Electronic structures of trans-dioxometal complexes
Patrick
Hummel
,
Jay R.
Winkler
and
Harry B.
Gray
*
California Institute of Technology, 1200 E. California Blvd., Mail Code 139-74, Pasadena, CA 91125, USA. E-mail: hbgray@caltech.edu
First published on 4th November 2005
Abstract
We have employed computational methods based on density functional theory to elucidate the effects of equatorial ligands on the electronic structures of trans-dioxometal complexes. In complexes with ammine (σ-only) equatorial donors, the 1A1 g(b2 g)2 → 1Eg(b2 g)1(eg)1 excitation energy increases with metal oxidation state: Mo(IV) < Tc(V) < Ru(VI) and W(IV) < Re(V) < Os(VI). Increasing transition energies are attributed to enhanced oxometal π-donor interactions in the higher valent central metals. But in complexes with cyanide equatorial donors, the 1A1 g(b2 g)2 → 1Eg(b2 g)1(eg)1 energy remains roughly independent of metal oxidation state, likely owing to the compensating increased π-donation from the π(CN) orbitals to the metal dxy orbitals as the oxidation state of the metal increases.
Introduction
Multiple bonds stabilize high oxidation states of oxometal units. The tetragonal d1 and d2 oxometal complexes, often incorrectly formulated as M![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O instead of M
O instead of M![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) O, have been studied for over 40 years.1–12 Of special interest here are trans-dioxometal complexes of the form [MO2L4]z (Fig. 1). We consider M = Mo, Tc, Ru, W, Re, Os; L = NH3, z = 2+, +, 0; L = CN−, z = 2−, 3−, 4−. The ground state is 1A1 g(b2 g)2, with a well established ligand field splitting pattern: b2 g(xy) < eg(xz, yz) < b1 g(x2-y2) < a1 g(z2). The lowest lying electronic excitations involve transitions from the nonbonding b2 g orbital to a π-antibonding eg orbital. The 1A1 g(b2 g)2
→
1Eg(b2 g)1(eg)1 excitation and the spin-forbidden 1A1 g(b2 g)2
→
3Eg(b2 g)1(eg)1 transition are distinct features in the absorption spectra of trans-dioxometal complexes. Radiative decay of the triplet excited state is often prominent as well.4–6
O, have been studied for over 40 years.1–12 Of special interest here are trans-dioxometal complexes of the form [MO2L4]z (Fig. 1). We consider M = Mo, Tc, Ru, W, Re, Os; L = NH3, z = 2+, +, 0; L = CN−, z = 2−, 3−, 4−. The ground state is 1A1 g(b2 g)2, with a well established ligand field splitting pattern: b2 g(xy) < eg(xz, yz) < b1 g(x2-y2) < a1 g(z2). The lowest lying electronic excitations involve transitions from the nonbonding b2 g orbital to a π-antibonding eg orbital. The 1A1 g(b2 g)2
→
1Eg(b2 g)1(eg)1 excitation and the spin-forbidden 1A1 g(b2 g)2
→
3Eg(b2 g)1(eg)1 transition are distinct features in the absorption spectra of trans-dioxometal complexes. Radiative decay of the triplet excited state is often prominent as well.4–6
![The structure of the [MO2L4]z complexes in question.](/image/article/2006/DT/b512299f/b512299f-f1.gif) | ||
| Fig. 1 The structure of the [MO2L4]z complexes in question. | ||
We now report a systematic investigation of the electronic structures of d2-trans-dioxometal complexes. In particular, we use density functional theory (DFT) and time-dependent density functional theory (TDDFT) to analyze the effects of equatorial ligand interactions on the Δ = E[eg(xz, yz)] − E[b2 g(xy)] energy gap as a function of metal oxidation state. For [MO2(NH3)4]z, we note a general increase in Δ with increase in metal oxidation state: Mo(IV) < Tc(V) < Ru(VI) and W(IV) < Re(V) < Os(VI). For [MO2(CN)4]z the values of Δ remain roughly independent of the oxidation state of the central metal.
Computational methods
All calculations reported herein were performed with the TURBOMOLE program package for ab initio electronic structure calculations.13 We used the TZVP basis set14 for all atoms in calculating the properties of these complexes. For each metal, we used the effective core potentials given by Andrae et al.15 to account for relativistic effects. We performed four different DFT calculations for each complex, each using a different exchange–correlation functional selected from B3LYP, PBE, BP86, and BLYP.16–24 The calculations were done using the COSMO continuum solvation model25 with dielectric constant 37.5. Each calculation was performed with an m3 gridsize.26 The geometry of each complex was optimized using TURBOMOLE's JOBEX program with generalized internal coordinates27 and the corresponding STATPT module. Energies of well-converged ground-state molecular orbitals were calculated with the DSCF module for semi-direct self-consistent-field evaluation. We then used these ground-state molecular orbitals to calculate the energies of the lowest-lying singlet → singlet transitions with the ESCF package for full TDDFT calculations.28,29 We have previously used similar methods in TURBOMOLE successfully to investigate the electronic structures of various inorganic complexes.30–32Results and discussion
The calculated ground-state bond distances and 1A1 g(b2 g)2 → 1Eg(b2 g)1(eg)1 excitation energies for twelve d2-trans-dioxometal complexes are in good agreement with experiment (Tables 1–5).6,33–38 The overall performance of the four exchange–correlation functionals is roughly the same. For the [MO2(NH3)4]z complexes, the experimental excitation energies are closest to those calculated with PBE, BP86, and BLYP. For [MO2(CN)4]z, the best results are obtained with B3LYP.| Expt | B3LYP | B-LYP | BP86 | PBE | |
|---|---|---|---|---|---|
| [MoO2(NH3)4] | 1.833 | 1.852 | 1.842 | 1.843 | |
| [TcO2(NH3)4]+ | 1.747 | 1.763 | 1.787 | 1.777 | 1.777 |
| [RuO2(NH3)4]2+ | 1.724 | 1.752 | 1.741 | 1.741 | |
| [WO2(NH3)4] | 1.855 | 1.873 | 1.863 | 1.862 | |
| [ReO2(NH3)4]+ | 1.765 | 1.790 | 1.812 | 1.803 | 1.803 |
| [OsO2(NH3)4]2+ | 1.74 | 1.752 | 1.777 | 1.768 | 1.768 |
| [MoO2(CN)4]4− | 1.834 | 1.844 | 1.867 | 1.857 | 1.856 |
| [TcO2(CN)4]3− | 1.779 | 1.805 | 1.795 | 1.795 | |
| [RuO2(CN)4]2− | 1.740 | 1.771 | 1.760 | 1.760 | |
| [WO2(CN)4]4− | 1.842 | 1.866 | 1.886 | 1.875 | 1.875 |
| [ReO2(CN)4]3− | 1.781 | 1.806 | 1.830 | 1.819 | 1.818 |
| [OsO2(CN)4]2− | 1.75 | 1.767 | 1.796 | 1.786 | 1.786 |
| Expt | B3LYP | B-LYP | BP86 | PBE | |
|---|---|---|---|---|---|
| [MoO2(NH3)4] | 2.303 | 2.322 | 2.287 | 2.283 | |
| [TcO2(NH3)4]+ | 2.158 | 2.214 | 2.236 | 2.205 | 2.201 |
| [RuO2(NH3)4]2+ | 2.153 | 2.182 | 2.155 | 2.152 | |
| [WO2(NH3)4] | 2.304 | 2.320 | 2.288 | 2.284 | |
| [ReO2(NH3)4]+ | 2.162 | 2.222 | 2.224 | 2.216 | 2.213 |
| [OsO2(NH3)4]2+ | 2.11 | 2.167 | 2.193 | 2.169 | 2.164 |
| [MoO2(CN)4]4− | 2.220 | 2.229 | 2.228 | 2.201 | 2.198 |
| [TcO2(CN)4]3− | 2.170 | 2.176 | 2.148 | 2.145 | |
| [RuO2(CN)4]2− | 2.124 | 2.145 | 2.113 | 2.110 | |
| [WO2(CN)4]4− | 2.177 | 2.224 | 2.226 | 2.202 | 2.198 |
| [ReO2(CN)4]3− | 2.124 | 2.178 | 2.181 | 2.160 | 2.157 |
| [OsO2(CN)4]2− | 2.139 | 2.151 | 2.128 | 2.125 |
| Expt | B3LYP | B-LYP | BP86 | PBE | |
|---|---|---|---|---|---|
| [MoO2(CN)4]4− | 1.169 | 1.181 | 1.183 | 1.183 | |
| [TcO2(CN)4]3− | 1.162 | 1.174 | 1.175 | 1.175 | |
| [RuO2(CN)4]2− | 1.157 | 1.169 | 1.169 | 1.169 | |
| [WO2(CN)4]4− | 1.169 | 1.182 | 1.183 | 1.183 | |
| [ReO2(CN)4]3− | 1.156 | 1.162 | 1.174 | 1.175 | 1.175 |
| [OsO2(CN)4]2− | 1.156 | 1.168 | 1.169 | 1.169 |
| B3LYP | B-LYP | BP86 | PBE | |
|---|---|---|---|---|
| [MoO2(NH3)4] | −3.66/0.50 | −2.30/−0.31 | −2.41/−0.43 | −2.30/−0.33 |
| [TcO2(NH3)4]+ | −6.79/−2.20 | −5.16/−2.88 | −5.26/−2.99 | −5.13/−2.88 |
| [RuO2(NH3)4]2+ | −10.11/−5.33 | −8.38/5.88 | −8.52/−6.00 | −8.40/−5.89 |
| [WO2(NH3)4] | −3.21/0.89 | −2.03/0.20 | −2.17/0.06 | −2.05/0.17 |
| [ReO2(NH3)4]+ | −6.25/−1.59 | −4.82/−2.31 | −4.96/−2.44 | −4.83/−2.32 |
| [OsO2(NH3)4]2+ | −9.65/−4.66 | −8.03/−5.26 | −8.18/−5.40 | −8.06/−5.29 |
| [MoO2(CN)4]4− | −3.33/0.88 | −2.34/0.01 | −2.63/−0.22 | −2.50/−0.08 |
| [TcO2(CN)4]3− | −5.67/−1.30 | −4.34/−2.04 | −4.60/−2.24 | −4.46/−2.10 |
| [RuO2(CN)4]2− | −7.96/−4.02 | −6.51/−4.60 | −6.74/−4.78 | −6.60/−4.64 |
| [WO2(CN)4]4− | −3.11/1.14 | −2.24/0.30 | −2.55/0.05 | −2.42/0.18 |
| [ReO2(CN)4]3− | −5.33/−0.87 | −4.18/−1.63 | −4.45/−1.83 | −4.31/−1.69 |
| [OsO2(CN)4]2− | −7.64/−3.45 | −6.29/−4.07 | −6.53/−4.27 | −6.40/−4.13 |
| Expt | B3LYP | B-LYP | BP86 | PBE | |
|---|---|---|---|---|---|
| [MoO2(NH3)4] | 2.33 | 2.27 | 2.28 | 2.26 | |
| [TcO2(NH3)4]+ | 2.53 | 2.71 | 2.56 | 2.58 | 2.57 |
| [RuO2(NH3)4]2+ | 2.73 | 3.00 | 2.78 | 2.82 | 2.81 |
| [WO2(NH3)4] | 2.55 | 2.47 | 2.49 | 2.48 | |
| [ReO2(NH3)4]+ | 2.83 | 2.93 | 2.79 | 2.81 | 2.80 |
| [OsO2(NH3)4]2+ | 3.25 | 3.02 | 3.06 | 3.06 | |
| [MoO2(CN)4]4− | 2.64 | 2.51 | 2.59 | 2.59 | |
| [TcO2(CN)4]3− | 2.71 | 2.51 | 2.58 | 2.58 | |
| [RuO2(CN)4]2− | 2.50 | 2.06 | 2.12 | 2.12 | |
| [WO2(CN)4]4− | 2.85 | 2.67 | 2.73 | 2.74 | |
| [ReO2(CN)4]3− | 3.17 | 2.96 | 2.72 | 2.80 | 2.80 |
| [OsO2(CN)4]2− | 2.98 | 2.80 | 2.36 | 2.41 | 2.42 |
As the oxidation state of the central metal atom increases, the eg(xz, yz) orbitals become more π-antibonding, owing to greater oxometal π-donor interactions. It follows that we expect a corresponding increase in the energies of the 1A1 g(b2 g)2 → 1Eg(b2 g)1(eg)1 excitations. This increase is observed for [MO2(NH3)4]z complexes, but not for [MO2(CN)4]z complexes, where the excitation energies remain roughly constant. Clearly for [MO2(CN)4]z there is some other effect that causes significant changes in the relative energies of the b2 g(xy) and eg(xz, yz) orbitals.
To probe this effect, we analyze average changes in orbital energies per unit increase in the charge of the central metal. The average change in E[eg(xz, yz)] is −2.78 eV for [MO2(NH3)4]z, and −2.26 eV for [MO2(CN)4]z. For E[b2 g(xy)], the average change is −2.89 eV for [MO2(NH3)4]z and −2.10 eV for [MO2(CN)4]z. Consistent with the trends in energies of the 1A1 g(b2 g)2 → 1Eg(b2 g)1(eg)1 excitation, the average change in E[eg(xz, yz)] is greater (less negative) than the average change in E[b2 g(xy)] for [MO2(NH3)4]z, but the average change in E[b2 g(xy)] is greater than the average change in E[eg(xz, yz)] for [MO2(CN)4]z. The energy changes cited here do not vary strongly with exchange–correlation functional or central metal.
The 1Eg(b2 g)1(eg)1 excited state of a [MO2(CN)4]z complex is stabilized by backbonding from the metal (dxz, dyz) orbitals to the cyanide π* orbitals. Thus one possible explanation for the differing trends in [MO2(NH3)4]z and [MO2(CN)4]z is that greater bonding overlap between the (dxz, dyz) and π*(CN) orbitals is achieved as metal oxidation state increases. However, our calculations are not in accord with this explanation. For [MO2(NH3)4]z, the average change in E[eg(xz, yz)] per unit increase in the charge of the central metal is 0.52 eV less than the average change for [MO2(CN)4]z, indicating a trend opposite of that postulated here. Also, the calculated electron density pictures of the LUMO orbitals (performed with MOLDEN39) indicate that there is slightly less bonding overlap between (dxz, dyz) and π*(CN) for the complexes with higher charge (see, e.g., Fig. 2).
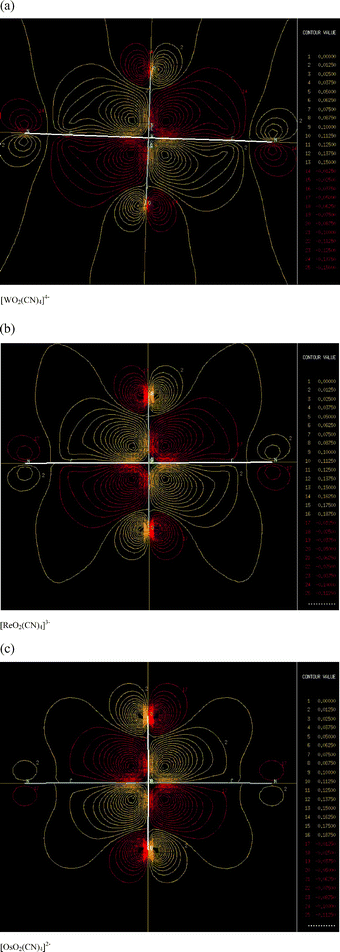 | ||
| Fig. 2 Electron density pictures of the eg(xz, yz) orbitals calculated with B-LYP for MO2(CN)4 with M = W, Re, and Os. | ||
Another attractive possibility is that increasing the charge of the central metal atom decreases the amount of dxy → π*(CN) backbonding, thereby destabilizing the b2 g(xy) orbital, and in turn decreasing Δ. While variations in backbonding may affect the orbital splitting, they do not account for the observed trends. The metal oxidation states in these complexes range from IV–VI, and there is very little backbonding in complexes with such high oxidation states. Also, there is little variation in the C–N bond distances in these complexes, suggesting only minor variations in π*(CN) populations. For [MO2(NH3)4]z, the average change in E[b2 g(xy)] per unit increase in the charge of the central metal atom is 0.79 eV less than the average change for [MO2(CN)4]z. Such a small change in backbonding cannot be the primary cause for the large difference noted here.
We are left with one last explanation: For σ-only equatorial ligands such as NH3, the b2 g(xy) orbital is virtually nonbonding. But for equatorial ligands such as CN−, there will be an antibonding interaction between the dxy orbital and cyanide π-orbitals. As the metal oxidation state increases, there will be more π-donation from the filled π(CN) orbitals to the metal dxy orbital, thereby destabilizing b2 g(xy). Since the extent of π-donation increases significantly per unit increase in metal oxidation state, this trend could easily account for the relative destabilization of b2 g(xy) in [MO2(CN)4]z compared to [MO2(NH3)4]z. We conclude that variations in cyanide donor interactions with metal oxidation state account for the observed trends in 1A1 g(b2 g)2 → 1Eg(b2 g)1(eg)1 energies.
Conclusion
For complexes of the form [MO2(NH3)4]z, the energy of the 1A1 g(b2 g)2 → 1Eg(b2 g)1(eg)1 excitation increases with metal oxidation state, reflecting increased π-donation from the oxygen p orbitals to the metal (dxz, dyz) orbitals. For complexes of the form [MO2(CN)4]z, these energies remain roughly constant, as there is relatively greater destabilization of b2 g(xy) when π-donors occupy equatorial positions.References
- C. J. Ballhausen and H. B. Gray, Inorg. Chem., 1962, 1, 111–122 CrossRef CAS.
- H. B. Gray and C. R. Hare, Inorg. Chem., 1962, 1, 363–368 CrossRef CAS.
- J. R. Winkler and H. B. Gray, Commun. Inorg. Chem., 1981, 1, 257–263 Search PubMed.
- J. R. Winkler and H. B. Gray, Inorg. Chem., 1985, 24, 346–355 CrossRef CAS.
- W. A. Nugent and J. M. Mayer, Metal–Ligand Multiple Bonds, Wiley-Interscience: New York, 1988 Search PubMed.
- V. M. Miskowski, H. B. Gray and M. D. Hopkins, Adv. Transition Met. Coord. Chem., 1996, 1, 159–186 Search PubMed.
- A. K. Mohammed and A. W. Maverick, Inorg. Chem., 1992, 31, 4441–4443 CrossRef CAS.
- I. Demachy and Y. Jean, Inorg. Chem., 1996, 35, 5027–5031 CrossRef CAS.
- R. A. Isovitsch, A. S. Beadle and F. R. Fronczek, Inorg. Chem., 1998, 37, 4258–4264 CrossRef CAS.
- E. Inego, E. Zangrando, S. Mestroni, G. Fronzoni, M. Stener and E. Alessio, J. Chem. Soc., Dalton Trans., 2001, 8, 1338–1346 RSC.
- J. K. Grey, I. S. Butler and C. Reber, Can. J. Chem., 2004, 82, 1083–1091 CrossRef CAS.
- J. K. Grey, I. S. Butler and C. Reber, Inorg. Chem., 2004, 43, 5103–5111 CrossRef CAS.
- R. Ahlrics, M. Bär, H. P. Baron, R. Bauernschmitt, S. Böcker, P. Deglmann, M. Ehrig, K. Eichkorn, S. Elliott, F. Furche, F. Haase, M. Häser, H. Horn, C. Hättig, C. Huber, U. Huniar, M. Kattannek, A. Köhn, C. Kölmel, M. Kollwitz, K. May, C. Ochsenfeld, H. Öhm, H. Patzelt, O. Rubner, A. Schäfer, U. Schneider, M. Sierka, O. Treutler, B. Unterreiner, M. von Arnim, F. Weigend, P. Weis and H. Weiss, TURBOMOLE V5-7, Quantum Chemistry Group, University of Karlsruhe, Karlsruhe, Germany ( 2004) Search PubMed.
- A. Schäfer, C. Huber and R. Ahlrichs, J. Chem. Phys., 1994, 100, 5829–5835 CrossRef CAS.
- D. Andrae, U. Haeussermann, M. Dolg, H. Stoll and H. Preuss, Theor. Chim. Acta, 1990, 77, 123–132 CAS.
- P. A. M. Dirac, Proc. R. Soc. London, Ser. A, 1929, 123, 714–733 CrossRef CAS.
- J. C. Slater, Phys. Rev., 1951, 81, 385–390 CrossRef CAS.
- S. H. Vosko, L. Wilk and M. Nusair, Can. J. Phys., 1980, 58, 1200–1211 CrossRef CAS.
- J. P. Perdew and Y. Wang, Phys. Rev. B, 1992, 45, 13244–13249 CrossRef.
- A. D. Becke, Phys. Rev. A, 1988, 38, 3098–3100 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B, 1988, 37, 785–789 CrossRef CAS.
- J. P. Perdew, Phys. Rev. B, 1986, 33, 8822–8824 CrossRef.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- A. Klamt and G. Schürmann, J. Chem. Soc., Perkin Trans. 2, 1993,(5), 799–805 RSC.
- O. Treutler and R. Ahlrichs, J. Chem. Phys., 1995, 102, 346–354 CrossRef CAS.
- M. von Arnim and R. Ahlrichs, J. Chem. Phys., 1999, 111, 9183–9190 CrossRef CAS.
- F. Furche, J. Chem. Phys., 2001, 114, 5982–5992 CrossRef CAS.
- F. Furche and R. Ahlrichs, J. Chem. Phys., 2002, 117, 7433–7447 CrossRef CAS.
- P. Hummel, J. Oxgaard, W. A. Goddard, III and H. B. Gray, J. Coord. Chem., 2005, 58, 41–45 CrossRef CAS.
- P. Hummel, J. Oxgaard, W. A. Goddard, III and H. B. Gray, Inorg. Chem., 2005, 44, 454–458.
- P. Hummel and H. B. Gray, Coord. Chem. Rev. Search PubMed , submitted.
- V. W. Day and J. L. Hoard, J. Am. Chem. Soc., 1968, 90, 3374–3379 CrossRef CAS.
- R. H. Fenn, A. J. Graham and N. P. Johnson, J. Chem. Soc. A, 1971, 2880–2883 RSC.
- J. M. Malin, E. O. Schlemper and R. K. Murmann, Inorg. Chem., 1977, 16, 615–619 CrossRef CAS.
- W. Depmeier and S. A. Mason, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1978, B34, 920–922 CrossRef CAS.
- M. E. Kastner, M. J. Lindsay and M. J. Clarke, Inorg. Chem., 1982, 21, 2037–2040 CrossRef CAS.
- A. Abou-Hamdan, A. Roodt and A. E. Merbach, Inorg. Chem., 1998, 37, 1278–1288 CrossRef CAS.
- G. Schaftenaar and J. H. Noordik, J. Comput.-Aided Mol. Des., 2000, 14, 123–134 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2006 |