Olefination and group transfer reactions of an electron deficient tantalum methylidene complex†
Sarah M.
Mullins
,
Robert G.
Bergman
* and
John
Arnold
*
Department of Chemistry, University of California, Berkeley, CA, USA 94720-1460
First published on 14th November 2005
Abstract
The reactivity of an electronically unsaturated tantalum methylidene complex [TolC(NSiMe3)2]2Ta(CH2)CH3 (1) supported by [TolC(NSiMe3)2] amidinate ligands is described. Electrophilic addition and olefination reactions of the Ta![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2 functionality are reported. Alkylidene 1 participates in group-transfer reactions not observed in sterically similar, but electronically saturated, analogues. Reactions with substrates containing unsaturated C–X (X = C, N, O) bonds yield [Ta]X compounds and vinylated organic products; carbon–sulfur cleavage reactions to produce tantalum thioformaldehyde and tantalum sulfido complexes.
CH2 functionality are reported. Alkylidene 1 participates in group-transfer reactions not observed in sterically similar, but electronically saturated, analogues. Reactions with substrates containing unsaturated C–X (X = C, N, O) bonds yield [Ta]X compounds and vinylated organic products; carbon–sulfur cleavage reactions to produce tantalum thioformaldehyde and tantalum sulfido complexes.
Introduction
Transition-metal methylidene complexes are important in both industrial and synthetic organic applications. For example, surface-bound methylene groups are proposed as intermediates in the Fischer–Tropsch reaction.1,2 Heteroatom (O, S, NR) abstraction reactions by methylidene complexes model hydrodesulfurization and hydrodeamination of petroleum feedstocks as well as the poisoning of catalyst surfaces.3 Molecular compounds that are analogous to the industrially important methylene-functionalized metal and metal oxide surface species provide insight into the structure and reaction mechanisms of these complicated heterogeneous systems.4 Finally, transition-metal methylidene complexes are useful reagents for organic transformations.5 Insertion of the carbene fragment into C–H bonds is one method for the functionalization of organic molecules.6 As a result of these applications, the chemistry of transition-metal methylidene complexes is now well established.While early metal methylidene complexes are unquestionably of great industrial and academic significance, their synthesis is often a challenge. In most early metal alkylidene complexes, the MCR2 moiety tends to be quite unstable to decomposition and dimerization. Accordingly, the isolation of such compounds often requires steric bulk at the metal, a high formal electron count, or substitution at the α-carbon atom. These strategies have been employed by Schrock and co-workers,7 who used electronic saturation to synthesize Cp2Ta(CH2)CH38,9 and a bulky substituent on the alkylidene carbon atom to prepare CpCl2Ta(CHCMe3).10 While the 18-electron Cp2Ta(CH2)CH3 reacts with some organic electrophiles,8 the less electron rich tantalum alkylidenes (e.g. 14e− CpCl2Ta(CHCMe3)) exhibit the most reactivity.7,11 Electronically unsaturated titanium methylidenes, which also exhibit a broad spectrum of reactivity with organic substrates, may be accessed from “masked” precursors, such as the aluminium alkyl-stabilized Tebbe reagent12 or titanacyclobutanes.13 Despite the potential increased reactivity of these derivatives compared to that of the saturated complexes, the chemistry of isolable electronically unsaturated transition-metal methylidene complexes remains largely unexplored.14–19
The α-carbon atoms of transition-metal methylidene complexes have been reported to exhibit either nucleophilic or electrophilic behavior. The electronic properties of these unsaturated ligands are highly sensitive to electronegativity of the metal, its oxidation state, and the overall charge of the complex.5 Generally, high oxidation state early metal carbenes exhibit nucleophilic behavior, while low oxidation state late metal carbenes generally act as electrophiles.20 However, these observations are far from rigid, and methylidenes are especially sensitive to the electron density at the metal center. For example, the late metal methylidene, Cp*(PMe3)Ir(CH2), exhibits nucleophilic behavior.21 The nucleophilic methylidene in Cp2Ta(CH2)CH3 stands in contrast to the electrophilic nature of the isoelectronic, cationic tungsten analogue, [Cp2W(CH2)CH3]+.22 Some methylidene complexes even exhibit amphiphilic behavior: an example is Cl(NO)PPh3OsCH2, which reacts with both electrophiles (H+) and nucleophiles (CO or CNR).23
Given the potential applications of electronically unsaturated transition-metal methylidenes, we were interested in exploring the reactivity afforded by the bis(amidinate)tantalum methylidene complex, [TolC(NSiMe3)2]2Ta(CH2)CH3 (1), the synthesis of which we have previously reported.24 The bis(trimethylsilyl)tolylamidinate ligand set enforces a cis geometry of the methylene and methyl ligands, which is important for potential migration processes, and provides steric bulk intermediate to that of bis(Cp) and bis(Cp*) ligand sets.25 The anionic bis(trimethylsilyl)tolylamidinate ligand is generally considered to be a four electron donor; this generates an electronically unsaturated (14e−) metal center in 1.
Our previous mechanistic work on the reaction of tantalum methylidene 1 with pyridine N-oxides demonstrates that the electron deficient metal center is critical to opening new reaction pathways (Scheme 1).26 The selective reactivity of 1 with pyridine N-oxides stands in contrast to the 18e− tantalocene analog, Cp2Ta(CH2)CH3, which is unreactive. Additional support for our belief that the electron count is important in this reaction comes from a recent report in which the 16e− “Cp2Ti(CH2)” was used to carry out an analogous pyridine N-oxide to methylpyridine transformation. This report also described the application of this methodology to a natural product synthesis.27,28
 | ||
| Scheme 1 | ||
These reactions encouraged us to determine whether the thermodynamic stability of early metal–heteroatom (O, NR, S) multiple bonds can be coupled with carbene transfer to selectively functionalize organic substrates. This report describes how the tantalum methylidene 1 participates not only in familiar electrophilic addition and olefination reactions, but also group transfer reactions not observed in sterically similar, but electronically saturated, analogues.
Results and discussion
Solid-state structure of 1
The X-ray structure of 1 provides a starting point for analysis of the steric characteristics of the bis(trimethylsilyl)tolylamidinate ligands. An ORTEP diagram of 1 is shown in Fig. 1. Selected crystal and refinement data are in Table 1; bond lengths and angles for 1 are given in Table 2. The dominance of the bis(trimethylsilyl)tolylamidinate ligands in the tantalum coordination sphere is not only evident visually, but is also reflected by the inability to crystallographically distinguish between the CH2 and CH3 ligands. Molecules of 1 have packed in a mixture of two enantiomers, resulting in a crystallographic C2 axis which bisects the (CH3)Ta(CH2) vector (despite the lack of molecular C2 symmetry). The observed Ta1–C1 bond length, 2.05 Å, is an average of the normal metal–carbon single and double bond lengths.5| 1 | 13 | 14 | 15 | |
|---|---|---|---|---|
| Reflections measured: hemisphere; refinement method: full-matrix least-squares on F2; hydrogen atoms: idealized positions. | ||||
| Empirical formula | C30H55N4Si4Ta | C39H64N5Si4Ta | C30H55N4Si4STa | C29H53N4Si4TaS |
| M r | 765.08 | 896.26 | 829.20 | 783.11 |
| T/°C | −118 | −103 | −105 | −107 |
| Crystal system | Monoclinic | Monoclinic | Monoclinic | Monoclinic |
| Space group | C2/c (no. 15) | P21/c (no. 14) | C2/c (no. 15) | C2/c (no. 15) |
| a/Å | 28.2182(1) | 17.9014(4) | 12.9195(3) | 28.2627(4) |
| b/Å | 8.7227(1) | 11.6015(2) | 18.8911(4) | 8.7781(2) |
| c/Å | 19.8359(1) | 23.0038(5) | 16.7243(2) | 19.8801(3) |
| V/Å3 | 3814.35(6) | 4459.0(2) | 3970.3(1) | 3819.2(1) |
| β/° | 128.625(1) | 111.041(1) | 103.423(1) | 129.253(1) |
| Z | 4 | 4 | 4 | 4 |
| D c/g cm−3 | 1.332 | 1.335 | 1.387 | 1.362 |
| Scan type/° | ω, 0.3 | ω, 0.3 | ω, 0.3 | ω, 0.3 |
| Frame collection time/s | 10 | 10 | ||
| 2θ range/° | 3–52.3 | 3–52.0 | 3–51.9 | 3–49.4 |
| µ/mm−1 | 30.26 | 26.00 | 30.14 | 30.77 |
| T max, Tmin | 0.640, 0.450 | 0.636, 0.517 | 0.817, 0.580 | 0.496, 0.223 |
| Crystal dimensions/mm | 0.20 × 0.20 × 0.30 | 0.12 × 0.25 × 0.30 | 0.10 × 0.12 × 0.12 | 0.20 × 0.15 × 0.25 |
| No. reflections measured | 10633 | 21557 | 9490 | 8561 |
| No. unique reflections | 3656 | 8314 | 3582 | 3355 |
| No. observations (I > 3σ) | 2920 | 4578 | 2562 | 2646 |
| No. variables | 177 | 442 | 173 | 181 |
| Data/parameter | 16.5 | 10.4 | 14.8 | 14.6 |
| R int (%) | 2.7 | 4.6 | 6.2 | 4.6 |
| R (%) | 3.0 | 2.7 | 4.2 | 3.4 |
| R w (%) | 3.7 | 3.0 | 5.2 | 3.9 |
| GOF | 1.42 | 0.93 | 1.47 | 1.4 |
| Largest diff. peak, hole/e Å−3 | 1.59, −0.94 | 0.76, −0.73 | 1.52, −1.13 | 0.72, −2.39 |
| Ta(1)–N(1) | 2.260(3) | N(1)–Ta(1)–N(2) | 60.4(1) |
| Ta(1)–N(2) | 2.191(3) | N(1)–Ta(1)–C(1) | 92.7(2) |
| Ta(1)–N(5) | 1.996(4) | N(2)–Ta(1)–C(1) | 102.6(2) |
| Ta(1)–C(1) | 2.049(5) | N(1)–C(8)–N(2) | 114.7(3) |
| N(1)–C(8) | 1.313(5) | ||
| N(2)–C(8) | 1.347(5) |
 | ||
| Fig. 1 ORTEP diagram of 1, showing 50% probability ellipsoids. Hydrogen atoms are omitted for clarity. | ||
A space-filling model of 1 shows that the bis(trimethylsilyl)tolylamidinate ligand set has a roughly rectangular shape and that the majority of the ligands' steric bulk is relatively distant from the methyl and methylidene groups (Fig. 2). Using CPK models, Teuben and co-workers determined the coordination aperture29 to be 84° for [PhC(NSiMe3)2]2M, compared to 103 and 72° for the metallocene analogues Cp2M and Cp*2M (M = Y).25 This result indicates that the steric bulk of the bis(tolylamidinate) ligand set is more similar to the bis(Cp*) set. A qualitative comparison of 1 and Cp2Ta(CH2)CH3 (Fig. 2) reveals that the open faces of the complexes are more similar than might be expected for two ancillary ligand sets of such different size and shape.
 | ||
| Fig. 2 Side and top views of space filling models for 1 and Cp2Ta(CH2)CH3. Carbon labels identify the CH2 and CH3 ligands. | ||
Reactions of 1 with electrophiles
The nucleophilic nature of the methylidene moiety of 1 is evident from its reactions with electrophilic alkylating reagents. The addition of 1 equiv. of methyl iodide to 1 results in the formation of the ethyl methyl iodo tantalum complex 2, presumably by initial attack of the Me+ fragment at the carbene carbon atom (Scheme 2). No reaction was observed between 1 and tert-butyl chloride or trimethylsilyl chloride.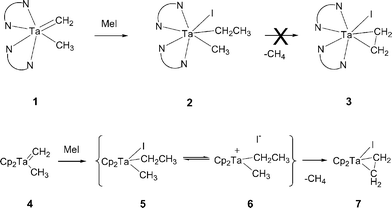 | ||
| Scheme 2 | ||
Differences in the reactions of methyl iodide between the analogous 14e− (1) and the 18e− (Cp2Ta(CH2)CH3) complexes provide evidence for the relatively electrophilic nature of the tolylamidinate-supported tantalum complex (Scheme 2). In the tantalocene example, iodide may dissociate from the ethyl methyl iodido intermediate (5–6). β-Hydride elimination followed by reductive elimination of methane forms the ethylene iodide tantalocene complex (7).8 No such dissociation or elimination reactions were observed for 2; no methane or ethylene was observed, even upon decomposition of 2. Presumably, the highly electrophilic tantalum favors more rapid attack of I− after the initial alkylation, and subsequently disfavors the iodide dissociation required for the conversion of 2 to 3.
Olefination reactions
Perhaps the most well-known reactions of early metal carbenes are those that yield organic molecules that contain unsaturated C–C bonds.30 Not surprisingly, the methylidene complex 1 also participates in a variety of these reactions. For example, 1 reacts with nitriles (8a,b) to form vinylimido tantalum complexes (11a,b) (Scheme 3). The vinyl group is clearly evident in the 1H NMR (δ 5.27 (d), 5.34 (d) ppm) and DEPT-135 (δ 105.3 ppm) experiments. | ||
| Scheme 3 | ||
The reaction is proposed to proceed first with nitrile coordination (9a,b), followed by formation of an azatantalacyclobutene (10a,b). Cycloreversion to the thermodynamically stable TaNR compound yields the product (11a,b) (Scheme 3). No intermediates were observed when the reaction was monitored by 1H NMR spectroscopy; however, similar mechanisms have been suggested for the reactions of other tantalum alkylidenes with nitriles. Schrock and co-workers found that the 14e− CpCl2Ta(CHtBu)10 and 10e− (tBuCH2)3Ta(CHtBu)31 undergo this reaction with nitriles, whereas electronically saturated Cp2Ta(CH2)CH38 and Cp2Ta(CHtBu)Cl32 do not react at all. This suggests that nitrile coordination may be the first step in all of these reactions.
Complex 1 reacts with certain aldehydes to yield olefinated organic products. For example, the addition of acetaldehyde to a benzene-d6 solution of 1 yields the tantalum oxo complex 1226 and propylene. However, no reaction is observed between 1 and carbonyl-containing substrates with larger substituents, such as tolualdehyde or benzophenone (Scheme 4). These [2 + 2] cycloaddition/reversion transformations are well-established for tantalum carbenes and phosphorus ylide complexes.33–35
 | ||
| Scheme 4 | ||
When a benzene-d6 solution of 1 was exposed to small alkenes (ethylene or propylene, 1 equiv. to 1 atm), complex mixtures of organic and organometallic products were obtained. Propylene and 1-butene were among the organic products identified by 1H NMR and GC-MS. Attempts to isolate the tantalum-containing product(s) were unsuccessful, even when the reaction was carried out in the presence of added Lewis base. Carbene fragment insertion into small alkenes was previously observed in pincer27 and cyclopentadienyl tantalum neopentylidene complexes.36
Reaction with isocyanide: Formation of a metal-bound ketenimine
Metal-ketenimine complexes are versatile building blocks for the synthesis of heterocyclic complexes. While carbene addition to an isocyanide is an established method for the synthesis of metal–ketenimine complexes, examples of this reaction with early metal carbenes have not been forthcoming.37 Consequently, we tested the behavior of 1 with isocyanides.The reaction of 1 with tert-butyl isocyanide yields multiple intractable products. In contrast, addition of 1 equiv. of 2,6-dimethylphenyl isocyanide to 1 yields the η2-ketenimine complex 13 (eqn (1)). The 1H NMR spectrum of complex 13 reveals two singlets, at δ 4.78 ppm (1H) and δ 5.99 ppm (1H). A 13C–1H HMQC experiment revealed that both protons correlate to a single resonance in the 13C NMR spectrum (δ 77.2 ppm) (Fig. 3). A DEPT-135 experiment confirmed that this carbon resonance is the CH2 group of the ketenimine. We presume that the two-bond coupling of these protons is too small to be observed.
 | (1) |

The X-ray structure of 13 establishes the regiochemistry of the ketenimine in the solid state and helps to explain the 1H NMR spectrum. An ORTEP diagram of 13 is presented in Fig. 4. Metrical information is in Table 3; selected crystal and refinement data are in Table 1. The complex has a distorted capped trigonal prismatic geometry (Fig. 5). C39 is the capping ligand; three of the amidinate nitrogen atoms (N1, N2, N4) and the ketenimine nitrogen (N5) form the rectangular face. The tantalum atom sits 0.09 Å below the plane defined by these four nitrogen atoms (average nitrogen deviation from least squares plane = 0.03 Å). The remaining amidinate nitrogen atom, N3, and the ketenimine carbon, C29, form the third vertices of the triangular faces. The C29–N5 bond distance (1.380(7) Å) is consistent with the presence of a single bond (lit. C(sp2)–N(sp2) = 1.355 Å).38 The very different chemical environments for the two methylene protons of the ketenimine moiety are apparent from this structure. Proton a is clearly oriented toward the trimethylsilyl group of the amidinate, whereas proton b points toward the aryl group of the ketenimine (Fig. 5).
| Ta(1)–N(1) | 2.123(4) | N(1)–Ta(1)–N(2) | 61.1(2) |
| Ta(1)–N(3) | 2.164(4) | N(2)–Ta(1)–N(4) | 91.0(2) |
| Ta(1)–N(5) | 1.996(4) | N(3)–Ta(1)–N(4) | 60.6(2) |
| Ta(1)–C(39) | 2.188(6) | N(4)–Ta(1)–N(5) | 87.9(2) |
| N(5)–C(29) | 1.379(7) | N(1)–Ta(1)–N(5) | 119.6(2) |
| C(29)–C(30) | 1.364(7) | N(3)–Ta(1)–C(29) | 92.3(2) |
| Ta(1)–N(2) | 2.296(4) | N(5)–C(29)–C(30) | 130.4(5) |
| Ta(1)–N(4) | 2.284(4) | Ta(1)–C(29)–N(5) | 66.1(3) |
| Ta(1)–C(29) | 2.106(5) | Ta(1)–N(5)–C(29) | 74.7(3) |
| N(1)–C(1) | 1.347(6) | N(1)–C(1)–N(2) | 114.5(4) |
| N(2)–C(1) | 1.329(6) | N(3)–C(15)–N(4) | 114.0(5) |
| N(3)–C(15) | 1.359(6) |
 | ||
| Fig. 4 ORTEP diagram of 13, showing 50% probability ellipsoids. Hydrogen atoms are omitted for clarity. | ||
 | ||
| Fig. 5 ORTEP diagram of the immediate coordination sphere of 13. Trimethylsilyl and aryl groups omitted for clarity. | ||
Variable-temperature 1H NMR spectra of 13 reveal that while multiple intramolecular fluxional processes occur, the methylene group orientation is preserved in solution. At room temperature, fast exchange of the bis(trimethylsilyl)tolylamidinate ligands is evident from the presence of a single trimethylsilyl resonance and a single tolyl methyl group resonance. At −70 °C, two tolyl methyl group resonances (c, d; see Fig. 6) are observed, suggesting slow exchange between amidinate ligands. Three trimethylsilyl resonances (e, f, g) are observed in a 18H : 9H : 9H integration ratio. This implies that rotation about the internal C2 axis39 of one amidinate ligand is slow, while for the second amidinate the rotation barrier is more rapid. The line drawing of 13, based on the X-ray structure (Fig. 6), shows the two different chemical environments of the amidinate ligands. The large 2,6-dimethylphenyl substituent on the ketenimine hinders the rotation of the amidinate containing N3 and N4, whereas the N1, N2 amidinate is unencumbered and can rotate freely.40 Hindered rotation of the 2,6-dimethylphenyl group is also observed at low temperature, leading to two methyl resonances (h, i), and is consistent with the slow exchange-limit structure. A 1H NMR spectrum at the slow exchange limit is shown in Fig. 6.
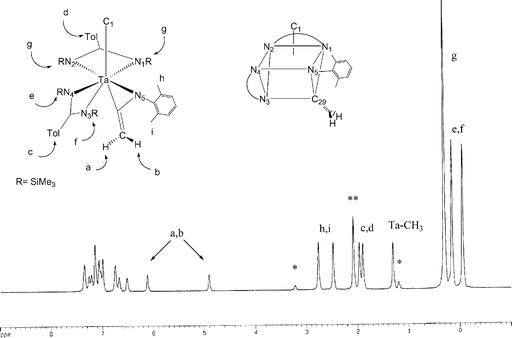 | ||
| Fig. 6 1H NMR spectrum of 13 (toluene-d8) at 203 K (* = residual diethyl ether, ** = solvent). | ||
Sulfur transfer reactions
As part of an exploration of carbon-sulfur bond cleavage reactions, we investigated the reactivity of 1 towards several sulfur-containing organic compounds. Tantalum methylidene 1 abstracts sulfur from propylene sulfide to yield the η2-thioformaldehyde compound 14 and propylene (eqn (2)). The methylene protons of 14 appear as a singlet resonance at δ 2.87 ppm (2H), in the 1H NMR spectrum. A resonance at δ 71.1 ppm in the 13C{1H} NMR spectrum was identified as the methylene carbon of the thioformaldehyde ligand by a DEPT-135 experiment.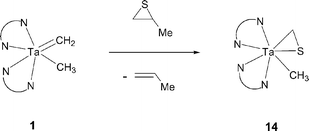 | (2) |
Complex 14 crystallizes in the centrosymmetric space group C2/c. The η2-thioformaldehyde ligand is bound as a metallathiacyclopropane, with the sulfur in the inside position. An ORTEP diagram is shown in Fig. 7. Selected crystal and refinement data are in Table 1; metrical parameters are given in Table 4. As in the X-ray structure of 1, the superposition of enantiomers produces a crystallographic-C2 axis, which leads to disorder in refinement of the –CH2S and –CH3 groups. The sulfur atom of the thioformaldehyde (S1), which was disordered over two positions, was refined at half-occupancy. The carbon atoms of the methylene and the methyl group were refined as one full occupancy carbon (C12); see X-ray details for further information.
| Ta(1)–S(1) | 2.295(5) | S(1)–Ta(1)–N(1) | 129.5(2) |
| Ta(1)–N(1) | 2.281(6) | S(1)–Ta(1)–N(2) | 104.8(2) |
| Ta(1)–N(2) | 2.156(6) | S(1)–Ta(1)–C(15) | 77.0(3) |
| Ta(1)–C(15) | 2.23(1) | N(1)–Ta(1)–N(2) | 61.0(2) |
| S(1)–C(15) | 1.93(1) | N(1)–Ta(1)–C(15) | 148.0(3) |
| N(1)–C(1) | 1.31(1) | N(2)Ta(1)–C(15) | 98.4(3) |
| N(2)–C(1) | 1.355(9) | Ta(1)–S(1)–C(15) | 63.0(3) |
| Ta(1)–C(15)–S(1) | 66.6(4) |
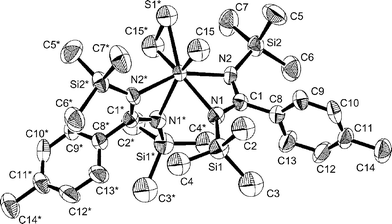 | ||
| Fig. 7 ORTEP diagram of 14, showing 50% thermal probability ellipsoids. Hydrogen, S1, C16/C16*, C17/C17*, and C18/C18* atoms (disordered sulfur atom and trimethylsilyl group) are omitted for clarity. | ||
Compound 14 is one of only a few η2-thioformaldehyde complexes known for early metals (Ta,41 Zr42 and Ti43). In addition, 14 is thermally robust; no changes were observed upon prolonged heating at 135 °C. This behavior contrasts to that of other tantalum thioaldehydes, Cp2Ta(SCHR)H, which undergo facile migrations to the more thermodynamically stable terminal sulfido alkyl complexes, Cp2Ta(S)CH2R.44
Carbon–sulfur bond cleavage of a different kind occurs in the reaction of 1 with triphenylmethanethiol, to form the terminal sulfido complex 15 and triphenylethane (eqn (3)). The 1H and 13C{1H} NMR spectra of the tantalum product are similar to those for the terminal tantalum oxo analog, 12. The electron impact mass spectrum shows the molecular ion at m/z 782.
 | (3) |
The structure of the terminal tantalum–sulfido complex was confirmed by X-ray crystallography. An ORTEP diagram is shown in Fig. 8. Selected crystal and refinement data are given in Table 1; metrical parameters are in Table 5. Like 1 and 14, the sulfido complex 15 crystallizes in the centrosymmetric space group, C2/c. Again, the superposition of the two enantiomers produces a crystallographic C2 axis. The sulfido and methyl groups are disordered over two positions; S1 and C15 were each refined at half-occupancy. The Ta1–S1 bond length is 2.22 Å, similar to other reported terminal Ta–S multiple bonds.45,46
| Ta(1)–S(1) | 2.222(10) | S(1)–Ta(1)–N(1) | 153.7(2) |
| Ta(1)–N(1) | 2.251(4) | S(1)–Ta(1)–N(2) | 92.8(2) |
| Ta(1)–N(2) | 2.144(3) | S(1)–Ta(1)–C(15) | 95.4(7) |
| Ta(1)–C(15) | 2.27(4) | N(1)–Ta(1)–N(2) | 61.1(1) |
| N(1)–C(1) | 1.320(5) | N(1)–C(1)–N(2) | 114.6(4) |
| N(2)–C(1) | 1.337(6) |
 | ||
| Fig. 8 ORTEP diagram of 15, showing 50% thermal probability ellipsoids. Hydrogen atoms, C15* and S1* are omitted for clarity. | ||
Several possible mechanisms for the formation of 15 and triphenylethane are outlined in Scheme 5. In mechanism A, the methylidene deprotonates the thiol to form the symmetrical intermediate 16. β-Elimination reaction yields 15 and triphenylethane. In mechanism B, Ph3CSH reacts as an electrophile (Ph3C+), attacking the nucleophilic methylidene to form intermediate 17. β-Hydride transfer from the thiolate group yields 15 and triphenylethane. For pathway C, Ph3C+ abstracts the methide group to yield triphenylethane and a tantalum methylidene thiolate intermediate (18). In the final step, 18 rearranges to the thermodynamically preferred tautomer (15).
 | ||
| Scheme 5 | ||
While no intermediates were observed in this reaction, the three proposed mechanisms proceed through distinct intermediates that are distinguishable by performing a deuterium tracer experiment. The addition of 1 equiv. of triphenylmethanethiol (73%-d1) to 1 yields a mixture of deuterium-incorporated products [TolC(SiMe3)2]2Ta(S)CH2D (0.442 H), Ph3CCH3 (1.09 H), [TolC(SiMe3)2]2Ta(S)CH3 (1.15 H) and Ph3CCH2D (0.438 H).47 When the relative integration values of these products are corrected to account for the incompletely deuterated starting material, a nearly statistical ratio of products is revealed (TaCH3 : TaCH2D = 1.07 : 1.00, Ph3CCH3 : Ph3CCH2D = 1.02 : 1.00). These data are consistent with the intermediacy of 16 in mechanism A, which is also the only pathway expected to give a mixture of deuterated products. Furthermore, the observed isotopic distribution (i.e. lack of d2 or d3 products) suggests that the protonation reaction which forms 16 is irreversible.
Elimination reactions from tantalum(V) thiolate complexes have been reported. When β-hydrogens are present on the thiolate ligand, β-hydride elimination to thioformaldehyde is favored.41 In the case of a tertiary substituted thiolate group, carbon-sulfur cleavage is preferred.48,49 The latter is consistent with the proposed mechanism for the reaction between 1 and Ph3CSH.
Oxygen abstraction reactions
Carbene complex 1 does not react with the oxygen analogs of the sulfur reagents discussed above (propylene oxide, Ph3COH). We previously reported on the reactions of 1 with pyridine N-oxides and nitrones.26Summary and conclusion
The bis(amidinate) ancillary ligands on the tantalum methyl methylidene complex 1 produce an electronically unsaturated (14e−) tantalum center that has open space similar to that of early metal bent metallocenes. Tantalum methylidene 1 is a stable, isolable complex, but is reactive toward a range of electrophilic substrates. Reactions with substrates containing unsaturated C–X (X = C, N, O) bonds yield [Ta]X compounds and vinylated organic products. Methylidene 1 participates in carbon-sulfur cleavage reactions to produce tantalum thioformaldehyde and tantalum sulfido complexes. The electrophilic character of the 14e− tantalum center is important in this chemistry. The range of reactions reported herein for 1 expands the scope of transformations known for early metal methylidene complexes.
Experimental
General considerations
Standard Schlenk line and glove box techniques were used throughout. Hexanes was distilled from purple Na/benzophenone under nitrogen. Toluene and hexamethyldisiloxane (HMDSO) were distilled from Na under nitrogen. Benzene and pentane were dried by passage through activated alumina and then degassed with a nitrogen purge.50 C6D6 was vacuum transferred from Na/benzophenone. Propylene sulfide (Aldrich) was first dried over molecular sieves, transferred under static vacuum to a Teflon-sealed flask and stored at 5 °C. Pyridine N-oxide and 2-methylpyridine N-oxide (Aldrich) were dried over molecular sieves in toluene and the crude solid sublimed before use. 2-Ethylpyridine N-oxide was synthesized starting from ethylpyridine via the literature procedure.51 2,2′-Dipyridyl N-oxide52 was prepared according to literature procedures. Methyl iodide was stored over Cu wire. 1,3,5-Trimethoxybenzene (Aldrich) was sublimed before use. Ph3CSH (Aldrich), 2,6-dimethylphenyl isocyanide (Fluka), and CH3OTf (Aldrich) were used as received. Acetonitrile and p-tolylnitrile were distilled from CaH2. Melting points were determined in sealed capillaries and are uncorrected. 1H and 13C{1H} NMR spectra were recorded at ambient temperatures, unless otherwise noted. IR samples were prepared as mineral oil mulls and taken between KBr plates. Elemental analysis and mass spectral data were determined by the College of Chemistry, University of California, Berkeley, CA. Single-crystal X-ray determinations were performed at CHEXRAY, University of California, Berkeley, CA.(a) Typical procedure: Reaction with pyridine N-oxide. In the glove-box, [TolC(NSiMe3)2]2Ta(CH2)CH3 (0.075 g, 0.098 mmol) and pyridine N-oxide (0.0093 g, 0.098 mmol) were weighed into a tared vial. C6D6 (0.5 mL) was added to dissolve the reagents. After 14 h, 1H and 13C{1H} NMR spectra were obtained of this reaction mixture. These spectra contained peaks matching authentic samples of 12 and 2-methylpyridine.26 Conversion by 1H NMR: 99%.
(b) Reaction with 2-methylpyridine N-oxide. 1H and 13C{1H} NMR spectra matched those for an authentic sample of 2,6-dimethylpyridine. Conversion by 1H NMR: 99%.26
(c) Reaction with 2-ethylpyridine N-oxide. The 1H NMR spectrum contained peaks consistent with 6-ethyl-2-methylpyridine. Conversion by 1H NMR: 90%. Relevant 1H NMR (300 MHz): δ 1.26 (t, J = 7 Hz, 2H, CH2CH3), 2.42 (s, 3H, CH3), 2.79 (q, J = 7 Hz, 3H, CH2CH3), 6.19 (m, 1H, Ar), 6.27 (m, 1H, Ar), 6.60 (m, 1H, Ar).
(d) Reaction with 2,2′-dipyridyl N-oxide. The 1H NMR spectrum contained peaks consistent with 6-methyl-2,2′-dipyridyl. Conversion by 1H NMR: 95%. Relevant 1H NMR (300 MHz): δ 2.45 (s, 3H, CH3), 6.67 (m, 2H, Ar), 7.24 (m, 2H, Ar), 8.53 (m, 1H, Ar), 8.64 (d, J = 8 Hz, 1H, Ar), 8.75 (d, J = 8 Hz, 1H, Ar).
General experimental details for X-ray structure determinations
Crystals were mounted onto glass fibers using Paratone N hydrocarbon oil and were transferred to a Siemens SMART diffractometer/CCD area detector,54 centered in the beam, and cooled by a nitrogen-flow low-temperature apparatus. Preliminary orientation matrix and cell constants were determined by collection of 60 10 s frames, followed by spot integration and least squares refinement. A hemisphere of data was collected using ω scans of 0.3° counted for a total of 10 s per frame. The raw data were integrated by the program SAINT.55 Data analysis was performed using XPREP.56 For 13, and 14, an absorption correction was applied using XPREP; for 1 and 15, the correction was applied using SADABS.57 The unit cell parameters and statistical analysis of the intensity distribution were used for space group determination.58 The data were corrected for Lorentz and polarization effects, but no correction for crystal decay was applied. Unique equivalent reflections were merged. The structures were solved by direct methods59 and expanded using Fourier techniques.60 Except as noted, all non-hydrogen atoms were refined anisotropically and hydrogen atoms were included as fixed contributions but not refined. The quantity minimized by the least-squares program was Σw(|Fo| − |Fc|),2 where w is the weight of a given observation. The weighting scheme was based on counting statistics and included a factor (p = 0.030) to downweight the intense reflections. The analytical forms of the scattering factor tables for the neutral atoms were used,61 and all scattering factors were corrected for both the real and imaginary components of anomalous dispersion.62 All calculations were performed using the teXsan63 crystallographic software package of Molecular Structure Corp. Detailed information regarding disordered atoms in individual structure solutions is given below.For 1, the complex resides on a crystallographic C2 axis that passes through Ta1. Therefore, the tantalum-bound methylene and methyl groups of 1 are disordered; C1 was refined anisotropically at full occupancy to account for both of these ligands.
As in 1, 14 also resides on a crystallographic C2 axis that passes through Ta1; C15 represents the disordered Ta–(CH2S) and Ta–CH3 carbon atoms; it was refined isotropically at full occupancy. The sulfur atom, S1, was disordered over two sites; it was refined isotropically at half-occupancy. Carbon atoms C2–C4 and C16–C18 of 14 are part of a disordered trimethylsilyl group; each atom was refined isotropically at half-occupancy.
Like in 1 and 14, complex 15, resides on a crystallographic C2 axis that passes through Ta1. The Ta–CH3 carbon atom, C15, is also positionally disordered; it was refined isotropically at half-occupancy. The sulfur atom, S1, was disordered in a similar manner; it was refined anisotropically at half-occupancy.
CCDC reference numbers 283319–283322.
For crystallographic data in CIF or other electronic format see DOI: 10.1039/b512741f
Acknowledgements
This work was supported by grants from the National Science Foundation (CHE-0345488 to R. G. B.) and (CHE-0072819 to J. A.). We thank Dr Fred Hollander for expert assistance with the X-ray solution and refinement.References
- P. M. Maitlis, H. C. Long, R. Quyoum, M. L. Turner and Z.-Q. Wang, Chem. Commun., 1996, 1 RSC.
- M. J. Overett, R. O. Hill and J. R. Moss, Coord. Chem. Rev., 2000, 206–207, 581 CrossRef CAS.
- J. P. Collman, L. S. Hegedus, J. R. Norton and R. G. Finke, in Principles and Applications of Organotransition Metal Chemistry, Mill Valley, CA, 1987 Search PubMed.
- J. Toyir, M. Leconte, G. P. Niccolai and J. M. Basset, J. Catal., 1995, 152, 306 CrossRef CAS.
- W. A. Nugent and J. M. Mayer, Metal–Ligand Multiple Bonds, John Wiley & Sons, New York, 1988 Search PubMed.
- M. P. Doyle and M. N. Protopopova, Tetrahedron, 1998, 54, 7919 CrossRef CAS.
- R. R. Schrock, Acc. Chem. Res., 1979, 12, 98 CrossRef CAS.
- R. R. Schrock and P. R. Sharp, J. Am. Chem. Soc., 1978, 100, 2389 CrossRef CAS.
- R. R. Schrock, J. Am. Chem. Soc., 1975, 97, 6577 CrossRef CAS.
- C. D. Wood, S. J. McLain and R. R. Schrock, J. Am. Chem. Soc., 1979, 101, 3210 CrossRef CAS.
- R. R. Schrock, Science, 1983, 219, 13 CrossRef CAS.
- F. N. Tebbe, G. W. Parshall and G. S. Reddy, J. Am. Chem. Soc., 1978, 100, 3611 CrossRef CAS.
- K. A. Brown-Wensley, S. L. Buchwald, L. Cannizzo, L. Clawson, S. Ho, D. Meinhardt, J. R. Stille and R. H. Grubbs, Pure Appl. Chem., 1983, 55, 1733 CrossRef CAS.
- M. D. Fryzuk, X. Gao, K. Joshi, P. A. MacNeil and R. L. Massey, J. Am. Chem. Soc., 1993, 115, 10581 CrossRef CAS.
- M. D. Fryzuk, S. A. Johnson and S. J. Rettig, Organometallics, 1999, 18, 4059 CrossRef CAS.
- A. H. Lui, R. C. Murray, J. C. Dewn, B. D. Santarsiero and R. R. Schrock, J. Am. Chem. Soc., 1987, 109, 4282 CrossRef CAS.
- L. Andrews, H. G. Cho and X. Wang, Inorg. Chem., 2005, 44, 4834 CrossRef CAS.
- J. J. Eisch and A. A. Adeosun, Eur. J. Org. Chem., 2005, 993 CrossRef CAS.
- A. E. Enriquez, P. S. White and J. L. Templeton, J. Am. Chem. Soc., 2001, 123, 4992 CrossRef CAS.
- R. H. Crabtree, The Organometallic Chemistry of the Transition Metals, John Wiley and Sons, New York, 1994 Search PubMed.
- D. P. Klein and R. G. Bergman, J. Am. Chem. Soc., 1989, 111, 3079 CrossRef CAS.
- J. C. Hayes, P. Jernakoff, G. A. Miller and N. J. Cooper, Pure Appl. Chem., 1984, 56, 25 CrossRef CAS.
- A. F. Hill, W. R. Roper, J. M. Waters and A. H. Wright, J. Am. Chem. Soc., 1983, 105, 5939 CrossRef CAS.
- D. Y. Dawson and J. Arnold, Organometallics, 1997, 16, 1111 CrossRef CAS.
- R. Duchateau, C. Van Wee, A. Meetsma, P. van Duijen and J. Teuben, Organometallics, 1996, 15, 3539.
- S. M. Mullins, R. G. Bergman and J. Arnold, Organometallics, 1999, 18, 4465 CrossRef CAS.
- K. C. Nicolaou, A. E. Koumbis, S. A. Snyder and K. B. Simonsen, Angew. Chem., Int. Ed., 2000, 39, 2529 CrossRef CAS.
- K. C. Nicolaou, S. A. Snyder, X. H. Huang, K. B. Simonsen, A. E. Koumbis and A. Bigot, J. Am. Chem. Soc., 2004, 126, 10162 CrossRef CAS.
- The coordination aperture is defined as the angle between the two planes through the metal center that touch the inner van der Waals surface of the ligand system.
- T. Takeda, Bull. Chem. Soc. Jpn., 2005, 78, 195 CrossRef CAS.
- R. R. Schrock and J. D. Fellmann, J. Am. Chem. Soc., 1978, 100, 3359 CrossRef CAS.
- R. R. Schrock, L. W. Messerle, C. D. Wood and L. J. Guggenberger, J. Am. Chem. Soc., 1978, 100, 3793 CrossRef CAS.
- R. R. Schrock, J. Am. Chem. Soc., 1976, 98, 5399 CrossRef CAS.
- G. Wittig, J. Organomet. Chem., 1975, 100, 279 CrossRef CAS.
- G. Wittig and G. Geissler, Justus Liebigs Ann. Chem., 1954, 580, 44.
- S. J. McLain, C. D. Wood and R. R. Schrock, J. Am. Chem. Soc., 1976, 98, 3519.
- R. Aumann, Angew. Chem., Int. Ed. Engl., 1988, 27, 1456 CrossRef.
- D. R. Lide, in Handbook of Chemistry and Physics, CRC Press, Boca Raton, FL, 1996 Search PubMed.
- Rotation around the internal C2 axis has been observed in amidinate complexes. See: L. A. Koterwas, J. C. Fettinger and L. R. Sita, Organometallics, 1999, 18, 4183 Search PubMed.
- The assignments of the individual tolyl methyl (c, d) and trimethylsilyl groups (e, f, g) are not conclusively known, but are instead based on the X-ray structure.
- J. E. Nelson, G. Parkin and J. E. Bercaw, Organometallics, 1992, 11, 2181 CrossRef CAS.
- S. L. Buchwald, R. B. Nielson and J. C. Dewan, J. Am. Chem. Soc., 1987, 109, 1590 CrossRef CAS.
- J. W. Park, L. M. Henling, W. P. Schaefer and R. H. Grubbs, Organometallics, 1990, 9, 1650 CrossRef CAS.
- G. Parkin, E. Bunel, B. J. Burger, M. S. Trimmer, A. van Asselt and J. E. Bercaw, J. Mol. Catal., 1987, 41, 21 CrossRef CAS.
- M. G. B. Drew, D. A. Rice and D. M. Williams, J. Chem. Soc., Dalton Trans., 1984, 845 RSC.
- G. Parkin, Prog. Inorg. Chem., 1998, 47, 1 CAS.
- The sums of TaCH3 and TaCH2 integrations relative to trimethoxybenzene internal standard are: 3.35 H (start) and 3.12 H (finish).
- H. Kawaguchi and K. Tatsumi, Organometallics, 1997, 16, 307 CrossRef CAS.
- K. Tatsumi, A. Tahara and A. Nakamura, J. Organomet. Chem., 1994, 471, 111 CrossRef CAS.
- P. J. Alaimo, D. W. Peters, J. Arnold and R. G. Bergman, J. Chem. Educ., 2001, 78, 64 CrossRef CAS.
- L. Kaczmarek, R. Balicki and P. Nantka-Namirski, Chem. Ber., 1992, 125, 1965 CrossRef CAS.
- D. Wenkert and R. B. Woodward, J. Org. Chem., 1983, 48, 283 CrossRef CAS.
- D. Plant, D. S. Tarbell and C. Whiteman, J. Am. Chem. Soc., 1955, 77, 1572 CrossRef CAS.
- SMART: Area-Detector Software Package, Siemens Industrial Automation, Inc., Madison, WI, 1995 Search PubMed.
- SAINT: SAX Area-Dectector Integration Program, V4.024, Siemens Industrial Automation, Inc., Madison, WI, 1995 Search PubMed.
- XPREP: (v 5.03) Part of the SHELXTL Crystal Structure Determination Siemens Industrial Automation, Inc., Madison, WI, 1995 Search PubMed.
- G. M. Sheldrick, SADABS: Siemens Area Detector ABSorption correction program, 1996. Advance copy, private communication Search PubMed.
- D. T. Cromer and J. T. Waber, International Tables for X-ray Crystallography, The Kynoch Press, Birmingham, England, 1974, vol. IV, table 2.2 A Search PubMed.
- SIR92: A. Altomare, M. C. Burla, Camalli, M. Cascarano, C. Giacovazzo, A. Guagliardi and G. Polidori, J. Appl. Crystallogr., 1993, 26, 343 Search PubMed.
- DIRDIF92: P. T. Beurskens, G. Admiraal, G. Beurskens, W. P. Bosman, S. Garcia-Granda, R. O. Gould, J. M. M. Smits and C. Smykalla, The DIRDIF program system, Technical Report of the Crystallography Laboratory, University of Nijmegen, The Netherlands, 1992 Search PubMed.
- D. C. Creagh and J. H. Hubbell, International Tables for Crystallography, Vol C, ed. A. J. C. Wilson, Kluwer Academic Publishers, Boston, MA, 1992, Table 4.2.4.3, pp. 200–206 Search PubMed.
- J. A. Ibers and W. C. Hamilton, Acta Crystallogr., 1964, 17, 781 CrossRef.
- teXsan: Crystal Structure Analysis Package, Molecular Structure Corporation, The Woodlands, TX, 1985 and 1992 Search PubMed.
Footnote |
| † In memory of our friend and colleague Professor Ian Rothwell. |
| This journal is © The Royal Society of Chemistry 2006 |