Metal-based antitumour drugs in the post genomic era
Paul J.
Dyson
a and
Gianni
Sava
bc
aInstitut des Sciences et Ingénierie Chimiques, Ecole Polytechnique Fédérale de Lausanne (EPFL), CH-1015, Lausanne, Switzerland. E-mail: paul.dyson@epfl.ch
bCallerio Foundation Onlus, Via A. Fleming 22-31, 34127, Trieste, Italy. E-mail: gsava@fc.units.it
cDipartimento di Scienze Biomediche, Università di Trieste, Via L. Giorgieri 7-9, 34127, Trieste, Italy
First published on 28th March 2006
Abstract
The discovery of new metal-based antitumour drugs, whether cisplatin derivatives or those based on other metals, has been largely based on cell viability assays (IC50 values) and compounds that bind to DNA. This approach has been applied for more than 30 years during which time very few new drugs have entered clinical use. In this article we discuss what the future holds for metal-based drugs, in particular anti-metastasis drugs, in these enlightened times of the post genomic era.
 Paul J. Dyson Paul J. Dyson | Paul Dyson is the director of the Laboratory of Organometallic and Medicinal Chemistry (since 2002) at the Swiss Federal Institute of Technology in Lausanne. Previously he was based in the UK conducting research at the University of York, Imperial College and the Universities of Edinburgh and Cambridge. |
 Gianni Sava Gianni Sava | Gianni Sava is the scientific director of the Callerio Foundation Onlus (www.callerio.org), a non-profit scientific organization active in biomedical research, since 1992, is professor of pharmacology in the Faculty of Pharmacy and, since 2003, head of the Department of Biomedical Sciences at the University of Trieste. |
Inorganic compounds have had an enormous impact on medicine,1 in particular in the treatment of cancer. Rosenberg's discovery of cisplatin2 and it's subsequent clinical use has become so well known that it barely needs mentioning here. Cisplatin continues to be used in 50–70% of all cancer patients, usually in combination with other drugs, with particularly good activity against testicular, ovarian, oropharyngeal, bronchogenic, cervical and bladder carcinomas, lymphoma, osteosarcoma, melanoma and neuroblastoma.3 Although cisplatin is arguably the most successful anticancer drug in the world it is not without problems, notably it exhibits a high general toxicity leading to undesirable side-effects, although minimised by careful administration protocols, and also it is inactive against many cancer cell lines and metastasis (secondary) cancers.4 The challenge to inorganic chemists to develop platinum based anticancer drugs that are superior to cisplatin has been met with considerable enthusiasm over the last 30 or so years and literally thousands of compounds have been prepared, based on well conceived ideas to improve efficacy, and subsequently screened. Nevertheless, from these endeavours only two further compounds have entered world-wide clinical use, these being carboplatin and oxaliplatin, although three others are used in the clinic in a limited number of countries and around 10 other platinum drugs are currently undergoing clinical trials.
While the ratio of compounds prepared to compounds in use may be no worse than that of organic drugs, the current design strategies are clearly not working efficiently. The highly complicated multifactor problem of drug resistance, for example, has barely been tackled, although two recent examples which show promise involve using the Pt(IV) unit as a scaffold onto which organic compounds of known therapeutic action have been attached (Fig. 1). Lippard reported a Pt(IV)–oestrogen complex, formed by conjugating an oestrogen-derivative via a succinate linker onto a trans-Pt(IV) carboxylate structure, that was able to sensitize oestrogen-receptor(+) mammalian tumour cells, used to both target cancer cells and overcome resistance.5 The second example uses ethacrynic acid, also tethered to the Pt(IV) scaffold, a known inhibitor of glutathione-S-transferase (GST), which is over-expressed in many cisplatin resistant cancer cell lines and is implicated in various deactivation pathways.6 The ethacrynic acid moiety blocks the active site of the GST enzyme while still coordinated to the Pt(IV) centre, which presumably activates the Pt–O bond releasing cisplatin to exert its cytotoxic effect whilst preventing GST catalysed elimination of cisplatin from the cell.
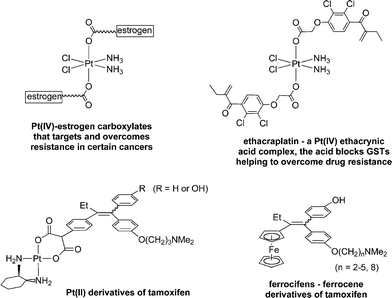 | ||
| Fig. 1 Examples of metal-based anticancer compounds that target and/or overcome resistance in certain cancers. | ||
In a slightly different strategy that had been developed by Jaouen, organometallic fragments are tethered to selective oestrogen receptor modulators (SERMs) used in the adjuvant treatment of hormone-dependent cancers such as breast cancer.7 The standard antioestrogen is hydroxytamoxifen, which for reasons of bioavailability, is administered in its non-hydroxylated form, tamoxifen (a third of hormone-dependent tumours do not respond to tamoxifen and another third acquire resistance). Tamoxifen has been coupled to platinum, but was found to be only slightly cytotoxic towards hormone-dependent breast cancers.8 However, a number of organometallic tamoxifen derivatives have been prepared that show considerable promise, of which the ferrocenyl derivatives (the so-called ferrocifens—Fig. 1) are in the most advanced stage of development ready for entry into clinical trials.
The ferrocenyl moiety imparts a number of important functions onto the tamoxifen unit, in particular, facilitating uptake by cells and also being involved in cytotoxicity, possibly involving oxidation of the iron centre to the ferricinium ion, thus initiating Fenton-type reactivity.9 Accordingly, the approach has been subsequently applied to other types of diseases, notably, Brocard and co-workers have modified the chloroquine series of drugs, to produce a ferrocenyl derivative called ferroquine.10 Ferroquine has proven effective against chloroquine resistant malaria parasites,11 which represents a major and growing problem. Ferroquine is now completing phase II clinical trials. In general, the strategy of attaching an organometallic fragment to a known organic drug is becoming increasingly recognised as a method to endow the drug with new properties, which include overcoming drug resistance.12
Returning to cancer, with the surgical removal of primary tumours being a highly successful procedure, the need for effective drugs to treat metastasis tumours is becoming increasingly important. Very few selective drugs have been identified that target metastasis, whether organic, inorganic or organometallic compounds. Remarkably, an inorganic drug based on ruthenium, ImH[trans-ImDMSORuCl4] (NAMI-A) that failed the usual screens of putative anticancer agents, such as the National Cancer Institute (NCI, USA) screen against a panel of 60 cell lines, has been shown to prevent the development and growth of pulmonary metastases in all the solid tumours on which it has been tested: rodent transplantable tumours of Lewis lung carcinoma,13 MCa mammary carcinoma,14 TS/A mammary adenocarcinoma,15 and human tumours engrafted in the nude mouse.16 Such behaviour is observed in the absence of activity against primary tumours, and recently, this intriguing compound has successfully completed Phase I Clinical Trials.17
The activity of NAMI-A (see Fig. 2) is most effective in the lungs where its half-life of elimination is about eight times longer than that in the primary tumour mass, probably due to the high content of collagen to which NAMI-A binds efficiently. The mechanism of metastasis control seems to be attributable to the combined effects of anti-angiogenic and anti-invasive properties of NAMI-A on tumour cells and on blood vessels. The control of angiogenesis, confirmed in the chick allantoic membrane and in the eye cornea model in the rabbit,18 is pronounced and is probably due to the scavenging properties of NAMI-A on the NO produced by the endothelial cells. At the molecular level NAMI-A blocks the mitogen activated phosphokinase/extracellular regulated kinase mitogen activated protein kinase/extracellular signal regulated kinase pathway, in endothelial cells, probably due to inhibition of a membrane PKC, leading to arrest of c-myc transcription, capspase-3 activation and apoptosis.19 Concerning the anti-invasive effects, it has been proven that NAMI-A reduces the spontaneous invasion of matrigel by tumour cells and this effect is accompanied by a dose-dependent reduction of gelatinase release by the same cells and activation of adhesion molecules such as beta-integrins.20 CD44 (a cell surface glycoprotein involved in cell/cell and cell/matrix interactions) is another cell membrane receptor modulated by NAMI-A to either increase TIL (tumour infiltrating lymphocytes) recruitment, change cell shape or reduce metastasis growth.21 The CD44 interaction, besides the control of cell cytoskeleton via F-actin condensation together with beta-integrins, is of great importance given its role in the pathway of c-Met (a receptor protein tyrosyne kinase involved in tumour malignancy), an oncogene associated with metastasis. The ligand exchange processes and redox changes of NAMI-A that have been demonstrated in vitro22 have yet to be correlated to the various biological effects described above.
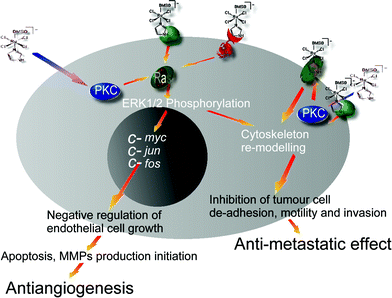 | ||
| Fig. 2 Schematic view of NAMI-A targets on cancer and endothelial cells showing the pharmacological consequences. RAS proteins are associated with transforming genes identified as human homologs of the murine sarcoma virus oncogenes v-Ha-ras or v-Ki-ras. PKC is protein kinase C involved in the regulation of proliferation and differentiation. c-myc, c-jun, c-fos are immediate early oncogenes involved in cycling. MMP are matrix metallo proteinases. | ||
It is worth noting that a closely related compound, indazolium trans-[tetrachlorobis(1H-indazole)ruthenate(III)] (KP1019), induces apoptosis in colorectal carcinoma cells and it is currently in Phase I Clinical Trials.23
NAMI-A undergoes a complicated series of ligand exchange reactions in the physiological environment and the somewhat more stable (at least in the sense of ligand exchange) organoruthenium compounds were therefore identified as potential antitumour drugs,24 although other medicinal applications are also feasible.25 One of the lead compounds in question, [RuCl2(η6-toluene)(pta)] (RAPTA-T, where pta = 1,3,5-triaza-7-phospha-adamantane—Fig. 3) was shown to exhibit pH dependent DNA binding properties in vitro. A subsequent study employing an extensive series of RAPTA compounds with modified arene ligands showed them to be only poorly toxic towards cancer cells and completely non-toxic to non-tumourigenic (healthy) cells in vitro. Like NAMI-A, RAPTA failed the NCI screening process.26In vivo RAPTA-T was found to inhibit lung metastases in CBA mice bearing the MCa mammary carcinoma, reducing their weight, although again with only mild effects on primary tumours. Like NAMI-A, extremely low general toxicity combined with excellent clearance rates was observed.
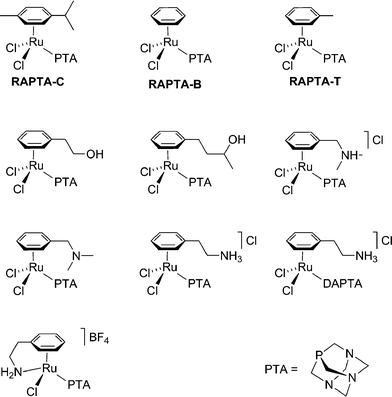 | ||
| Fig. 3 Some example of RAPTA compounds that have been evaluated in vitro and/or in vivo. | ||
The comparatively recent study of the RAPTA compounds means that their biological mechanism of activity is less well understood than that of NAMI-A. However, a number of parameters have been varied in the RAPTA series of compounds, using approaches characteristic of medicinal chemistry, in order to test various hypotheses regarding their mode of action or to improve their efficacy. As mentioned above, pH dependent DNA binding led to the proposal of a mechanism centring on activation by protonation. To further test such a mechanism the methylated-pta+ analogues were prepared as protonated model compounds, which proved to be equally toxic to the cancer and non-tumourigenic cell lines in vitro, suggesting that such a process could be in operation. The pKa of RAPTA estimated by NMR titration indicates protonation is unlikely under the physiological environment, unless another major modification to the compound takes place. Such a modification could occur as it was found that on binding to a 14-mer oligonucleotide the arene ligand may be lost,27 which drastically alters the pKa of the system. In an attempt to increase the general toxicity of these compounds, functional groups with potential hydrogen bonding substituents were tethered to the arene, since this should increase their tendency towards DNA binding, but this actually resulted in a decrease in toxicity towards the cancer cell line and an increase in toxicity to the non-tumourigenic cells.28 The replacement of the arene for other isolelectronic ligands is also possible without major changes to activity/selectivity in vitro,29 although the corresponding η5-C5H5 complex was not cytotoxic, perhaps due to its lower lipophilicity.30 Overall, it would appear that the pta ligand is responsible for the selective drug action, and while other ruthenium–arene compounds that do not utilise the pta ligand have been screened as anticancer drugs, some showing in vitro cytotoxicity comparable to that of cisplatin,31 as far as we are aware none show selectivity towards metastasis tumours.
It is remarkable that two such different compounds, NAMI-A (a ruthenium(III) coordination complex) and RAPTA-T (a ruthenium(II) organometallic compound) exhibit such similar antitumour behaviour, both being inactive towards primary tumours, but active towards secondary metastasis tumours. Both compounds also show very low general toxicity which makes them highly attractive as drugs, from a fundamental stance it will be highly interesting to study their mechanism of action which, as it unfolds, will identify new drug targets for metastasis. When the drug targets are identified effective assays can be developed, and since both drugs display very poor IC50 values in cell studies in vitro, new assays are urgently required. Currently, we postulate that the low general toxicity of these drugs could be connected to an internalisation → externalisation mechanism, in which the drugs enter the diseased cells and are then modified by a cellular defence mechanism against xenobiotics, e.g. glutathione-S-transferase catalysed conjugation of glutathione, the first step in the mercapturic acid pathway.32 Following expulsion, perhaps by multi-drug resistance proteins, the modified ruthenium compound remains attached to the cell membrane where it exerts its pharmacological effect.33 In order to substantiate such a mechanism extensive studies are required, and whether this proposal turns out to be proved or not, it seems likely that protein targets are more important than DNA in the activity of NAMI-A and RAPTA-T. In fact, we have recently shown in an in vivo study on a lung metastasis model that when NAMI-A is co-administered with cisplatin a superior activity is observed, ca. 60% of all animals were cured, which represents, as far as we are aware, the most successful treatment reported for a metastasis tumour.34 The study was combined with a laser ablation inductively coupled plasma mass spectrometric analysis that probed the interaction of the two drugs with plasma proteins, which indicated that the two drugs maintain their individual binding sites on plasma proteins without any interference from each other. The implication of plasma proteins in metal drug delivery, including for example the transferrin cycle, has attracted much interest in the scientific community.35
To conclude, the two antimetastasis compounds focused on here indicate that the discovery of new DNA-binding drugs is perhaps less important in future drug discovery programmes and the dominance of IC50 values as a first screen for putative drug should be treated with caution—what is essential is that cells showing the appropriate targets are employed. It is imperative that new assays for potential drug candidates are devised and that methods are required to rapidly locate drug interactions with key protein targets, such as mass spectrometric methods,36 which could prove to be valuable in establishing the interactions of these compounds with the targets responsible for cell invasion and metastasis. The interaction between various metal drugs with a limited range of proteins, notably albumin and transferrin, has been studied in great detail and it is time, in light of the current methods available to study the proteome, that metal binding studies are expanded to a much wider range of proteins and that the methods currently available to identify protein targets are utilised to their full potential.37 Finally, metal drugs may not be directly active, but may interact with the proteins that regulate apoptosis, therefore increasing the activity of organic compounds that induce apoptosis. Such compounds could prove to be particularly important where the proteins are responsible for drug resistance. To conclude, if we capitalise on the enormous expertise acquired on metal-based inorganic and organometallic drugs in these last thirty years, focussing on how these drugs could match and pharmacologically interact with protein targets that are selective for tumour malignancy, it is not unreasonable to expect that innovative agents that modify cell behaviour and cancer growth will be discovered
Acknowledgements
The authors wish to thank Enzo Alessio, University of Trieste, for the significant contribution to the growth of the chemical knowledge on ruthenium anticancer complexes. They thank the Swiss Cancer League (ligue Suisse contre le cancer), the EPFL, Swiss National Science Foundation, LINFA laboratory and the COST D20 Action for financial support.References
- (a) For example see: B. K. Keppler, Metal Complexes in Cancer Chemotherapy, Wiley-VCH, Weinheim, 1993 Search PubMed; (b) N. Farrell, Uses of Inorganic Chemistry in Medicine, Royal Society of Chemistry, Cambridge, UK, 1999 Search PubMed; (c) N. Farrell, Bioorganometallics, ed. G. Jaouen, Wiley-VCH, Weinheim, 2005 Search PubMed.
- B. Rosenberg, L. VanCamp and T. Krigas, Nature, 1965, 205, 698 CAS.
- (a) J. Reedijk, Chem. Commun., 1996, 801 RSC; (b) E. Wong and C. M. Giandomenico, Chem. Rev., 1999, 99, 2451 CrossRef CAS and references cited therein.
- D. Wang and S. J. Lippard, Nat. Rev. Drug Discovery, 2005, 4, 307 CrossRef CAS.
- K. R. Barnes, A. Kutikov and S. J. Lippard, Chem. Biol., 2004, 11, 557 CrossRef CAS.
- W. H. Ang, I. Khalaila, C. S. Allardyce, L. Juillerat-Jeanneret and P. J. Dyson, J. Am. Chem. Soc., 2005, 127, 1382 CrossRef CAS.
- A. Vessières, S. Top, W. Beck, E. Hillard and G. Jaouen, Dalton Trans., 2006, 529 RSC.
- S. Top, E. B. Kaloun, A. Vessières, G. Leclercq, I. Laïos, M. Ourevitch, C. Deuschel, M. J. McGlinchey and G. Jaouen, ChemBioChem, 2003, 4, 754 CrossRef CAS.
- E. Hillard, A. Vessières, L. Thouin, G. Jaouen and C. Amatore, Angew. Chem., Int. Ed., 2006, 45, 285–290 CrossRef.
- For a recent study see: L. Delhaes, H. Abessolo, C. Biot, L. Berry, P. Delcourt, L. A. Maciejewski, J. S. Brocard, D. Camus and D. Dive, Parasitol. Res., 2001, 87, 239 Search PubMed.
- C. Biot, G. Glorian, L. A. Maciejewski and J. S. Brocard, J. Med. Chem., 1997, 40, 3715 CrossRef CAS.
- C. S. Allardyce, A. Dorcier, C. Scolaro and P. J. Dyson, Appl. Organomet. Chem., 2005, 19, 1 CrossRef CAS.
- G. Sava, I. Capozzi, K. Clerici, G. Gagliardi, E. Alessio and G. Mestroni, Clin. Exp. Metastasis, 1998, 16, 371 CrossRef CAS.
- G. Sava, A. Bergamo, S. Zorzet, B. Gava, C. Casarsa, M. Cocchietto, A. Furlani, V. Scarcia, B. Serli, E. Iengo, E. Alessio and G. Mestroni, Eur. J. Cancer, 2002, 38, 427 CrossRef CAS.
- A. Bergamo, R. Gagliardi, V. Scarcia, A. Furlani, E. Alessio, G. Mestroni and G. Sava, J. Pharmacol. Exp. Ther., 1999, 289, 559 CAS.
- G. Sava, S. Zorzet, C. Turrin, F. Vita, M. R. Soranzo, G. Zabucchi, M. Cocchietto, A. Bergamo, S. DiGiovine, G. Pezzoni, L. Sartor and S. Garbisa, Cancer Res., 2003, 9, 1898 CAS.
- J. M. Rademaker-Lakhai, D. van den Bongard, D. Pluim, J. H. Beijnen and J. H. M. Schellens, Clin. Cancer Res., 2004, 10, 3717 CAS.
- (a) A. Vacca, M. Bruno, A. Boccarelli, M. Coluccia, D. Ribatti, A. Bergamo, S. Garbisa, L. Sartor and L. G. Sava, Br. J. Cancer, 2002, 86, 993 CrossRef CAS; (b) L. Morbidelli, S. Donnini, S. Filippi, L. Messori, F. Piccioli, P. Orioli, G. Sava and M. Ziche, Br. J. Cancer, 2003, 88, 1481.
- (a) G. Pintus, B. Tadolini, A. M. Posadino, B. Sanna, M. Debidda, F. Bennardini, G. Sava and C. Ventura, Eur. J. Biochem., 2002, 269, 5861 CrossRef CAS; (b) B. Sanna, M. Debidda, G. Pintus, B. Tadolini, A. M. Posadino, F. Bennardini, G. Sava and C. Ventura, Arch. Biochem. Biophys., 2002, 403, 209 CrossRef CAS.
- G. Sava, F. Frausin, M. Cocchietto, F. Vita, E. Podda, P. Spessotto, A. Furlani, V. Scarcia and G. Zabucchi, Eur. J. Cancer, 2004, 40, 1383 CrossRef CAS.
- S. Pacor, S. Zorzet, M. Cocchietto, M. Bacac, M. Vadori, C. Turrin, B. Gava, A. Castellarin and G. Sava, J. Pharmacol. Exp. Ther., 2004, 310, 737 CrossRef CAS.
- (a) M. Bacac, A. C. G. Hotze, K. van der Schilden, J. G. Haasnoot, S. Pacor, E. Alessio, G. Sava and J. Reedijk, J. Inorg. Biochem., 2004, 98, 402 CrossRef CAS; (b) B. Serli, E. Zangrando, E. Iengo, G. Mestroni, L. Yellowlees and E. Alessio, Inorg. Chem., 2002, 41, 4033 CrossRef CAS.
- S. Kapitza, M. Pongratz, M. A. Jakupec, P. Heffeter, W. Berger, L. Lackinger, B. K. Keppler and B. Marian, J. Cancer Res. Clin. Oncol., 2005, 131, 101 CrossRef CAS.
- C. S. Allardyce, P. J. Dyson, D. J. Ellis and S. L. Heath, Chem. Commun., 2001, 1396 RSC.
- C. S. Allardyce, P. J. Dyson, D. J. Ellis, P. A. Salter and R. Scopelliti, J. Organomet. Chem., 2003, 668, 35 CrossRef CAS.
- C. Scolaro, A. Bergamo, L. Brescacin, R. Delfino, M. Cocchietto, G. Laurenczy, T. J. Geldbach, G. Sava and P. J. Dyson, J. Med. Chem., 2005, 48, 4161–4171 CrossRef CAS.
- A. Dorcier, P. J. Dyson, C. Gossens, U. Rothlisberger, R. Scopelliti and I. Tavernelli, Organometallics, 2005, 24, 2114 CrossRef CAS.
- C. Scolaro, T. J. Geldbach, S. Rochat, A. Dorcier, C. Gossens, A. Bergamo, M. Cocchietto, I. Tavernelli, G. Sava, U. Röthlisberger and P. J. Dyson, Organometallics, 2006, 25, 756 CrossRef CAS.
- B. Serli, E. Zangrando, T. Gianferrara, C. Scolaro, P. J. Dyson, A. Bergamo and E. Alessio, Eur. J. Inorg. Chem., 2005, 3423 CrossRef CAS.
- D. N. Akbayeva, L. Gonsalvi, W. Oberhauser, M. Peruzzini, F. Vizza, P. Brüggeller, A. Romerosa, G. Sava and A. Bergamo, Chem. Commun., 2003, 264 RSC.
- Y. K. Yan, M. Melchart, A. Habtemarian and P. J. Saddler, Chem. Commun., 2005, 4764 RSC.
- D. Townsend and K. Tew, Oncogene, 2003, 22, 7369 CrossRef CAS.
- F. Frausin, V. Scarcia, M. Cocchietto, A. Furlani, B. Serli, E. Alessio and G. Sava, J. Pharmacol. Exp. Ther., 2005, 313, 227 CAS.
- I. Khalaila, A. Bergamo, F. Bussy, G. Sava and P. J. Dyson, Int. J. Oncol., 2006 Search PubMed , in press.
- (a) For examples which consider KP1019 in this context see: A. V. Rudnev, S. S. Aleksenko, O. Semenova, C. G. Hartinger, A. R. Timerbaev and B. K. Keppler, J. Sep. Sci., 2005, 28, 121 Search PubMed; (b) M. Pongratz, P. Schluga, M. A. Jakupec, V. B. Arion, C. G. Hartinger, G. Allmaier and B. K. Keppler, J. Anal. At. Spectrom., 2004, 19, 46 RSC.
- I. Khalaila, C. S. Allardyce, C. Verma and P. J. Dyson, ChemBioChem, 2005, 6, 1788 CrossRef CAS.
- C. S. Allardyce, P. J. Dyson, F. R. Abou-Shakra, H. Birtwhistle and J. Coffey, Chem. Commun., 2001, 2708 RSC.
| This journal is © The Royal Society of Chemistry 2006 |