irrespective of the filter types, except for the PTIO + TEA filters, with which recoveries varied from 63 to 99% (this variation was not improved by changing the sample volume (1 L or 10 L) or the matrix (N2 or air)). However, when SO2 coexisted with NOx in the air samples, SO2 recoveries decreased in all cases, although SO2 detected in the 2nd alkaline filters, which were installed behind the 1st filters, was just trace (<5%).
These results suggested that SO2 was collected quantitatively on all alkaline filters, but when SO2 coexisted with NOx it was partly transformed to S-species, which were not recoverable by the analytical method (ion chromatography) used. Such a reaction might be promoted when concentrated standard gases prepared in dry N2 matrix are used. Sickles and Hodson16 also reported that a portion of the SO2 retained by nylon filters was unrecoverable. To convert the unrecoverable S-species on alkaline filters to SO42−, the oxidative digestion method37 was applied as the post-sampling treatment. When the air samples contained SO2 and NOx, SO2 recoveries were low (38–69%) by the H2O2 extraction method (described above), but improved to 72–100% when the oxidative digestion method was employed (Table 3). On the other hand, recoveries from air containing SO2 alone (without NOx) were quantitative in both methods. These results suggested that the intermediate oxidized S-species are formed on the alkaline filters during sampling.
| Alkaline solutions impregnated into filters | Recovery of SO2 (%) | |||
|---|---|---|---|---|
| From SO2 alonea | From SO2 + NOxb | |||
| H2O2 extraction | Oxidative digestion | H2O2 extraction | Oxidative digestion | |
| Values: means (standard deviations), replicates. The quartz fiber filters (Pallflex 2500QAST, 47 mm id) were used.a 10 ppmv SO2 in 10 L N2.b 10 ppmv SO2 + 40 ppmv NO + 20 ppmv NO2 in 10 L N2.c Not tested. | ||||
| 0.5 M Na2CO3 | 96.7 (2.1), 5 | 96.8 (2.1), 3 | 49.7 (8.3), 4 | 83.7 (3.0), 10 |
| 1 M Na2CO3 | —c | — | 64.0 (3.9), 3 | 84.6 (10.6), 3 |
| 1 M NaOH | 99.4 (—), 2 | — | 64.6 (6.9), 3 | 90.2 (7.8), 10 |
Effect of humidity on SO2 recovery
Recovery of SO2 from air with different humidities was also examined (Fig. 2). Humid air samples were prepared as follows: ambient air (humid; 10 L) was cleaned by passing it through the PTIO + TEA filters, and then mixed with the artificially polluted air (dry; 10 L, N2 matrix) through a Y-type joint tube. The resulting air samples had relative humidities of 18–35% (temperature: 27–29 °C), on the assumption that the humidities were half those of the ambient air.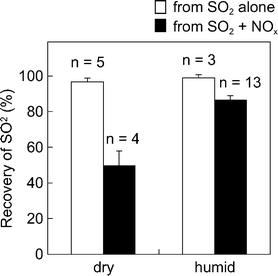 | ||
| Fig. 2 Recoveries of SO2 at different humidities. Relative humidity: dry, ca. 0%; humid, 25.2 ± 5.4% (18–35%). Concentrations of pollutants: SO2, 5–10 ppmv; NO, 20–40 ppmv; NO2, 10–20 ppmv. Filter: Na2CO3 filter of 47 mm id. | ||
Recoveries of SO2 from dry air with SO2 and NOx were ca. 50%, whereas those from humid air were ca. 87% (range: 83–92%). This could indicate that moisture accelerates the oxidation of SO2 collected on the Na2CO3 filters to SO42−, even when NOx coexists. Lewin38 and Sickles et al.10 also reported that humidity was an important parameter in the determination of SO2 by the FP method.
Recovery of NOx by PTIO + TEA filters
Recovery of NOx from artificially polluted air samples was studied as a function of NOx concentration, species of matrix gas, and number and size of filters (Table 4). Recoveries were calculated from the amounts of NO2− and also from the total amounts of NO2− and NO3− that were analyzed.| Artificially polluted air samples | Filters | Recovery of NOx (%) | ||||||
|---|---|---|---|---|---|---|---|---|
| Pollutants | Conc./ppmv | Volume/L | Matrix | Size/mm id | Sheets | As NO2− | As NO2− + NO3− | Replicates |
| Values: means (standard deviations).a 40 ppmv NO + 20 ppmv NO2 + 10 ppmv SO2.b 400 ppmv NO + 200 ppmv NO2 + 100 ppmv SO2.c Each filter was impregnated with 0.5 ml of 0.1 M PTIO + 0.5 M TEA (acetone solution).d Each filter was impregnated with 0.1 ml of 0.1 M PTIO + 0.5 M TEA (acetone solution). | ||||||||
| NO | 40 | 10 | N2 | 47c | 6 | 87.6 (3.6) | 97.4 (5.3) | 11 |
| NO | 40 | 10 | Air | 47 | 6 | 103.0 (4.2) | 104.2 (4.5) | 8 |
| NO | 20 | 10 | Air | 47 | 5 | 101.3 (2.8) | 103.5 (2.7) | 4 |
| NO | 20 | 10 | Air | 47 | 4 | 91.5 (5.6) | 93.6 (5.4) | 4 |
| NO | 20 | 10 | Air | 25d | 4 | 70.7 (9.5) | 73.8 (9.7) | 22 |
| NO2 | 20 | 10 | N2 | 47 | 6 | 87.7 (5.1) | 104.7 (9.9) | 10 |
| NO2 | 200 | 1 | N2 | 47 | 6 | 101.2 (2.3) | 104.7 (2.0) | 6 |
| NOx + SO2 | Lowa | 10 | N2 | 47 | 6 | 103.3 (1.9) | 107.4 (1.2) | 4 |
| NOx + SO2 | Highb | 1 | N2 | 47 | 6 | 99.3 (6.1) | 102.3 (7.4) | 5 |
NOx was recovered quantitatively using the 6 piled sheets of PTIO + TEA filters. When 40 ppmv NO in the 10 L bag with N2 was sampled, the amount of NO collected on each filter decreased exponentially from the 1st to 6th filters, with 90% of the NO being recovered as NO2− and the remainder as NO3−. However, most (ca. 99%) NO was recovered as NO2− when NO was prepared in an air matrix (with moisture). As shown in Fig. 3, NO is oxidized to NO2 by PTIO, and then the NO2 is disproportionated with TEA and H2O to form NO2− and NO3−.39,40 Therefore, conversion of NO to NO2− may be promoted by moisture. At the lower NO loadings, 5 filter sheets were sufficient for quantitative collection, but the recoveries decreased if the number of filters was reduced further or if smaller filter sizes were used. This may be attributed to the slow reaction between NO and PTIO, and thus a portion of NO may have escaped without oxidative collection. Recoveries were also decreased when coarse filters were used.
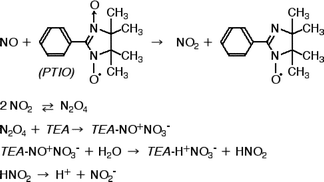
NO2 was collected more easily, and thus by less filters than NO, probably because the reaction between NO2 and TEA was rapid. For example, NO2 in air samples (20 ppmv in 10 L N2) was collected quantitatively on the 1st to 3rd filters. Sickles et al.33 also recovered NO2 almost quantitatively (87 ± 9%, n = 33) on glass fiber filters with TEA from air containing 5–400 ppbv NO2.
NOx was also recovered quantitatively from air samples with NOx and SO2, and most (>96%) of the NOx was recovered as NO2−. SO2 did not interfere with the NOx determination. Similar results have also been reported by Sickles et al.33
Elimination of PTIO from the PTIO + TEA filters extract
In NOx analysis using PTIO, extracts are usually determined by colorimetric or flow injection methods.5,34 However, after elimination of the co-extracted PTIO, they can be analyzed more conveniently by ion chromatography. Saito5 removed PTIO from the extracts by extraction with ethyl ether, whereas we used chloroform extraction followed by filtration through a C18-cartridge. As a result, 10 mmol L−1 of PTIO in the extracts was reduced to 40 ± 2 nmol L−1 after extraction, and then to 21 ± 1 nmol L−1 after filtration (n = 15, elimination rate: 99.8%; PTIO concentration was determined colorimetrically at 345 nm). Recoveries of 3.0 mg L−1 of NO2−, NO3− and SO42−, spiked in the filter extracts, were 100%, 105% and 99%, respectively (n = 5), after treatment. Smaller amounts (0.3 mg L−1) of NO2− and SO42− were also recovered quantitatively without any effect from the treatment, although NO3− recovery was affected by the impurities of PTIO when NO3− was present at a low concentration.Field measurements
Atmospheric SOx and NOx were collected simultaneously on Na2CO3 (1 sheet) and the PTIO + TEA (6 sheets) filters, respectively, installed in that order, at NIES and Mt. Tsukuba (ca. 20 km away from NIES). Concentrations of SO2 recovered from Na2CO3 filters that were placed behind an additional quartz fiber filter (2500QAST, Pallflex) (this filter can remove particulate SO42−) were 85 ± 12% (n = 5, SO2 concentrations were 0.9–1.9 ppbv) of those measured by the UV fluorescence method, though the difference was not significant (p > 0.05). A portion of the SO2 might have been adsorbed on the quartz fiber filter.14,31 In addition, although the conversion of SO2 to unrecoverable S-species on the Na2CO3 filter seems to be insignificant when moist and dilute ambient air is sampled (see Fig. 2), the reaction might have occurred to a slight extent, because the sampling periods were very dry. However, this small underestimation may not be significant in field measurements.Most of the NOx collected on the PTIO + TEA filters was recovered as NO2− (NO3− was trace), probably because the ambient air contained moisture. Distributions of NO2− recovered from the 1st to 7th filters are shown at different concentrations of NOx in ambient air (Fig. 4). More than 98% of the NO2− was detected in the PTIO + TEA filters (the 2nd to 7th filters). Trace NO2− and NO3−, recovered from the Na2CO3 filter (the 1st filter), might have been nitrous acid and particulate nitrate + nitric acid, respectively.10,33 In other FP and passive sampling methods using TEA as absorbent, most NO2 is retained as NO2− (with trace NO3−) on the filters.33,39,41
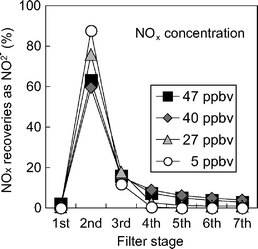 | ||
| Fig. 4 Distribution of NOx recovered from each filter at different atmospheric concentrations of NOx. 1st filter: Na2CO3 filter, 2nd–7th filters: PTIO + TEA filters. Sampling sites: NIES and Mt. Tsukuba. Period: January–April 2004. | ||
When ambient SOx was sampled, it was not detected from the PTIO + TEA filters (these can also collect SOx) behind the Na2CO3 filter, or from the other Na2CO3 filter which was tentatively placed behind the first Na2CO3 filter. The side reaction (i.e., from SO2 to unrecoverable S-species) on the Na2CO3 filter was usually negligible under moist conditions. Also, almost quantitative recoveries of ambient SO2 using similar alkaline filters have been reported.9 In the case of ambient NOx, it was recovered mostly from the first PTIO + TEA filter, and recoveries decreased exponentially at the subsequent filters (those from the last filter were negligible) (see Fig. 4). In addition, as described later (Fig. 5), good correlation was observed between the ambient pollutant concentrations obtained by our FP method and those by other instrumental methods. Therefore, even under field diluted conditions we expected good recoveries of SOx and NOx, similar to those observed for standard gases with high pollutants.
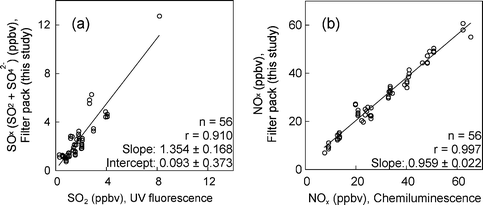 | ||
| Fig. 5 Relationships between (a) daily average concentrations of SOx obtained by the current FP method and those of SO2 obtained by UV fluorescence, and (b) daily average concentrations of NOx obtained by the current FP method and those obtained by chemiluminescence. Values of slope and intercept: mean ±95% confidence interval. Period: January 2004–February 2005. | ||
Comparison of our data with that obtained by other instrumental methods
Daily average concentrations of SOx and NOx obtained by our FP method were compared with those obtained by other instrumental methods (Fig. 5). The data were plotted without weighting because: (1) They had been already averaged (i.e., daily average values); and (2) The regression lines in Fig. 5 were almost the same as the new regression lines, calculated using the averages of the data allocated to the sub-divided concentration ranges.In Fig. 5(a), the pollutants analyzed were different between our FP and the instrumental methods (i.e., SOx in the former and SO2 in the latter); thus, linear regression that did not pass the origin was employed. SOx concentrations obtained by our method correlated well with those obtained for SO2 (not SOx) by the UV fluorescence method, but were always ca. 30% higher, probably due to particulate SO42− also being accounted for in this method, as our sampler was not equipped with a particle collection filter and/or an impactor. The intercept did not differ significantly from zero; suggesting that the SO42− fractions were relatively small and changed with SO2 (i.e., were absent when SO2 was absent). High concentrations of SOx, affected by volcanic gases discharged and transported from Miyakejima Island (located 240 km SSW of NIES) were sometimes observed. This was supported by back trajectory analysis using METEX.42
In Fig. 5(b), the pollutants analyzed were the same between both methods; therefore, linear regression including the origin was employed. NOx concentrations obtained by our method were in good agreement with those obtained by chemiluminescence, showing a regression line with a slope of about one.
During the study period, temperature and relative humidity varied widely over ranges of 2.8–29.5 °C (average: 13.0 ± 8.7 °C) and 29–83% (59 ± 15%), respectively. Therefore, our FP method may enable simultaneous determination of atmospheric concentrations of SOx and NOx, without significant effects from temperature or humidity.
Reproducibility and determination limits
Mean relative standard deviations of the results (13 sets) obtained by the three FP samplers collocated at the same sites were 6 ± 3% for SOx and 5 ± 4% for NOx. Those for nitrous acid and particulate nitrate + nitric acid (NO2− and NO3−, respectively, recovered from the Na2CO3 filter) were 26 ± 27% and 13 ± 12%, respectively, and increased with decreasing pollutant concentrations. The determination limit of SOx was defined as that of ion chromatography (0.05 mg L−1 as SO42−; corresponding to 0.2 ppbv in air) because the filter blanks were small (0.02 ± 0.04 ppbv). On the other hand, the filter blanks for NOx were relatively high (0.6 ± 0.3 ppbv), and the determination limit was defined as the value (1.0 ppbv), which was about three times as large as the standard deviation. Since battery power and the efficacy of the PTIO reagent can last for 2 days, and the determination limits of SOx and NOx can be lowered approximately to 0.1 ppbv and 0.5 ppbv, respectively; over a 2-day sampling period, we should be able to obtain reasonable data for SOx and NOx levels using our FP method at all sites in Japan.Acknowledgements
The authors thank Dr M. Nishikawa of NIES for providing data from the Air Monitoring Station.References
- A. Bytnerowicz, M. Tausz, R. Alonso, D. Jones, R. Johnson and N. Grulke, Environ. Pollut., 2002, 118, 187 CrossRef CAS.
- A. Bytnerowicz, O. Badeab, F. Popescuc, R. Musselmand, M. Tanaseb, I. Barbue, I. Fraczekf, N. Gembasub, A. Surdub, F. Danescub, D. Postelnicug, R. Cenusae and C. Vasile, Environ. Pollut., 2005, 137, 546 CrossRef CAS.
- R. M. Cox, Environ. Pollut., 2003, 126, 301 CrossRef CAS.
- T. Takamatsu, H. Sase and J. Takada, Can. J. For. Res., 2001, 31, 663 CrossRef.
- K. Saito, J. Jpn. Soc. Atmos. Environ., 2003, 38, 145 Search PubMed (in Japanese).
- R. H. Brown, J. Environ. Monit., 2000, 2, 1 RSC.
- S. V. Krupa and A. H. Legge, Environ. Pollut., 2000, 107, 31 CrossRef CAS.
- M. S. Erduran and S. G. Tuncel, J. Environ. Monit., 2001, 3, 661 RSC.
- S. Leppänen, P. Anttile, H. Lättilä and U. Makkonen, Atmos. Environ., 2005, 39, 2683 CrossRef.
- J. E. Sickles II, L. L. Hodson and L. M. Vorburger, Atmos. Environ., 1999, 33, 2187 CrossRef.
- ADORC (ADORC prepared for EANET), Technical document for filter pack method in East Asia, Acid Deposition and Oxidant Research Center, Niigata, Japan, 2003 Search PubMed.
- U.S. EPA (MACTEC Engineering and Consulting, Inc. prepared for U.S. EPA), Clean air status and trends network (CASTNet) 2003 annual report, U.S. EPA Office of Air and Radiation Clean Air Markets Division, Washington, DC, USA, 2004 Search PubMed.
- EMEP, EMEP/CCC-Report 1/95 (revision 2001), EMEP manual for sampling and chemical analysis, Norwegian Institute for Air Research, Kjeller, Norway, 2001 Search PubMed.
- B. R. Appel, Y. Tokiwa, M. Haik and E. L. Kothny, Atmos. Environ., 1984, 18, 409 CrossRef CAS.
- T. A. Pakkanen, R. E. Hillamo, M. Aurela, H. V. Andersen, L. Grundahl, M. Ferm, K. Persson, V. Karlsson, A. Reissell, O. Royset, I. Floisand, P. Oyola and T. Ganko, J. Aerosol Sci., 1999, 30, 247 CrossRef CAS.
- J. E. Sickles II and L. L. Hodson, Atmos. Environ., 1999, 33, 2423 CrossRef.
- M. Possanzini, A. Febo and A. Liberti, Atmos. Environ., 1983, 12, 2605 CrossRef CAS.
- P. Koutrakis, C. Sioutas, S. T. Ferguson, J. M. Wolfson, J. D. Mulik and R. M. Burton, Environ. Sci. Technol., 1993, 27, 2497 CAS.
- P. Beddoes, Atmos. Environ., 1967, 1, 595 CrossRef CAS.
- K. W. Brown, G. B. Wiersma and C. W. Frank, J. Environ. Qual., 1981, 10, 52.
- A. Gupta, R. Kumar, K. M. Kumari and S. S. Srivastava, Atmos. Environ., 2003, 37, 4837 CrossRef CAS.
- P. Demokritou, I. G. Kavouras, S. T. Ferguson and P. Koutrakis, Aerosol Sci. Technol., 2001, 35, 741 CAS.
- M. Harper and B. S. Muller, J. Environ. Monit., 2002, 4, 648 RSC.
- D. Mark and J. H. Vincent, Ann. Occup. Hyg., 1986, 30, 89 CrossRef CAS.
- C. Misra, M. Singh, S. Shen, C. Sioutas and P. A. Hall, J. Aerosol Sci., 2002, 33, 1027 CrossRef CAS.
- C. J. Tsai, C. H. Huang, S. H. Wang and T. S. Shih, Aerosol Sci. Technol., 2001, 35, 611 CAS.
- Y. Wang, A. Allen, D. Mark and R. M. Harrison, J. Environ. Monit., 1999, 1, 423 RSC.
- A. Naemura, A. Tsuchiya, Y. Fukuoka, K. Nakane, H. Sakugawa and H. Takahashi, Jpn. J. Biometeor., 1996, 33, 131 Search PubMed.
- R. Gasche and H. Papen, J. Geophys. Res., 1999, 104, 18505 CrossRef CAS.
- X. K. Xu and K. Inubushi, Soil Sci. Plant Nutr., 2005, 51, 683 Search PubMed.
- S. Batterman, I. Osak and C. Gelman, Atmos. Environ., 1997, 31, 1041 CrossRef CAS.
- A. Bytnerowicz, D. M. Olszyk, P. J. Dawson and L. Morrison, J. Environ. Qual., 1989, 18, 268 CAS.
- J. E. Sickles II, P. M. Grohse, L. L. Hodson, C. A. Salmons, K. W. Cox, A. R. Turner and E. D. Estes, Anal. Chem., 1990, 62, 338 CrossRef CAS.
- Ogawa & Company, USA, Inc., NO, NO2, NOx and SO2 sampling protocol using the Ogawa passive sampler, 1998.
- M. Nishikawa, personal communication.
- T. Takamatsu, Trans. Res. Inst. Oceanochem., 1999, 12, 43 Search PubMed (in Japanese).
- T. Takamatsu, Int. J. Environ. Anal. Chem., 1996, 62, 303 CrossRef CAS.
- E. Lewin and B. Zachau-Christiansen, Atmos. Environ., 1977, 11, 861 CrossRef CAS.
- R. Cee and J. C. Ku, Analyst, 1994, 119, 57 RSC.
- K. Hirano, H. Maeda and K. Matsuda, Simultaneous determination method of NO, NO2 and SO2 in ambient air by use of diffusional sampling device for short-term integrated samplers, YERI-Report 128, Yokohama Environmental Science Research Institute, Yokohama, Japan, 1997 (in Japanese) Search PubMed.
- D. Krochmal and A. Kalina, Atmos. Environ., 1997, 31, 3473 CrossRef.
- J. Zeng, M. Katsumoto, R. Ide, M. Inagaki, H. Mukai and Y. Fujinuma, Data analysis and graphic display system for atmospheric research using PC, CGER Report M014-2003, National Institute for Environmental Studies, Tsukuba, Japan, 2003 Search PubMed.
| This journal is © The Royal Society of Chemistry 2006 |