New polyethyleneglycol-functionalized texaphyrins: synthesis and in vitro biological studies†‡
Wen-Hao Weia, Zhong Wangb, Toshihisa Mizuno§a, Cecilia Cortezb, Lei Fub, Mint Sirisawadb, Louie Naumovskib, Darren Magda*b and Jonathan L. Sessler*a
aDepartment of Chemistry and Biochemistry, Institute for Cellular and Molecular Biology, 1 University Station-A5300, The University of Texas at Austin, Austin, TX 78712-0165. E-mail: Sessler@mail.utexas.edu
bPharmacyclics, Inc., 995 East Arques Avenue Sunnyvale, CA 94085. E-mail: dmagda@pcyc.com
First published on 24th January 2006
Abstract
The synthesis of four new analogues of motexafin gadolinium (MGd), a gadolinium(III) texaphyrin complex in clinical trials for its anticancer properties, is described. These new derivatives contain either 1,2-diaminobenzene or 2,3-diaminonaphthalene subunits as the source of the imine nitrogens and bear multiple 2-[2-(2-methoxyethoxy)ethoxy]ethoxy (PEG) groups, on either meso aryl or beta-pyrrolic substituents, to increase their water solubility. All four analogues were found to be more active in vitro than the parent system MGd as judged from cell proliferation assays using the PC3 and A549 cell lines.
Introduction
Xcytrin® (MGd, motexafin gadolinium, 1; Fig. 1) has completed two Phase III clinical trials as an agent for treating brain metastases in conjunction with whole brain radiation therapy, with strong positive trends being seen in both trials for patients with non-small cell lung cancer.1–3 Recent preclinical and early stage clinical trials have provided support for the notion that this gadolinium texaphyrin derivative may also have utility as a stand-alone anticancer agent.4 One proposed mechanism of action involves the concept of redox cycling,5–7 wherein a reduced texaphyrin species, formed by reaction with endogenous electron rich species such as ascorbate or glutathione, serves to form reactive oxygen species including superoxide and peroxide that are known to act as apoptosis triggering agents.8–10 A similar process of electron capture followed by air-based oxidation could also contribute to the induced expression of metallothioneins and altered zinc homeostasis.11 To the extent such redox-based modes of action are important, it provides an incentive to make and test new texaphyrin derivatives whose electronic properties are modified compared to 1. In this paper, we report the synthesis and preliminary in vitro biological analysis of a new series of gadolinium texaphyrins in which the electron-rich dialkoxydiaminobenzene derivative used in 1 is replaced by diaminonaphthalene and diaminobenzene subunits (compounds 2–5; Fig. 1). The diaminonaphthalene-derived systems 2 and 4 were found to display similar in vitro activity in PC3 and A549 cell models, compared to the corresponding diaminobenzene systems 3 and 5. Both sets of new compounds were found to be more active in vitro than the parent system 1.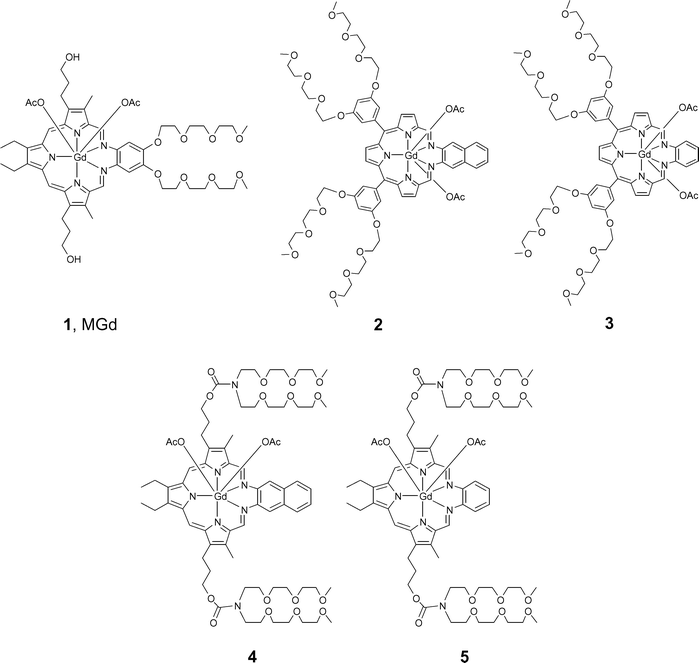 | ||
| Fig. 1 Motexafin gadolinium (MGd, 1) and new targets prepared in the course of the present study. | ||
Results and discussion
Synthesis
A naphthalene-containing texaphyrin was synthesized previously via the direct condensation of a tripyrrane dialdehyde with diaminonaphthalene.12–13 The product, however, proved to have very low solubility in aqueous media. Thus, a new analogue, 2, bearing multiple 2-[2-(2-methoxyethoxy)ethoxy]ethoxy (PEG) groups to increase water solubility, was targeted for synthesis. Its preparation is summarized in Schemes 1 and 2. The first stage in the synthesis consisted of preparing the diformyltripyrrane 7. Towards this end, meso-diaryltripyrrane 6 was prepared (Scheme 1) by condensing pyrrole (3–5 equiv.) with 3,5-bis[2-[2-(2-methoxyethoxy)ethoxy]ethoxy]-benzaldehyde (1 equiv.)14 in the presence of trifluoroacetic acid (TFA, 5% equiv.) according to the procedure of Lee and Lindsey.15,16 After purification, the product was isolated in 15–23% yield. While this purified tripyrrane proved stable when stored in the freezer for over two weeks, it darkened rapidly, turning from pink to dark brown, when dissolved in common organic solvents and left to stand on the bench top unprotected from air or light. Thus, as a general rule, this intermediate was converted directly into the more stable diformyl precursor 7; this was performed by subjecting it to Vilsmeier formylation (DMF/POCl3) in accordance with standard protocols.17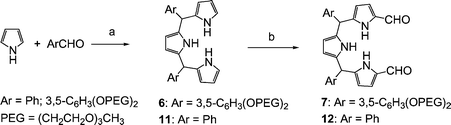 | ||
| Scheme 1 Reagents and conditions: (a) 5% TFA, rt, 0.5 h, 23% for 6, 24% for 11; (b) 1) DMF/POCl3, rt, 1 h, under Ar, 2) 0.5 N aq. NaOH, 85 °C, 8 h, 72% for 7, 38% for 12. | ||
![Reagents and conditions: (a) 37% HCl, MeOH, reflux, overnight; (b) Et3N, Gd(OAc)3·4H2O, air [O2], MeOH, reflux, 9% without DBU and 23% with DBU for 2.](/image/article/2006/DT/b515636j/b515636j-s2.gif) | ||
| Scheme 2 Reagents and conditions: (a) 37% HCl, MeOH, reflux, overnight; (b) Et3N, Gd(OAc)3·4H2O, air [O2], MeOH, reflux, 9% without DBU and 23% with DBU for 2. | ||
Once in hand, intermediate 7 was subject to acid-catalyzed condensation with 2,3-diaminonaphthalene under conditions of high dilution. This gave the so-called “sp3-texaphyrin” 8. The structure of this nonaromatic macrocycle was confirmed by CI-MS and HRMS. Unfortunately, compound 8 proved to be unstable, decomposing completely when attempts were made to purify it by column chromatography over silica gel or alumina, or using a tC18 cartridge. This instability stands in contrast to what is true for most meso-free sp3-texaphyrins, which are generally quite stable and which can be separated by column chromatography over various supports with nearly quantitative recovery.18,19 A possible explanation for the instability of 8 is that the two meso-aryl groups introduce strain that make the imine bonds more susceptible to solvolysis. While not an exact analogy, it is noteworthy that several meso-aryl substituted porphyrins have been reported that have distorted non-planar conformations.20,21
Fortunately, we found that intermediate 8 could be used directly in the next step without a need for purification. Specifically, treatment of 8 with 2 equiv. of Gd(OAc)3·4H2O and 7.5 equiv. of Et3N in the presence of air in methanol under reflux was found to effect ring oxidation and metal insertion. This produced a green solution of the crude gadolinium texaphyrin complex, 2. This target species was purified by column chromatography over neutral alumina followed by passage through a reversed-phase tC18 cartridge column (Waters) to afford the desired meso-diaryl substituted gadolinium texaphyrin 2 in pure form. The overall yield was 23% based on 7 with DBU and 9% without DBU. The UV-vis spectrum of compound 2 recorded in methanol is characterized by a Soret-like band at 466 nm and Q-type band at 840 nm. Thus, compared to MGd 1, the Soret-type band is blue-shifted, while the Q-type band is red-shifted (the λmax of MGd, 1, are 470 nm and 740 nm, respectively, in this same solvent).
An analogous compound 3, which has the same meso-PEG substitution, but a simple benzene ring in place of the naphthalene structure, was also synthesized; it was prepared from dialdehyde 7 and 1,2-phenylenediamine via intermediate 9 as shown in Scheme 3. The overall yield for the two steps was 26% without DBU. As might be expected, the spectral features of compound 3 (λmax = 451 and 747 nm in methanol) are blue shifted compared to those of 2 (λmax = 466 and 840 nm, also in methanol).
![Reagents and conditions: (a) 37% HCl, MeOH, reflux, overnight; (b) Et3N, Gd(OAc)3·4H2O, air [O2], MeOH, reflux, 26% for 3, 38% for 10, (c) anion exchange with NaNO3, 100%.](/image/article/2006/DT/b515636j/b515636j-s3.gif) | ||
| Scheme 3 Reagents and conditions: (a) 37% HCl, MeOH, reflux, overnight; (b) Et3N, Gd(OAc)3·4H2O, air [O2], MeOH, reflux, 26% for 3, 38% for 10, (c) anion exchange with NaNO3, 100%. | ||
To the best of our knowledge, compounds 2 and 3 represent the first texaphyrins bearing meso substitutents. In both cases, the associated meso-3,5-bis[2-[2-(2-methoxyethoxy)ethoxy]-ethoxy]phenyl functionality imparts sufficient water-solubility to allow for testing under typical in vitro assay conditions (vide infra). Such solubility features were considered critical since they would allow for direct comparisons with MGd 1 in cell culture. Nonetheless, it was also recognized that the presence of these meso substituents might make compounds 2 and 3 less than ideal electronic analogues of 1; they could engender deviations from ring planarity,20–23 which, in turn, could lead to altered interactions with biological targets.24–26
In light of this potential concern, we prepared a simpler analog of 3, namely, the meso-phenyl substituted system 10. The thought was that this model system would prove amenable to characterization by single crystal X-ray diffraction analysis, allowing the structural effect of the meso-aryl substitution to be more accurately assessed. Compound 10 was synthesized using a procedure analogous to that used to prepare 2 and 3. Specifically, a TFA catalyzed condensation between benzaldehyde and pyrrole was used to obtain the meso-diphenylsubstituted tripyrrane 11. This intermediate was then formylated to yield the corresponding tripyrrane dialdehyde 12 (Scheme 1). This dialdehyde was then condensed with 4,5-dimethoxy-1,2-phenylenediamine to give 13 (Scheme 3). Following standard oxidative metal insertion and purification, target 10 was obtained in the form of its acetate salt. The nitrate form 14, which we considered more suitable for growing X-ray diffraction quality single crystals based on our prior experience,19 was obtained via anion exchange with nitrate (Scheme 3). Both derivatives (i.e., 10 and 14) were characterized by ESI-HRMS and UV/Vis spectroscopy, while complex 14 was further characterized by single-crystal X-ray diffraction analysis.
The two water soluble meso-substituted texaphyrins 2 and 3 were tested in an in vitro assay using the A549 cell line, and compared with MGd, 1. The results are shown in Fig. 2. Compounds 2 and 3 showed dramatically higher activities than MGd towards A549 cells. Compound 2 was also tested in Ramos lymphoma cell line in tissue culture. It induced apoptosis as demonstrated by annexin V-positive cells at 2.5 µM but not 1 µM (Fig. 3). Compound 2 also activated caspases at 2.5 µM, demonstrating that compound 2 induced apoptosis (data not shown).
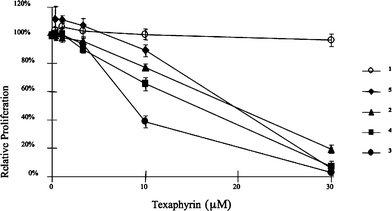 | ||
| Fig. 2 Antiproliferative activity of motexafin gadolinium (MGd, 1) and analogues 2–5 in A549 lung cancer cells. A549 cultures were treated with texaphyrins for 24 h, medium was replaced with fresh medium, and viable cells were quantified after 72 h by tetrazolium salt reduction. | ||
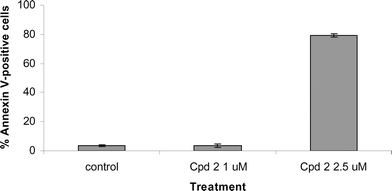 | ||
| Fig. 3 Cytotoxicity of compound 2 as determined by annexin V-positive cells. Ramos cells were treated with compound 2 at 1 or 2.5 µM for 24 h. Annexin V-positive cells were quantified by flow cytometry. Similar cytotoxic effects were seen when normalized caspase-3 activity was monitored (data not shown). | ||
These promising results prompted our interest to synthesize other related Gd-Tex derivatives, and try to understand the key structural characteristics as well as physicochemical properties that lead to the increased biological activity.
Thus, compounds 4 and 5 were synthesized utilizing a slightly different strategy starting from tripyrrane dialdehyde 15, an intermediate used in the synthesis of MGd. As shown in Scheme 4, reaction of 15 with disuccinimidyl carbonate in the presence of diisopropylethylamine (DIEA), followed by reaction with bis[2-[2-(2-methoxyethoxy)ethoxy]ethyl]amine, gave the side-chain PEGylated tripyrrane dialdehyde 16. Column chromatography over silica gel yielded this intermediate in 62% yield in purified form. An HCl-catalyzed condensation of dialdehyde 16 with 2,3-diaminonaphthalene gave the non-aromatic macrocycle 17, which was used directly without purification. Finally, Gd insertion/oxidation in methanol at reflux in the presence of DIEA and O2 furnished the Gd complex 4. The overall yield from 16 was 36%. Using the diformyl tripyrrane 16 as a common intermediate, compound 5 was also readily synthesized in 55% yield. As with 2 and 3, compounds 4 and 5 displayed good solubility in aqueous media.
![Reagents and conditions: (a) N,N′-disuccinimidyl carbonate, N,N-diisopropylethylamine (DIEA), DMSO, rt, then diPEG amine, 62%; (b) 2,3-diaminonaphthalene, HCl, MeOH, 45 °C; (c) Gd(OAc)3·4H2O, DIEA, air [O2], MeOH, reflux, 36%.](/image/article/2006/DT/b515636j/b515636j-s4.gif) | ||
| Scheme 4 Reagents and conditions: (a) N,N′-disuccinimidyl carbonate, N,N-diisopropylethylamine (DIEA), DMSO, rt, then diPEG amine, 62%; (b) 2,3-diaminonaphthalene, HCl, MeOH, 45 °C; (c) Gd(OAc)3·4H2O, DIEA, air [O2], MeOH, reflux, 36%. | ||
Crystal structure of 14
The single crystal X-ray structure of 14 is shown in Fig. 4, with important bond lengths and angles listed in Table 1. As can be inferred from an inspection of Fig. 4, 14 consists of a 1 : 1 metal–ligand complex in which the Gd(III) cation is found to be 10-coordinate. Five of the donor atoms are provided by the nitrogen atoms present within the pentaaza texaphyrin core; four donor atoms come from the coordinated bidentate nitrate anions, while the last donor atom is derived from a methanol solvent molecule. The two phenyl groups linked to the meso-positions adopt a nearly orthogonal orientation with regard to the plane of the five nitrogen atoms. Presumably as a consequence of the resulting steric interactions, the pyrrole ring between the two phenyl substituents deviates from planarity, being bent “up” toward the two NO3− anions. The two imine nitrogens are also raised such that the macrocycle as a whole exists in a “ruffled” non-planar conformation.| a The pyrrolic nitrogens are numbered N1, N8 and N23; and the imine nitrogens are numbered N13 and N20. X-ray Experimental for (C36H26N5O2)Gd(NO3)2-CH3OH-0.5C6H6. Green prisms were grown by slow evaporation from methanol–benzene, monoclinic, P21/c (No. 2), Z = 4 in a cell of dimensions: a = 17.9235(2) Å, b = 11.9870(1) Å, c = 18.7840(2) Å, β = 114.244(1)°, V = 3679.79(6) Å3, Mr = 912.98, ρcalc = 1.648 g cm−3, µ = 1.871 mm−1, F(000) = 1832. A total of 14460 reflections were measured, 8372 unique (Rint = 0.017), on a Nonius Kappa CCD using graphite monochromatized Mo Kα radiation (λ = 0.71073 Å) at −120 °C. The structure was refined on F2 to an Rw = 0.0741, with a conventional R = 0.0278 (6989 reflections with Fo > 4[σ(Fo)]), and a goodness of fit = 1.14 for 491 refined parameters. A molecule of benzene was found to be disordered about a crystallographic inversion center at 0, ½, ½. The molecule was poorly resolved and its contribution to the scattering was removed using the utility SQUEEZE in PLATON98. For full details see the Supporting Information. | |||||||
|---|---|---|---|---|---|---|---|
| Gd–N1 | 2.486(2) | Gd–N20 | 2.565(2) | Gd–O1A | 2.484(2) | Gd–O2B | 2.5529(18) |
| Gd–N8 | 2.4142(19) | Gd–N23 | 2.424(2) | Gd–O2A | 2.786(3) | Gd–O1C | 2.5498(18) |
| Gd–N13 | 2.540(2) | Gd–O1B | 2.5500(19) | ||||
| N1–Gd–N8 | 75.68(6) | N20–Gd–N23 | 65.33(6) | O1A–Gd1–O2A | 47.15(8) | O2B–Gd1–O2A | 63.59(7) |
| N8–Gd–N13 | 66.02(7) | N23–Gd–N1 | 74.46(7) | O1A–Gd1–O1B | 67.09(7) | O1A–Gd1–O1C | 138.61(7) |
| N13–Gd–N20 | 61.96(6) | O1B–Gd1–O2B | 49.90(6) | ||||
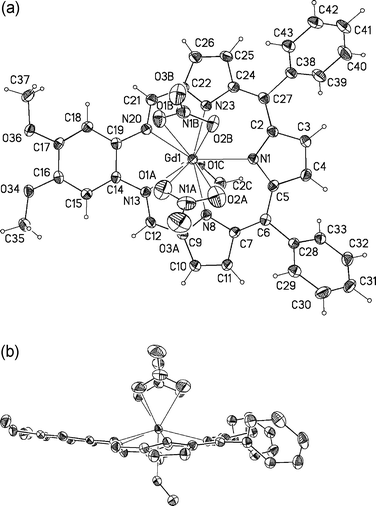 | ||
| Fig. 4 (a) Top (a) and side views of 14 showing the atom labeling scheme. Displacement ellipsoids are scaled to the 50% probability level. The Gd3+ ion is 0.698(1) Å out of the plane defined by the five nitrogen atoms of the texaphyrin macrocycle. | ||
CCDC reference number 284060.
For crystallographic data in CIF or other electronic format see DOI: 10.1039/b515636j
In vitro biological study
Once in hand, the four new Gd-Tex derivatives 2–5 were tested for anti-proliferative activity in vitro using two cell lines (PC3 and A549), with representative data being shown in Fig. 2 and 5. From these data, we infer that the new naphthalene and benzene ring-containing texaphyrins all display higher in vitro activity than MGd. These compounds displayed significant activity in the 10–20 µM concentration range, while MGd, 1, showed little activity even at 30 µM. In preliminary studies, analogue 4 was also found to display significant cytotoxicity in Ramos cells at concentrations of 5 µM and above (data not shown).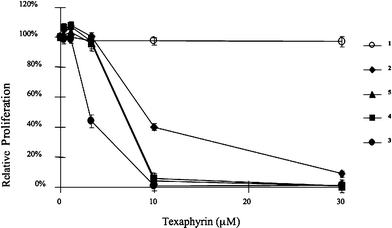 | ||
| Fig. 5 Antiproliferative activity of motexafin gadolinium (MGd, 1) and analogues 2–5 in PC3 prostate cancer cells. PC3 cultures were treated with texaphyrins for 24 h, medium was replaced with fresh medium, and viable cells were quantified after 72 h by tetrazolium salt reduction. | ||
Electrochemical study
Because it is believed that Gd-Tex derivatives affect cell metabolism, at least in part, through redox cycling, we became interested in measuring the redox potentials of this new series of texaphyrins, and in establishing a possible correlation between their biological activity and electrochemical properties. Accordingly, the redox potentials of 2–5 were determined in DMF solution using tetrabutylammonium perchlorate as the supporting electrolyte. The E1/2 values were calculated from cyclic voltammograms and are summarized in Table 2. The redox potential of MGd, 1, was also determined under the same conditions and is included for comparison.| 1 (MGd) | 2 | 3 | 4 | 5 | |
|---|---|---|---|---|---|
| a Recorded in anhydrous DMF using 0.2 M tetrabutylammonium perchlorate as the supporting electrolyte. | |||||
| 1E1/2/mV | −329 | −130 | −122 | −107 | −203 |
| 2E1/2/mV | −802 | −559 | −597 | −443 | −678 |
Comparing the order of biological activity to that of the redox potentials, one can see that both the antiproliferative activity and redox potentials of the new derivatives fall in a relatively narrow range, which is distinct from that of MGd. Moreover, the new derivatives with their more positive redox potentials display a higher level of in vitro antiproliferative activity. By contrast, no clear correlation between the presence or absence of the meso substitutents was observed. On this basis we conclude that limited differences in planarity do not have a direct effect on the biological activity. However, since such distortions could affect the observed redox behavior, an indirect effect cannot be ruled out.
Since MGd has been shown to redox cycle in the presence of ascorbate to generate cytotoxic H2O2,6 we sought to determine if compound 2 would display enhanced cytotoxicity in the presence of ascorbate. As shown in Fig. 6, treatment of Ramos cells with sub-toxic doses of 2 and ascorbate resulted in elevated levels of annexin V-positive cells and decreased growth. Cytotoxicity and growth inhibition was reversible in the presence of catalase, leading us to propose, in analogy to MGd, that the combination of 2 and ascorbate serves to generate H2O2via redox cycling.
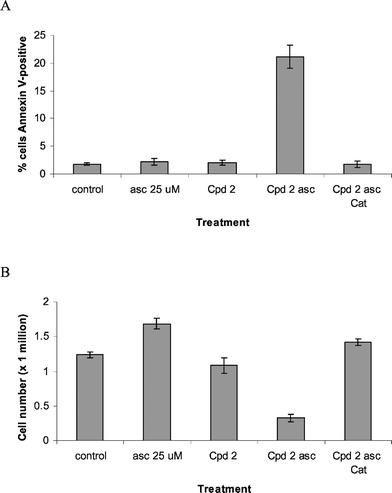 | ||
| Fig. 6 Effect of compound 2 on annexin V-binding and cell growth in Ramos. (A) Cells were treated with ascorbate (asc) 25 µM, compound (Cpd) 2 at 1 µM, the combination or the combination with catalase (Cat) 260 units ml−1 for 24 h. Apoptotic cells were determined by annexin V-binding. (B) Cells were treated as above for 4 days and cell numbers were measured by a Coulter counter. | ||
Conclusion
In summary, we have developed two general synthetic methods that can be applied to prepare a wide range of water-soluble, bioassay-compatible texaphyrin derivatives by simply using different aromatic diamines. Employing these two methods, four novel texaphyrins were synthesized, and cell culture studies showed these compounds possessed higher in vitro antiproliferative activity than MGd. Such preliminary structure–activity analyses lead us to suggest that the biological activity of MGd (1) and its analogues (e.g., 2–5) is closely correlated with the redox potential of the compounds. Detailed studies to clarify the mechanism of texaphyrin activity and efforts to assess these compounds in animal tumor models are currently underway.Experimental
All starting materials were purchased and used without further purification unless otherwise noted. 3,5-Di[2-[2-(2-methoxyethoxy)ethoxy]ethoxy]benzaldehyde,14 5,10-diphenyltripyrrane 11,15,16 1,2-diamino-4,5-dimethoxybenzene,19 and bis[2-[2-(2-methoxyethoxy)ethoxy]ethyl]amine27 were prepared according to literature methods. Proton and 13C NMR spectra were recorded using a 250 MHz Varian, 300 MHz GE Tacmag spectrometer, 400 MHz Varian MERCURY or 500 MHz Varian INOVA. UV/Vis spectra were taken on Beckman DU-640B or Agilent 8453 Spectrophotometer. Column chromatography was run using ICN-Silitech 32–63 D60 Å silica gel or Sorbent Technologies std. activity 50–200 µ neutral alumina. Sep-pak reverse-phase tC18 cartridge columns were purchased from Waters.Syntheses
General procedure for the preparation of meso-diarylsubstituted gadolinium texaphyrins 3 and 10
To a solution of tripyrrane dialdehyde 7 or 12 (1 equiv.) and the appropriate diamino precursor (1 equiv.) in MeOH (1 mmol 500 ml−1), was added 37% HCl (3 equiv.) at room temperature under argon. The solution was heated at reflux for 10 h. At this juncture, CI-MS analysis showed that the sp3-texaphyrin 9 or 13 was present as the dominant organic species. CI-MS: m/z 1155 [M + 1]+ for 9 and 565 [M + 1]+ for 13. Thus, Gd(OAc)3·4H2O (1.5 equiv.) and triethylamine (7.5 equiv.) were added to the reaction mixtures, which were then heated to reflux with stirring for an additional 24 h under air. UV/vis spectra were taken, and once the expected aromatic texaphyrin-like spectral features were observed, the respective reactions were deemed complete. The green solutions obtained in this way were allowed to cool to room temperature before the solvent was removed under reduced pressure. The resulting green solids were then dried in vacuo for several hours. The targeted complexes, either 3 or 10, were purified by column chromatography over neutral alumina. After the initial red/brown fraction was discarded, the green complexes were gradually eluted off the respective chromatography columns using 10–30% MeOH–CH2Cl2 as the eluent. The appropriate fractions were combined and washed with 3 × 50 ml of 0.1 M aqueous NaOAc with the organic layers being saved. After concentrating the organic solutions, the desired complexes were further purified using a tC18 cartridge column. Without DBU, this afforded pure meso-diarylsubstituted gadolinium-texaphyrin 3 and 10 in yields of 24 mg (26%) and 28 mg (38%), respectively.In vitro biological activity
The proliferation of exponential phase cultures of A549 and PC3 cells was assessed by tetrazolium salt reduction.28 In brief, A549 lung cancer cells were seeded on 96 well microtitre plates at 2000 cells well−1 (4000 cells well−1 for PC3) and allowed to adhere overnight in RPMI 1640 medium supplemented with 20 mM HEPES, 2 mM L-glutamine (Invitrogen), 10% heat inactivated fetal bovine serum (Hyclone), and antibiotics (200 U cm−3 penicillin and 200 µg cm−3 streptomycin). Stock solutions of 2, 3, 4, and 5 were formulated in DMSO, then diluted in a medium for secondary stocks of 90 µM. Stock solutions were serially diluted 1 : 3 on Rows B–F, whereupon plates were incubated at 37 °C under a 5% CO2/95% air atmosphere. The medium was replaced with fresh medium after 24 h of treatment. After a total of 72 h, the medium was exchanged for fresh medium (150 mm3 well−1) supplemented with the tetrazolium dye, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT, Sigma Chemical, 0.5 mg cm−3). The plates were incubated at 37 °C for approximately 2 h, whereupon the medium was removed and isopropyl alcohol (100 mm3 well−1) was added. Microplates were vortexed briefly and well absorbances at 560–650 nm were measured using a microplate reader (Molecular Devices, Sunnyvale, CA). The average absorption of wells in the absence of cells was subtracted from each well as background absorbance. Plate absorbances were normalized to wells containing untreated cells to allow plate-to-plate comparison. Data shown at each concentration of test article is the average of data from 10 wells. Error bars represent the associated standard deviation.For Ramos lymphoma assays, cell numbers were determined using a model Z2 counter (Beckman-Coulter, Miami, FL). Annexin V-fluorescein isothiocyanate (FITC) binding was assayed with a FACSCalibur instrument (Becton-Dickinson, San Jose, CA) using reagents from Biosource (Camarillo, CA) per manufacturer's protocol. Caspase-3 activity was assayed using the EnzChek Caspase-3 Assay Kit #2 (Molecular Probes, Eugene, OR) as per the manufacturer's protocol.
Electrochemistry
Electrochemical measurements were carried out at room temperature with a Cypress System 1090 apparatus operated in cyclic voltammetry mode. All reported electrochemical half-wave potentials were measured using a glassy carbon working electrode and a Ag/AgCl reference electrode and referenced to such. Platinum wire (0.5 mm diameter) was employed as an auxiliary electrode. Each experiment was performed on a 0.5–1.5 mM solution of texaphyrin complex in 1 mL of anhydrous DMF, using 0.2 M tetrabutylammonium perchlorate as the supporting electrolyte.The potential was scanned between 0 and −1100 mV. The electrochemical potentials presented in this paper were obtained using a 50 mV s−1 scan rate.
Acknowledgements
We would like to thank Pharmacyclics, Inc. and the NIH (grant CA 68682 to J. L. S.) for support of this work.References
- M. P. Mehta, W. R. Shapiro, M. J. Glantz, R. A. Patchell, M. A. Weitzner, C. A. Meyers, C. J. Schultz, W. H. Roa, M. Leibenhout, J. Ford, W. Curran, S. Phan, J. A. Smith, R. A. Miller and M. F. Renschler, J. Clin. Oncol., 2002, 20, 3445–3453 CrossRef CAS.
- M. P. Mehta, P. Rodrigus, C. H. J. Terhaard, A. Rao, J. Suh, W. Roa, L. Souhami, A. Bezjak, M. Leibenhaut, R. Komaki, C. Schultz, R. Timmerman, W. Curran, J. Smith, S.-C. Phan, R. A. Miller and M. F. Renschler, J. Clin. Oncol., 2003, 21, 2529–2536 CrossRef CAS.
- Pharmacyclics Announces Results from Phase 3 Smart Trial of Xcytrin® for Lung Cancer Brain Metastases, Press Release, Pharmacyclics, Inc., Dec. 19, 2005 Search PubMed.
- Drug Dev. Res., 2004, 5, 52-57 Search PubMed.
- D. Magda, N. Gerasimchuk, P. Lecane, R. A. Miller, J. E. Biaglow and J. L. Sessler, Chem. Commun., 2002, 2730–2731 RSC.
- D. Magda, C. Lepp, N. Gerasimchuk, I. Lee, J. L. Sessler, A. Lin, J. E. Biaglow and R. A. Miller, Int. J. Radiat. Oncol. Biol. Phys., 2001, 51, 1025–1036 CrossRef CAS.
- R. A. Miller, K. W. Woodburn, Q. Fan, I. Lee, D. Miles, G. Duran, B. Sikic and D. Magda, Clin. Cancer Res., 2001, 7, 3215–3221 CAS.
- J. Chandra, A. Samali and S. Orrenius, Free Radical Biol. Med., 2000, 29, 323–333 CrossRef CAS.
- G. Kroemer, P. Petit, N. Zamzami, J. L. Vayssiere and B. Mignotte, FASEB J., 1995, 9, 1277–1287 Search PubMed.
- J. M. Mates and F. M. Sanchez-Jimenez, Int. J. Biochem. Cell Biol., 2000, 32, 157–170 CrossRef CAS.
- D. Magda, P. Lecane, R. A. Miller, C. Lepp, D. Miles, M. Mesfin, J. E. Biaglow, V. V. Ho, D. Chawannakul, S. M. W. Karaman and J. G. Hacia, Cancer Res., 2005, 65(9), 3837–3845 CrossRef CAS.
- J. Waluk, G. Hemmi, J. L. Sessler and J. Michl, J. Org. Chem., 1991, 56, 2735–2742 CrossRef CAS.
- B. G. Maiya, T. E. Mallouk, G. Hemmi and J. L. Sessler, Inorg. Chem., 1990, 29, 3738–3745 CrossRef CAS.
- J. M. Davis, Ph.D. Thesis, University of Texas at Austin, 2001. An experimental procedure for this compound is included in the Electronic Supporting Information Search PubMed.
- H. Lee and C.-J. S. Lindsey, Tetrahedron, 1994, 50, 11427–11440 CrossRef CAS.
- B. J. Littler, M. A. Miller, C.-H. Hung, R. W. Wagner, D. F. O'Shea, P. D. Boyle and J. S. Lindsey, J. Org. Chem., 1999, 64, 1391–1396 CrossRef CAS.
- R. Guiland, M. A. Aukauloo, C. Tardieux and E. Vogel, Synthesis, 1995, 1480 CrossRef CAS.
- J. L. Sessler, T. D. Mody, D. A. Ford and V. Lynch, Angew. Chem., Int. Ed. Engl., 1992, 31, 453–455.
- J. L. Sessler, T. D. Mody, G. W. Hemmi and V. Lynch, Inorg. Chem., 1993, 32, 3175–3187 CrossRef CAS.
- G. A. Spyroulias, A. Despotopoulos, C. P. Raptopoulou, A. Terzis and A. G. Coutsolelos, Chem. Commun., 1997, 783–784 RSC.
- M. O. Senge, Porphyrin Handbook, 2000, 1, 239–347 Search PubMed.
- R. Harada, H. Okawa and T. Kojima, Inorg. Chim. Acta, 2005, 358, 489–496 CrossRef CAS.
- S. Meyer, M. C. Hoehner, V. Lynch and J. L. Sessler, J. Porphyrins Phthalocyanines, 1999, 3, 148–158 CrossRef CAS.
- E. Sigfridsson and U. Ryde, J. Biol. Inorg. Chem., 2003, 8, 273–282 CAS.
- T.-T. Chen, X.-H. Huang, Y.-Y. Feng, J. Y. Du, C.-P. Liu, W. Xing and T.-H. Lu, Wuji Huaxue Xuebao, 2003, 19, 941–944 Search PubMed.
- L. G. Marzilli, G. Petho, M. Lin, M. S. Kim and D. W. Dixon, J. Am. Chem. Soc., 1992, 114, 7575–7577 CrossRef CAS.
- C. Selve, J.-C. Ravey, M.-J. Stebe, C. El Moudjahid, E. M. Moumni and J.-J. Delpuech, Tetrahedron, 1991, 47, 411–428 CrossRef CAS.
- T. Mosmann, J. Immunol. Methods, 1983, 65, 55–63 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Cyclic voltammograms and X-ray experimental data for 14. See DOI: 10.1039/b515636j |
| ‡ Dedicated to the memory of Ian Rothwell. |
| § Current address: Department of Material Science, Graduate School of Engineering, Nagoya Institute of Technology, 466-8555 Japan. |
| This journal is © The Royal Society of Chemistry 2006 |