(N-Benzyl-bis-N′,N″-salicylidene)-cis-1,3,5-triaminocyclohexane copper(II): a novel catalyst for the aerobic oxidation of benzyl alcohol
Alison K.
Nairn
a,
Stephen J.
Archibald
a,
Rajiv
Bhalla
a,
Bruce C.
Gilbert
a,
Elizabeth J.
MacLean
b,
Simon J.
Teat
b and
Paul H.
Walton
*a
aDepartment of Chemistry, University of York, Heslington, York, UK YO10 5DD. E-mail: phw2@york.ac.uk; Tel: +44 1904 432500
bCCLRC Daresbury Laboratory, Daresbury, Warrington, Cheshire, UK WA4 4AD. E-mail: e.j.maclean@dl.ac.uk; Tel: +44 1925 603193
First published on 28th October 2005
Abstract
Reaction of Cu(BF4)2·6H2O with the N3O2 donor ligand H2L (where H2L = N-benzyl-N′,N″-di-tert-butyl-disalicyl-triaminocyclohexane) results in the formation of a novel CuIIL complex, 1. X-Ray crystallography of 1 shows the CuII centre coordinated by two phenolate oxygens and two imine nitrogens in a distorted square plane with an elongated bond to the amine nitrogen (2.512 Å) in the axial position. EPR spectroscopy of 1 gives g values of g1 = 2.277, g2 = 2.100, g3 = 2.025, and A1 = 15.6 mT which are consistent with the distorted square pyramidal coordination environment determined from the X-ray structure. UV/visible and electrochemical analysis of 1 shows that it undergoes two reversible processes assigned to the successive oxidation of the phenolate oxygens to phenoxyl radicals, the first at E½ = 0.89 V (ΔE = 81 mV, vs. Ag/AgCl) and the second at E½ = 1.13V (ΔE = 84 mV, vs. Ag/AgCl). Chemical oxidation of 1 results in the formation of a species, assigned as [1]+˙ which is EPR silent due to antiferromagnetic coupling between the CuII centre and the bound phenoxyl radical. The oxidised species catalyses the oxidation of benzyl alcohol to benzaldehyde.
Introduction
The fungal enzyme galactose oxidase (GOase)1,2 catalyses the selective oxidation of primary alcohols to aldehydes with the concomitant reduction of O2 to H2O2.3 The overall oxidation process requires two electrons and the enzyme does this with the use of both a CuI/CuII and a tyrosyl/tyrosinate redox couple.4,5 In recent years there has been considerable focus on the preparation of small molecule models of the active site of GOase.6,7 These complexes are of inherent interest as potential catalysts for selective oxidation chemistry.We have previously prepared and studied a range of coordinatively saturated FeIII complexes prepared from N3O3 donor ligands based on salicyl derivatives of 1,3,5-cis,cis-triiminocyclohexane ligands.8 The FeIII complexes exhibit reversible ligand-based oxidation processes to give bound phenoxyl radical species at remarkably low oxidation potentials. The complexes are, however, coordinatively saturated at the metal centre, limiting their ability to act as oxidation catalysts. We have now developed this ligand system further with the preparation of a novel unsymmetrical 1-amino-3,5-diiminocyclohexane-based N3O2 ligand.9 Our objective is to couple the stable ligand-based redox properties with a redox-active coordinatively unsaturated metal site (as in GOase), thus providing a novel chemical system capable of oxidation catalysis. Herein we describe the preparation and characterisation of a copper complex using this ligand system, including the EPR, X-ray structure, UV/visible studies, chemical oxidation and catalytic studies. We show that the complex catalyses the oxidation of benzyl alcohol to benzaldehyde. Such catalytic behaviour suggests that the complex offers the potential to act as an effective oxidation catalyst in other reactions.
Results and discussion
Preparation of the novel N3O2 donor ligand is carried out by a simple Schiff base condensation reaction of two equivalents of a 3,5-derivatised salicylaldehyde (to give the phenolate moiety) with the novel primary amine, I, to give the ligand H2L (N-benzyl-N′,N″-di-tert-butyl-disalicyl-triaminocyclohexane, Fig. 1). Derivatisation in these positions of the phenolate group has been shown to enhance the stability of any subsequent radical species.8,10 Complexation of the H2L ligand with CuII(BF4)2·6H2O occurs readily to give the corresponding CuIIL complex (1), in 70% yield (Fig. 1). 1 is purified by column chromatography on neutral alumina to give the product as a green complex which can be crystallised by slow evaporation of an alcohol solution of the complex.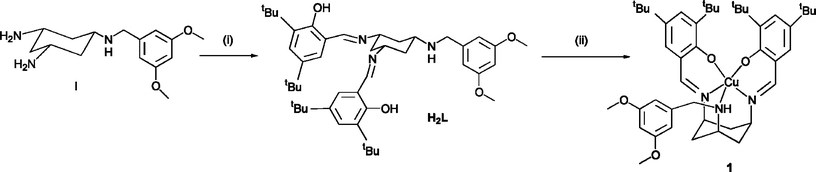 | ||
| Fig. 1 Preparation of the N3O2 donor ligand and the corresponding CuIIL complex (1). Conditions: (i) 2 equivalents of 3,5-di-tBu-salicylaldehyde, MeOH; (ii) Cu(BF4)2·6H2O, MeOH. | ||
The X-ray structure of 1 shows a five-coordinate copper(II) ion bound by two imine nitrogen atoms and the amine from the tach and two phenolate oxygen atoms giving a distorted square pyramidal coordination geometry (Fig. 2). The copper to imine nitrogen atom bond lengths [av. 1.991(4) Å] and the copper to phenolate oxygen atom bond lengths [1.936(3) Å and 1.994(4) Å] are within the expected ranges and provide the square base around the copper, the bond to the amine group [2.512(4) Å] is elongated and in the axial position. The sixth coordination site is unoccupied. Structurally characterised unsymmetrical tach complexes are limited to only a few examples.111 is the first example of a pentacoordinating tach-based ligand.
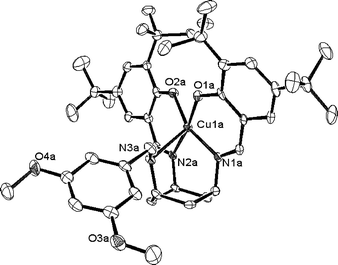 | ||
| Fig. 2 ORTEP12 view (50% probability ellipsoids) of the X-ray crystal structure of 1. Selected bond lengths (Å) and angles (°): Cu(1A)–O(1A) 1.936(3), Cu(1A)–N(1A) 1.991(4), Cu(1A)–N(3A) 2.512(4), Cu(1A)–O(2A) 1.994(4), Cu(1A)–N(2A) 1.991(4); O(1A)–Cu(1A)–O(2A) 84.41(15), O(2A)–Cu(1A)–N(1A) 159.06(17), O(2A)–Cu(1A)–N(2A) 90.37(16), O(1A)–Cu(1A)–N(3A) 97.39(14), N(1A)–Cu(1A)–N(3A) 80.21(16) O(1A)–Cu(1A)–O(2A) 84.41(15), O(1A)–Cu(1A)–N(1A) 89.82(16), O(1A)–Cu(1A)–N(2A) 171.99(16), N(1A)–Cu(1A)–N(2A) 97.16(17), O(2A)–Cu(1A)–N(3A) 120.45(15), N(2A)–Cu(1A)–N(3A) 80.01(15). | ||
The EPR spectrum (77 K) of 1 was recorded in frozen CH2Cl2 solution (Fig. 3). Computer simulation of the spectrum gives parameters of g1 = 2.277, g2 = 2.100, g3 = 2.025, and A1 = 15.6 mT, values that are typical of a CuII centre coordinated by three nitrogens and two phenolate oxygens in a distorted square-pyramidal environment {a (dx2−y2)1 electronic configuration}.13–18 The results suggest that the solution structure of the complex is very similar to that found in the solid state structure. When recording the EPR spectrum of the complex, it was noted that the appearance of the spectrum changed when a coordinating solvent, such as methanol, was employed. This suggests that the Cu centre can accommodate a sixth ligand.
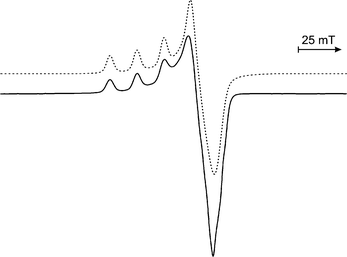 | ||
| Fig. 3 Frozen solution EPR spectrum of 1 recorded at 77 K in CH2Cl2 (—) and simulated spectrum (⋯⋯). | ||
The UV/visible spectrum of 1 in CH2Cl2 (Fig. 4) shows five different bands, three of which (230, 258, 326 nm, ε = 6000 to 170000 dm3 mol−1 cm−1) are assigned to ligand-based π to π* charge transfer transitions (in reference to the free ligand). A fourth band at 378 nm is assigned to a LMCT transition from the phenolate to the CuII metal centre (ε = 628 dm3 mol−1 cm−1).14,15,17 A fifth band at 594 nm (ε = 130 dm3 mol−1 cm−1) is assigned to a metal-based transition.
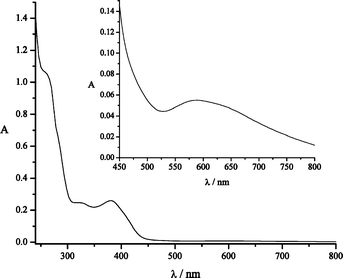 | ||
| Fig. 4 UV/visible spectrum of a CH2Cl2 solution of 1 (4 × 10−5 mol dm3) recorded at room temperature (4 × 10−4 mol dm3 inset). | ||
The cyclic voltammogram (CV) of 1 in CH2Cl2 (Fig. 5); shows no electrochemical processes in the negative region (between 0 and −1.8 V vs. Ag/AgCl, not shown). Two reversible processes on the CV timescale are apparent at positive potentials; the first at E½ = 0.89 V (ΔE = 81 mV, 0.34 V vs. FeCp2/FeCp2+) and the second at E½ = 1.13V (ΔE = 84 mV, 0.58 V vs. FeCp2/FeCp2+). These processes are observed at slightly lower potentials in MeCN, the first at E½ = 0.80 V (ΔE = 70 mV, 0.33 V vs. FeCp2/FeCp2+) and the second at E½ = 1.03 V (ΔE = 80 mV, 0.56 V vs. FeCp2/FeCp2+). Differential pulse experiments confirm that there is only one electron involved in each of the two processes.
![Cyclic voltammogram (298 K) of 1 (1 × 10−4 mol dm−3) in CH2Cl2 solution (0.5 mol dm−3 [NBu4]BF4) recorded at 200 mV s−1.](/image/article/2006/DT/b512296c/b512296c-f5.gif) | ||
| Fig. 5 Cyclic voltammogram (298 K) of 1 (1 × 10−4 mol dm−3) in CH2Cl2 solution (0.5 mol dm−3 [NBu4]BF4) recorded at 200 mV s−1. | ||
In reference to previous phenolate–tach complexes,8 both the first and second reversible oxidation processes observed in the CV of 1 are assigned to the production of first one and then a second phenoxyl-radical. It appears, therefore, that 1 undergoes two reversible ligand-based, phenolate to phenoxyl-radical redox processes, and no metal-based processes are observed within the redox range of the experiment.14,15,17 Another example of this type of redox behaviour was observed with a N3O2 triazacyclononane-based system prepared by Tolman and co-workers.16 In this case, both the ZnII and CuII complexes undergo analogous oxidation chemistry; the system also incorporates the same di-tert-butyl substitution pattern around the phenolate ring. Stack and co-workers also observed similar behaviour from a binapthyl salen-type N2O2-donor CuII complex.14,17
The first oxidation process observed for 1 can be carried out chemically by addition of CuII(CF3SO3)2 (E½ = 0.95 V vs. Ag/AgCl in MeCN, 0.40 V vs. FeCp2/FeCp2+).19 When the oxidant is added to a dilute pale green MeCN solution of 1 an instantaneous colour change to intense dark emerald green is observed. The UV/visible spectrum of the dark emerald green solution of [1]+˙ (Fig. 6) shows a strong absorption band at 390 nm (ε = 9860 dm3 mol−1 cm−1) which is assigned to a phenoxyl-radical to CuII LMCT8 in analogy to the electronic spectrum of the oxidised active form of GOase, which shows a distinctive feature at 445 nm (ε
≈ 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 dm3 mol−1 cm−1) likely to be due to a tyrosyl-radical to CuII LMCT band.20 Parallel EPR experiments on the oxidised [1]+˙ complex show that the addition of the oxidant to the complex results in the complete loss of the EPR signal. This loss of EPR activity strongly suggests that the bound phenoxyl-radical species, which is produced when the complex is oxidised, is anti-ferromagnetically coupled to the CuII centre.
000 dm3 mol−1 cm−1) likely to be due to a tyrosyl-radical to CuII LMCT band.20 Parallel EPR experiments on the oxidised [1]+˙ complex show that the addition of the oxidant to the complex results in the complete loss of the EPR signal. This loss of EPR activity strongly suggests that the bound phenoxyl-radical species, which is produced when the complex is oxidised, is anti-ferromagnetically coupled to the CuII centre.
![UV/visible spectrum of a MeCN solution of the chemically generated [1]+˙ species (1 × 10−4 mol dm−3) recorded at room temperature.](/image/article/2006/DT/b512296c/b512296c-f6.gif) | ||
| Fig. 6 UV/visible spectrum of a MeCN solution of the chemically generated [1]+˙ species (1 × 10−4 mol dm−3) recorded at room temperature. | ||
We have completed a preliminary study into the aerobic oxidation of benzyl alcohol to benzaldehyde using 1 as the catalyst. We have followed the method used by Stack and co-workers so that the two catalytic systems can be compared directly.14 Approximately 4% of the substrate was added to the reaction mixture in the deprotonated form (as the sodium salt, PhCH2ONa). The oxidation reactions were initiated by the final addition of the chemical oxidant, CuII(CF3SO3)2 (1 : 1 ratio to 1), to all the other components. The reactions were then monitored from this point onwards by GC, the reactions were run in air at room temperature. Control reactions without the presence of the catalyst show that the chemical oxidant, CuII(CF3SO3)2 can perform one pseudo-catalytic turnover. The amount of benzaldehyde produced by the CuII(CF3SO3)2 complex alone was taken into account when the turnovers numbers for the oxidation reactions were calculated. Analysis of the products by gas chromatography confirm the oxidation of benzyl alcohol to benzaldehyde is catalysed by 1 [average of 44 turnovers in 24 h (∼2% of substrate), 160 turnovers in 144 h (∼8% of substrate) at 295 K]. Oxidation of benzyl alcohol is likely to occur on the coordinated alcoholate anion rather than directly on the alcohol itself. We note, however, that up to 8% of the substrate is oxidised, greater than the 4% of PhCH2ONa which is added to the reaction. This shows that whilst PhCH2ONa is necessary to allow coordination of the substrate to the complex, benzyl alcohol is the species which is the principal substrate for the oxidation reaction. Tests carried out in the absence of air show much reduced oxidation activity indicating that the oxidant of 1 to give the active [1]+˙ species is molecular oxygen, although we cannot rule out the possibility that hydrogen peroxide—which may be produced as a product of the reaction—also plays an oxidative role.
There are several examples of oxidation catalysts based on GOase which aerobically oxidise primary alcohols to aldehydes. Copper complexes of thio-bis(2,4-di-tBu-phenol), seleno-bis(2,4-di-tBu-phenol), bis(2-hydroxy-3,5-di-tBu-phenyl)amine and bis(2-hydroxy-3,5-di-tBu-phenyl)phenylenediamine by Wieghardt and co-workers incorporate features seen in the active site of GOase, such as coordinated phenoxyl radicals, but all but the last are not direct structural models of the active site.6g,21 These complexes act as active catalysts for the aerobic oxidation of a range of different primary alcohols, the copper complexes which incorporate sulfur, however, also oxidise secondary alcohols and carry out oxidative C–C coupling reactions. The complexes incorporating selenium-based ligands show better selectivity, oxidising benzyl alcohol to benzaldehyde with 95 turnovers in 24 h.6g
The activity of 1 is similar to the analogous bis(di-tert-butyl-salicyl-imine)-binaphthyl CuII complex described by Stack and co-workers which gives 40 turnovers in 20 h under the same conditions.14 A bis(2-hydroxy-3,5-di-tBu-phenyl)ethylene-diamine copper complex, prepared by Pierre and co-workers, has been evaluated for the catalytic oxidation of primary alcohols ROH to RCHO (where R is Me, Et, Pr and Bu).22 The complex is selective for the aerobic oxidation of primary alcohols and is reported to give a maximum number of turnovers of 30. Later work by the same group showed that the copper complex of an unsymmetrical pyridyl-bis(2-hydroxy-3-tBu-phenyl)amine ligand catalyses the oxidation of benzylalcohol with approximately 300 turnovers in 24 h.6d
1 compares favourably with existing aerobic catalysts, and—compared to others—1 is notable for its longevity under catalytic conditions. Future work will concentrate on the development of this ligand system and the application of the complexes as catalysts in a wide range of oxidation processes.
Conclusions
We have demonstrated the design and synthesis of a new Cu(II) complex of a N3O2 donor ligand based on a bis-salicyl derivative of cis,cis-1,3,5-triaminocyclohexane, using a new synthetic method to prepare the unsymmetrical ligands. We suggest that this complex exhibits reversible redox chemistry involving the successive oxidation of the two coordinated phenolate groups to give two bound phenoxyl radicals. The complex has also been found to be an active catalyst and is effective for the oxidation of benzyl alcohol to benzaldehyde.Experimental
General procedures
Solvents for synthesis were supplied by Fisons Ltd., or Fischer Scientific International Company. Deuterated solvents were supplied by Aldrich Chemical company and Goss Scientific. All other reagents were supplied by either Aldrich Chemical Company Ltd., Lancaster Chemicals Ltd., Avocado Research Chemicals Ltd. or Fluka Ltd. All the reagents were used without further purification unless stated otherwise. FT-NMR spectra were recorded using a JEOL EX270 MHz spectrometer, referencing of the peaks was carried out using the residual protons in the solvent. Infrared spectra were recorded using a Mattson Sirius Research Series FTIR Spectrometer as KBr discs (pressed under 7 tonnes pressure). The data obtained were processed using Microsoft Win 1st software. Mass spectra were recorded on a Fisons Instruments Autospec. using a 0 to 650 °C temperature range. Elemental analyses were performed at the micro analytical laboratory at the University of Manchester. EPR spectra were recorded on a JEOL JES-RE1X Spectrometer (equipped with an X-band Gunn diode). Low temperature spectra were recorded with the use of a liquid N2 dewar (at 77 K). UV/visible spectra were recorded on a Hitachi U-3000 spectrometer using 1 cm3 quartz cells.Cyclic voltammetry was performed using a standard three-electrode configuration with platinum working (0.5 mm diameter disk) and counter electrodes and a Ag/AgCl reference which gave the FeCp/FeCp+ couple at 0.55 V (ΔE = 80 mV) using an EG & G potentiostat. All measurements were made in a nitrogen-purged solution of either CH2Cl2/0.5 mol dm−3 [n-Bu4N][BF4] or MeCN/0.2 mol dm−3 [n-Bu4N][BF4], over a range of scan rates (from 50 to 500 mV).
Synthesis
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–), 1533 (w), 1466 (m), 1417 (m), 1362 (m), 1254 (w), 1207 (m), 1156 (s), 1069 (s), 839 (w), 770 (w), 739 (w).
N–), 1533 (w), 1466 (m), 1417 (m), 1362 (m), 1254 (w), 1207 (m), 1156 (s), 1069 (s), 839 (w), 770 (w), 739 (w).
Crystallography
Crystals of CuIIL were grown by slow evaporation of a propan-2-ol solution of the complex. The X-ray diffraction data were measured at Station 9.8 of the CCLRC Daresbury Laboratory Synchrotron Radiation Source using a Bruker AXS SMART CCD diffractometer, ω rotation with narrow frames (synchrotron radiation, λ = 0.6883 Å). Crystal structure analysis for (CuIIL): C48H70CuN3O5, M = 832.61, green plates, crystal dimensions 0.07 × 0.06 × 0.02 mm, triclinic, a = 13.2972(12), b = 15.5508(14), c = 23.798(2) Å, α = 99.359(2)°, β = 102.082(2)°, γ = 104.520(2)°, cell volume = 4536.4(7) Å3, T = 150(2) K, space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , Z = 4, 34089 reflections collected, 12962 unique, Rint = 0.0806. The structure was solved using direct-methods in SHELXS and refined using SHELXL, R1 = 0.0621 (for 8884 reflections with I > 2σ(I)), wR2 = 0.1684 (all data).
, Z = 4, 34089 reflections collected, 12962 unique, Rint = 0.0806. The structure was solved using direct-methods in SHELXS and refined using SHELXL, R1 = 0.0621 (for 8884 reflections with I > 2σ(I)), wR2 = 0.1684 (all data).
CCDC reference number 282729.
For crystallographic data in CIF or other electronic format see DOI: 10.1039/b512296c
Oxidation chemistry
The catalytic oxidation reactions were carried out in air using 2 mL of MeCN as a solvent at room temperature. 1,2-Dichlorobenzene was used as an internal standard. In each experiment to 100% of PhCH2OH (1.94 mol dm−3) there was 0.05% of 1 (0.97 mmol dm−3) [and 0.05% of the initiator CuII(CF3SO3)2], 4% of PhCH2ONa and 5% of 1,2-dichlorobenzene present. 20 µL aliquots of the reaction were removed after 2, 24, 48, 72 and 144 h and then quenched with 20 µL of Et2O. These small samples were then analysed by GC. Control reactions without the presence of the catalyst show that the chemical oxidant, CuII(CF3SO3)2 can perform one pseudo-catalytic turnover. The volume of benzaldehyde produced by the CuII(CF3SO3)2 complex alone was taken into account when the turnover numbers for the oxidation reactions were calculated.Gas chromatographic experiments were performed on an AMS94 apparatus fitted with a Alltach Econo-cap Carbowax column (30 m by 0.25 mm, internal diameter 0.25 µm). The data were obtained using a column and injector temperature of 200 °C, the run time for each experiment was 45 minutes. The data were processed using the JCL6000 software for Microsoft Windows (version 2.0, revision 26).
Acknowledgements
This work is kindly supported by the EPRSC. We would like to thank Dr Vic Young from the University of Minnesota for preparing suitable files of the twinned crystallographic data to be refined using his program ROTWIN.References
- J. W. Whittaker, Adv. Protein Chem., 2002, 60, 1–49 CAS; N. Ito, S. E. V. Phillips, K. D. S. Yadav and P. F. Knowles, J. Mol. Biol., 1994, 238, 794–814 CrossRef CAS.
- M. A. McGuirl and D. M. Dooley, Curr. Opin. Chem. Biol., 1999, 3, 138–144 CrossRef CAS.
- C. D. Borman, C. G. Saysell and A. G. Sykes, JBIC, J. Biol. Inorg. Chem., 1997, 2, 480–487 CrossRef CAS.
- J. W. Whittaker, in Metal Ions in Biological Systems, ed., H. Sigel and A. Sigel, Marcel Dekker, New York, USA, 1994, vol. 30, pp. 315–360 Search PubMed.
- F. Himo, L. A. Eriksson, F. Maseras and P. E. M. Seigbahn, J. Am. Chem. Soc., 2000, 122, 8031 CrossRef CAS.
- Recent examples include: (a) M. Vaidyanathan, M. Palaniandavar and R. S. Gopalan, Inorg. Chim. Acta, 2001, 324, 241–251 CrossRef CAS; (b) T. Kruse, T. Weyhermüller and K. Wieghardt, Inorg. Chim. Acta, 2002, 331, 81–89 CrossRef CAS; (c) Y. Shimazaki, S. Huth, S. Hirota and O. Yamauchi, Inorg. Chim. Acta, 2002, 331, 168–177 CrossRef CAS; (d) F. Thomas, G. Gellon, I. Gautier-Luneau, E. Saint-Aman and J.-L. Pierre, Angew. Chem., Int. Ed., 2002, 41, 3047–3050 CrossRef CAS; (e) R. J. M. Klein Gebbink, M. Watanabe, R. C. Pratt and T. D. P. Stack, Chem. Commun., 2003, 630–631 RSC; (f) A. Philibert, F. Thomas, C. Philouze, S. Hamman, E. Saint-Aman and J.-L. Pierre, Chem.-Eur. J., 2003, 9, 3803–3812 CrossRef CAS; (g) T. K. Paine, T. Weyhermüller, K. Wieghardt and P. Chaudhuri, Dalton Trans., 2004, 2092–2101 RSC; (h) L. Benisvy, A. J. Blake, D. Collison, E. S. Davies, C. D. Garner, E. J. L. McInnes, J. McMaster, G. Whittaker and C. Wilson, Chem. Commun., 2001, 1824–1825 RSC; (i) M. Taki, H. Hattori, T. Osako, S. Nagatomo, M. Shiro, T. Kitagawa and S. Itoh, Inorg. Chim. Acta, 2004, 357, 3369–3381 CrossRef CAS; (j) F. Michel, F. Thomas, S. Hamman, E. Saint-Aman, C. Bucher and J.-L. Pierre, Chem.-Eur. J., 2004, 10, 4115–4125 CrossRef CAS.
- (a) B. A. Jazdzewski and W. B. Tolman, Coord. Chem. Rev., 2000, 200-202, 633–685 CrossRef; (b) S. Itoh, M. Taki and S. Fukuzumi, Coord. Chem. Rev., 2000, 198, 3–20 CrossRef CAS; (c) J.-L. Pierre, Chem. Soc. Rev., 2000, 29, 251–257 RSC; (d) V. Mahadevan, R. J. M. K. Gebbink and T. D. P. Stack, Curr. Opin. Chem. Biol., 2000, 4, 228–234 CrossRef CAS; (e) W. Kaim, Dalton Trans., 2003, 761–768 RSC.
- A. K. Nairn, R. Bhalla, S. P. Foxon, X. Liu, L. J. Yellowlees, B. C. Gilbert and P. H. Walton, J. Chem. Soc., Dalton Trans., 2002, 1253–1255 RSC.
- S. J. Archibald, A. K. Nairn, P. Timmins and P. H. Walton, Tetrahedron Lett., 2005, 46, 6441–6443 CrossRef CAS.
- (a) B. Adam, E. Bill, E. Bothe, B. Goerdt, G. Haselhorst, K. Hildebrand, A. Sokolowski, S. Steenken, T. Wehyermüller and K. Wieghardt, Chem.-Eur. J., 1997, 3, 308–319 CrossRef CAS; (b) A. Sokolowski, E. Bothe, E. Bill, T. Wehyermüller and K. Wieghardt, Chem. Commun., 1996, 1671–1672 RSC; (c) J. Müller, T. Wehyermüller, E. Bill, P. Hildebrandt, L. Ould-Moussa, T. Glaser and K. Wieghardt, Angew. Chem., Int. Ed., 1998, 37, 616–619 CrossRef CAS; (d) A. Sokolowski, J. Müller, T. Wehyermüller, R. Schnepf, P. Hildebrandt, K. Hildebrand, E. Bothe and K. Wieghardt, J. Am. Chem. Soc., 1997, 119, 8889–8900 CrossRef CAS.
- (a) L. Cronin, B. Greener, S. P. Foxon, S. L. Heath and P. H. Walton, Inorg. Chem., 1997, 36, 2594–2600 CrossRef CAS; (b) H.-S. Chong, F. M. Torti, S. V. Torti and M. W. Brechbiel, J. Org. Chem., 2002, 67, 8072–8078 CrossRef CAS; (c) H.-S. Chong, S. V. Torti, R. Ma, F. M. Torti and M. W. Brechbiel, J. Med. Chem., 2004, 47, 5230–5234 CrossRef CAS.
- C. K. Johnson and M. K. Burnett, ORTEP-3 v. 1.0.2, Oak Ridge National Laboratory, Oak Ridge, TN, USA, 1998 Search PubMed.
- (a) A. Sokolowski, H. Leutbecher, T. Wehyermüller, R. Schnepf, E. Bothe, E. Bill, P. Hildebrandt and K. Wieghardt, JBIC, J. Biol. Inorg. Chem., 1997, 2, 444–453 CrossRef CAS; (b) S. Itoh, S. Takayama, R. Arakawa, A. Furuta, M. Komatsu, A. Ishida, S. Takamuku and S. Fukuzumi, Inorg. Chem., 1997, 36, 1407–1416 CrossRef CAS; (c) M. A. Halcrow, L. M. L. Chia, X. Liu, E. J. L. McInnes, L. J. Yellowlees, F. E. Mabbs and J. E. Davies, Chem. Commun., 1998, 2465–2466 RSC; (d) E. Bill, J. Müller, T. Wehyermüller and K. Wieghardt, Inorg. Chem., 1999, 38, 5795–5802 CrossRef CAS; (e) D. Zurita, I. Gautier-Luneau, S. Ménage, J.-L. Pierre and E. Saint-Aman, JBIC, J. Biol. Inorg. Chem., 1997, 2, 46–55 CrossRef CAS.
- Y. Wang, J. L. BuBois, B. Hedman, K. O. Hodgson and T. D. P. Stack, Science, 1998, 279, 537–540 CrossRef CAS.
- D. Zurita, C. Scheer, J.-L. Pierre and E. Saint-Aman, J. Chem. Soc., Dalton Trans., 1996, 4331–4336 RSC.
- J. A. Halfen, B. A. Jazdzewski, S. Mahaptra, L. M. Berreau, E. C. Wilkinson, L. Que, Jr. and W. B. Tolman, J. Am. Chem. Soc., 1997, 119, 8217–8227 CrossRef CAS.
- Y. Wang and T. D. P. Stack, J. Am. Chem. Soc., 1996, 118, 13097–13098 CrossRef CAS.
- J. A. Halfen, V. G. Young and W. B. Tolman, Angew. Chem., Int. Ed. Engl., 1996, 35, 1687–1690 CrossRef CAS.
- N. G. Connelly and W. E. Geiger, Chem. Rev., 1996, 96, 877–910 CrossRef CAS.
- (a) M. M. Whittaker and J. W. Whittaker, J. Biol. Chem., 1988, 263, 6074–6080 CAS; (b) M. M. Whittaker, C. A. Ekberg, J. Peterson, M. S. Sendova, E. P. Day and J. W. Whittaker, J. Mol. Catal. B: Enzym., 2000, 8, 3–15 CrossRef CAS.
- (a) P. Chaudhuri, M. Hess, U. Flörke and K. Wieghardt, Angew. Chem., Int. Ed., 1998, 37, 2217–2220 CrossRef CAS; (b) P. Chaudhuri, M. Hess, T. Weyhermüller and K. Wieghardt, Angew. Chem., Int. Ed., 1999, 38, 1095–1098 CrossRef CAS; (c) P. Chaudhuri, M. Hess, J. Müller, K. Hildenbrand, E. Bill, T. Weyhermüller and K. Wieghardt, J. Am. Chem. Soc., 1999, 121, 9599–9610 CrossRef CAS.
- E. Saint-Aman, S. Ménage, J.-L. Pierre, E. Defrancq and G. Gellon, New J. Chem., 1998, 393–394 RSC.
| This journal is © The Royal Society of Chemistry 2006 |