Catalytic alcohol oxidation by an unsymmetrical 5-coordinate copper complex: electronic structure and mechanism†
Ekaterina
Zueva
a,
Paul H.
Walton
b and
John E.
McGrady
*b
aDepartment of Inorganic Chemistry, Kazan State Technological University, 68 Karl Marx St., Kazan, 420015, Russia. E-mail: zueva_e@kstu.ru.; Fax: +7 8432 365768; Tel: +7 8432 725939
bDepartment of Chemistry, University of York, Heslington, York, UK YO10 5DD. E-mail: jem15@york.ac.uk; Fax: +44 1904 432516; Tel: +44 1904 434539
First published on 15th November 2005
Abstract
Density functional theory reveals the detailed mechanism of alcohol oxidation by a model copper complex, CuIIL, L = cis-1-(3′,5′-dimethoxy-benzylideneamino)-3,5-[2-hydroxy-(3′,5′-di-tert-butyl)benzylideneimino]cyclohexane. Despite the obvious structural and functional parallels between the title compound and the enzyme galactose oxidase, the details of the catalytic pathway are fundamentally different. In the enzyme, coordination of the substrate produces an active form containing a CuII centre and a tyrosyl radical, the latter being responsible for the abstraction of hydrogen from the substrate. In the model system, in marked contrast, the active form contains a CuII centre, but the ligand radical character is localised on the substrate (alcoholate) oxygen, rather than the phenolate ligand. The result is a significantly higher barrier to hydrogen-atom abstraction compared to the enzyme itself. The origin of these significant differences is traced to the rigid nature of the pentadentate ligand, which resists changes in coordination number during the catalytic cycle.
Introduction
In the previous article in this issue,1 one of us reported the synthesis and structure of a CuII complex of a pentadentate ligand, cis-1-(3′,5′-dimethoxy-benzylideneamino)-3,5-[2-hydroxy-(3′,5′-di-tert-butyl)benzylideneimino]cyclohexane (Fig. 1). The square pyramidal coordination about the Cu centre in this complex, along with the presence of two phenolate moieties, is reminiscent of the key structural features of galactose oxidase, a fungal enzyme that catalyses the stereospecific two-electron oxidation of D-galactose to the corresponding aldehyde with the concomitant reduction of molecular oxygen to hydrogen peroxide.2![Molecular structure of CuIIL, L = cis-1-(3′,5′-dimethoxy-benzylideneamino)-3,5-[2-hydroxy-(3′,5′-di-tert-butyl)benzylideneimino]cyclohexane.](/image/article/2006/DT/b512298h/b512298h-f1.gif) | ||
| Fig. 1 Molecular structure of CuIIL, L = cis-1-(3′,5′-dimethoxy-benzylideneamino)-3,5-[2-hydroxy-(3′,5′-di-tert-butyl)benzylideneimino]cyclohexane. | ||
The active site of galactose oxidase features a mononuclear copper centre in a distorted square-pyramidal geometry, with a tyrosinate residue in an axial position and two histidine imidazole units, one modified tyrosinate ligand, and exogenous water or acetate occupying the equatorial sites. The enzyme has been isolated in three distinct redox states: the fully oxidised CuII–tyrosyl radical form in which a cupric ion and an equatorial tyrosyl radical are antiferromagnetically coupled, the semi-reduced Cu(II)–tyrosine form, and the fully reduced Cu(I)–tyrosine form. The fully oxidised form that combines both inorganic and organic one-electron cofactors is the catalytically active species, bringing about the two-electron oxidation of alcohols.
The mechanism of galactose oxidation has been extensively debated by both experimentalists and theoreticians.3–5 In the first step of the consensus mechanism (Fig. 2), the substrate binds to the equatorial coordination site of the active CuII–tyrosyl radical form, replacing the exogenous water. Substrate binding is followed by transfer of the hydroxyl proton to the axial tyrosinate ligand. The rate-limiting step is then the abstraction of a hydrogen atom from the α-carbon atom of the alcoholate by the equatorial tyrosyl radical, yielding a transient ketyl intermediate which decays by electron transfer to a CuI species and the aldehyde product. There has been some debate over the existence of the ketyl intermediate, and the hydrogen atom abstraction and electron transfer steps may in fact occur simultaneously. Following release of the aldehyde, molecular oxygen binds to the CuI site and regenerates the active CuII–tyrosyl radical form, along with hydrogen peroxide.
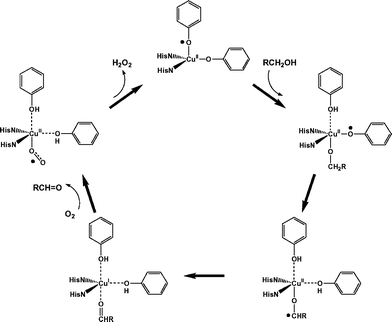 | ||
| Fig. 2 Consensus mechanism for alcohol oxidation by galactose oxidase. | ||
The key to the two-electron oxidative chemistry exhibited by galactose oxidase is clearly the presence of both inorganic (CuII) and organic (tyrosyl radical) cofactors in the active form of the enzyme. The role of the protein environment in stabilising the organic radical has been extensively debated,6,7 particularly the thioether substituent on the modified tyrosinate residue that has been shown to be essential for enzyme activity.8 It was originally proposed that the sulfur substituent stabilised the radical by delocalising the unpaired electron,9 but recent studies suggest that the electronic structure and reactivity are not significantly perturbed by the sulfide substituent,4,10 which may instead have a structural role.
Galactose oxidase and the closely related enzyme glyoxal oxidase11 are just two examples of the increasing diverse range of enzymes that are known to employ coordinated tyrosyl radicals as oxidising cofactors. As a result, there has been significant interest in the synthesis and characterization of structural and functional mimics. Structural and spectroscopic models for the semi-reduced (CuII),12–14 and fully reduced (CuI)15 forms of galactose oxidase have been reported, along with a number of stable O-bound phenoxyl radicals.16–28 In the majority of cases, the phenolate moieties are incorporated as part of multidentate ligands, preventing decoordination of the phenoxyl radicals during redox processes. Complexes with simple exogenous phenolate-containing ligands are, in contrast, very rare.27
Despite the large number of known CuII–phenoxyl radical species, only a very few are capable of stoichiometric or catalytic alcohol oxidation, in some cases under quite mild conditions, and with the concomitant conversion of molecular oxygen to hydrogen peroxide. Stack and coworkers24 have designed a series of four-coordinate complexes with a non-square planar N2O2 coordination sphere and shown that these model complexes catalyse the aerobic oxidation of alcohols with high turnover via a galactose oxidase-like mechanism. They have also confirmed that the thioether substituent on the phenyl ring is not necessary for catalytic reactivity, although its incorporation improves the number of turnovers. A computational analysis of the catalytic pathway has highlighted the key similarities and differences between the model system and the enzyme, and proposed modifications to the system that may lead to improved catalytic efficiency.5c In a series of papers,26 Wieghardt and coworkers have also reported systems that give efficient catalysis of aerobic alcohol oxidation. Significantly, other organic cofactors such as TEMPO, a stable nitroxyl radical, are also capable of participating in catalytic aerobic oxidation,29 apparently via mechanisms similar to galactose oxidase.
In the preceding paper in this issue, we showed that the copper complex shown in Fig. 1 is redox active (two reversible oxidations at 0.89 V and 1.13 V vs. Ag/AgCl) and, critically, is also capable of catalysing the aerobic oxidation of benzyl alcohol to benzaldehyde, albeit with dramatically reduced turnover rates compared to the native enzyme (0.0005 s−1vs. 800 s−1). The complex is therefore a functional, as well as a structural, mimic of the enzyme. Given the intense interest in understanding the mechanism of redox catalysis by enzymes and their mimics, we now present a computational analysis of the catalytic pathway based on density functional theory.
Computational methods
All calculations described in this paper were performed using the Gaussian0330 package. In the majority of cases, the ligand, L, was simplified by replacing the pendant 1,3-(MeO)2C6H4 arm, along with the tBu groups, with hydrogens. In order to test key conclusions, we have also done some test calculations where the bulky groups were included using the hybrid QM/MM methodology.31 In these cases, the quantum partition was as defined for the simplified model, and the remainder of the molecule was incorporated into the MM partition (UFF).32 Most calculations were performed using the B3LYP33b,c functional, in conjunction with the 6-31G** basis set for Cu, O, N and C and also the hydrogen atoms initially attached to the substrate (CH3OH). A 3-21G basis set was used for the remaining hydrogen atoms. Full geometry optimisations were performed without symmetry constraints, and stationary points were characterized as minima or transition states by vibrational analysis. In order to simplify the calculations, we have used methanol as substrate in place of benzyl alcohol. The overall reaction, CH3OH + O2 → CH2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O + H2O2, is marginally exothermic (ΔE = − 6.6 kJ mol−1) at the level of theory described above.
O + H2O2, is marginally exothermic (ΔE = − 6.6 kJ mol−1) at the level of theory described above.
Results and discussion
Electronic structure of CuL′, 21
The optimized structure of the doublet ground state of the model system, 21, is summarized in Fig. 3, along with its frontier orbital array. The copper centre shows the same approximate square pyramidal coordination noted in the X-ray structure with the amine nitrogen occupying the axial position. The frontier molecular orbitals of 21 are characteristic of a square pyramidal CuII complex, with the single vacancy in the valence shell (104a β), localized on the Cu dx2−y2 orbital, giving a net spin density of 0.71 on Cu. The optimized bond lengths are in generally good agreement with the crystallographic data (Table 1), with the exception of the axial Cu–N bond, which is some 0.25 Å shorter than the experimental value. If the bulky tBu and 3′,5′-dimethoxy-benzylidene groups are included in the computational model using the QM/MM methodology, the Cu–N bond length increases to 2.42 Å, indicating that steric interactions have a significant impact on the relatively weak axial coordination site. The stronger Cu–N and Cu–O bond lengths in the equatorial plane are, in contrast, much closer to experiment, and are essentially unaffected by the introduction of the bulky substituents. In the subsequent discussion of the intermediates in the catalytic cycle, the axial Cu–N bond is, in all cases, stronger. We therefore anticipate that steric effects will not exert such a large influence on the bond length.| CuIIL (expt) | 2 1 | 3 1 + | 4 1 2+ | |
|---|---|---|---|---|
| Cu–O(1) | 1.936 | 1.93 | 1.94 | 1.95 |
| Cu–O(2) | 1.994 | 1.93 | 1.94 | 1.98 |
| Cu–N(1) | 1.991 | 1.99 | 1.99 | 1.98 |
| Cu–N(2) | 1.991 | 2.02 | 1.99 | 2.02 |
| Cu–N(3) | 2.512 | 2.28 | 2.22 | 2.08 |
| O(1)–C | 1.304 | 1.29 | 1.29 | 1.28 |
| O(2)–C | 1.318 | 1.30 | 1.29 | 1.27 |
| Net spin densities Cu | 0.70 | 0.71 | 0.73 | |
| Phenolate rings | 0.15 | 1.54 | 2.55 |
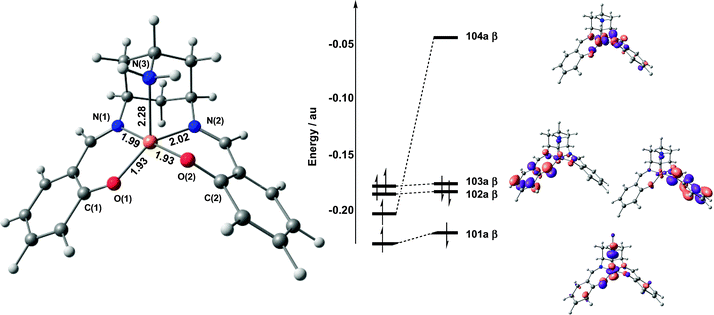 | ||
| Fig. 3 Optimised structure and frontier molecular orbital array for 21. Spin-α orbitals are shown on the left, spin-β on the right. | ||
One and two-electron oxidation of 21: structural and electronic consequences
As noted in the Introduction, the compound CuL undergoes two reversible oxidation processes in MeCN, at 0.89 V and 1.13 V (vs. Ag/AgCl) which, based on comparison with similar ligand systems, were assigned as ligand-based processes. Consistent with this hypothesis, one-electron oxidation of 1 gives a cationic species with a triplet (S = 1) ground state, 31+, with two vacancies in the spin-β manifold, one localized on Cu dx2−y2 (104a β) and the other delocalized over the two phenol rings (103a β) (Fig. 4). The net spin density on the Cu centre therefore remains essentially unchanged (0.71), with the additional unpaired electron on the two ligands. The structure of 31+ is almost indistinguishable from the neutral complex, 21, with marginal lengthening and contraction of the Cu–O and C–O bonds, respectively. Benisvy et al.28 have noted similar redox-induced changes in their crystallographic study of a copper-bound phenoxyl radical.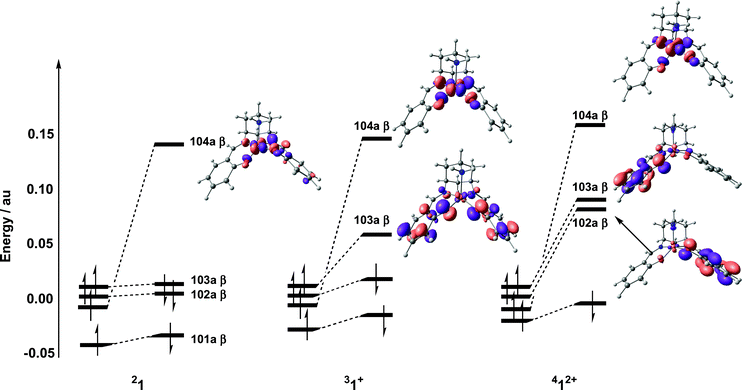 | ||
| Fig. 4 Comparison of frontier orbitals for 21, 31+ and 412+ (in each case the symmetric combination of spin-α phenol π* orbitals is taken as the energetic reference point). | ||
In the triplet ground state, the metal- and ligand-based electrons are ferromagnetically coupled. The corresponding antiferromagnetically coupled (broken symmetry) state (MS = 0) lies only 0.8 kJ mol−1 higher, but gradient corrected functionals such as BP8633d,f,g or BLYP33a,e reverse this order, suggesting that the magnetic coupling is intrinsically weak. The magnetic behaviour of coordinated radicals has been rationalised in terms of the Cu–O–C bond angle, α, and the dihedral angle, β, between the Cu equatorial plane and phenyl ring.23b These structural parameters determine the relative orientation of the two magnetic orbitals (metal-based dx2−y2 and ligand π*) and, hence, the extent of overlap between the two. The values of α (127°) and β (32°) in the triplet state lie in the range where ferro- and antiferromagnetic contributions to the exchange interaction are very similar (α = 127–130°, β = 23–30°), leading to almost isoenergetic high- and low-spin states.23c
Removal of a second electron leads to a dicationic species with a quartet ground state, 412+. The optimized structure (Table 1) and molecular orbital array (Fig. 4) of 412+ confirm that the second oxidation process is also ligand-based: of the three vacancies in the spin-β manifold, one (104a β) is metal-based while the other two (103a β and 102a β) are delocalized over the two phenol rings. We have also located two distinct broken-symmetry doublets (MS = 1/2), where the metal- and ligand-based electrons are antiferromagnetically coupled (either ααβ or αβα on O–Cu–O). These broken-symmetry states again lie less than 5 kJ mol−1 above the quartet ground state, further illustrating the very weak coupling between the metal- and ligand-based electrons. In summary, our calculations confirm that both oxidation processes are ligand-based, and that the magnetic coupling between the unpaired electrons on the coordinated phenoxyl radicals and copper ion is very weak. In the remainder of this paper, we will examine the potential role of 1 in the catalytic oxidation of alcohol.
Catalytic oxidation of CH3OH
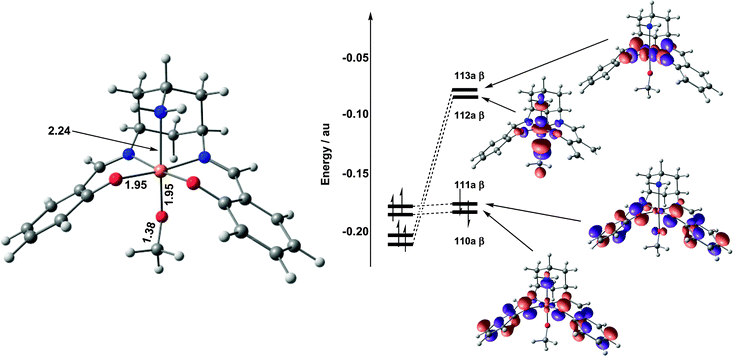 | ||
| Fig. 5 Optimised structure and frontier molecular orbital array for 32. | ||
The absence of significant unpaired spin density on either of the phenolate ligands following substrate binding offers a stark contrast to the situation in the active form of galactose oxidase, and will clearly have a significant impact on the subsequent hydrogen-atom abstraction step. Given the importance of this conclusion, and the continuing debate over the performance of density functional theory in modelling the balance between metal- and ligand-based redox processes,34 we have reassessed the electronic structure of 32 using a range of functionals (BP86, BLYP). In all cases, the outcome was unchanged: the methoxide adduct is best formulated as a mixture of CuIII–OMe and CuII–OMe˙ resonance forms, with negligible spin density on the phenolate rings.
Finally, we note that all attempts to dock a molecule of methanol (as opposed to methoxide ion) with 31+ were unsuccessful. The initial proton transfer step that is rapid and approximately thermoneutral in galactose oxidase4 must therefore be significantly endothermic in this case, consistent with the need for the conjugate base to initiate the reaction. The endothermicity of the proton transfer step has its origins in the very rigid ligand geometry, and in particular the CN double bonds which reduce the basicity of the phenolate groups and also prevent their rotation away from the metal centre following protonation. In the enzyme active site, in contrast, the protein backbone allows the axial tyrosine residue to decoordinate and also stabilises the phenol group through hydrogen bonding.
 | ||
| Fig. 6 Potential energy surface for methoxide oxidation by 31+. | ||
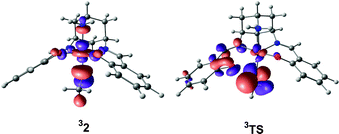 | ||
| Fig. 7 Singly-occupied, substrate-localised orbitals in 32 and 3TS. In each case, the orbital shown is the (vacant) spin-β component (112a β). | ||
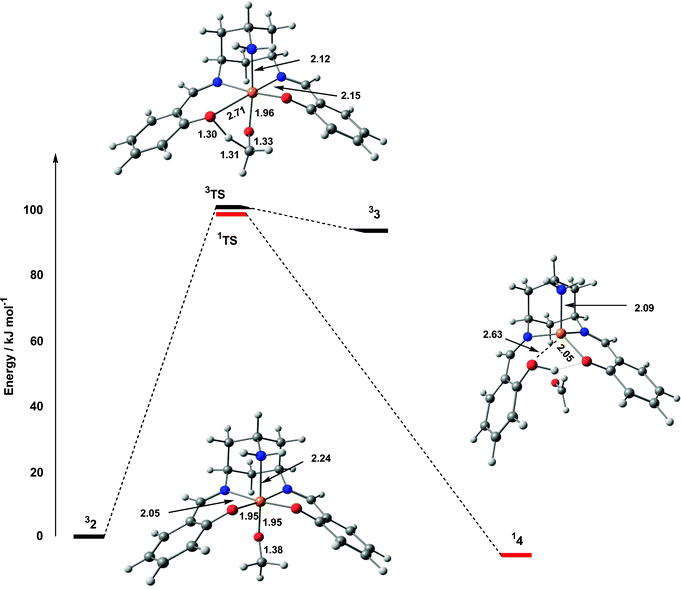
A number of authors have proposed the participation of a ketyl intermediate in the catalytic pathway of galactose oxidase, although recent computational results suggest that such a structure would be only transiently stable.4 Decomposition of the triplet transition state, 3TS, does indeed lead to a ketyl structure, 33, only 7.6 kJ mol−1 lower in energy, but there is no evidence for a similar species on the singlet potential energy surface. Instead, 1TS decays directly to a closed-shell singlet CuI species with a weakly coordinated aldehyde ligand (14). The bond to the protonated phenol ligand in 14 is also almost completely broken (2.63 Å), leaving the approximate tetrahedral coordination typical of CuI species. Stack has previously highlighted the ability of tripodal ligands to support tetrahedral geometries, and hence allow the CuII/I couple to participate in the oxidative process.24a
In summary, the oxidative part of the potential energy surface involves initial coordination of the alcoholate anion to 1+, leading to intramolecular electron transfer and formation of an adduct best described as a CuII–OMe˙ diradical. From this point, reorientation of the radical character leads to hydrogen-atom and electron transfer, and decay to a weakly bound CuI–aldehyde complex. The barrier to hydrogen-atom transfer is generally accepted as being the rate-limiting step in galactose oxidase, where computational estimates of the barrier (also using CH3OH as a model substrate) range from 46–67 kJ mol−1. The value of 100.0 kJ mol−1 in Fig. 6 therefore indicates a significantly reduced activity relative to galactose oxidase, consistent with the very low turnover rate.
2-Electron oxidation of 14 by O2
The oxidative component of the catalytic cycle is completed with the formation of a tetrahedral CuI species, 14, described above. The weakly coordinated formaldehyde molecule is readily displaced by O2, leading to a stable triplet dioxygen complex, 35, in a reaction that is exothermic by 19.4 kJ mol−1 (Fig. 8). The O–O bond length of 1.30 Å and spin densities of 0.56 and 1.33 on Cu and O2, respectively, clearly indicate that this species is best formulated as a CuII–superoxide species. However, 35 occupies a very shallow minimum on the potential energy surface, and a rotation about the Cu–O(O) bond results in the exothermic abstraction of a hydrogen atom from the decoordinated phenol ring. Homolytic cleavage of the Cu–O(O) bond then occurs without further barrier, leading to a CuII complex, weakly coordinated to a hydroperoxyl radical, HOO˙ (36).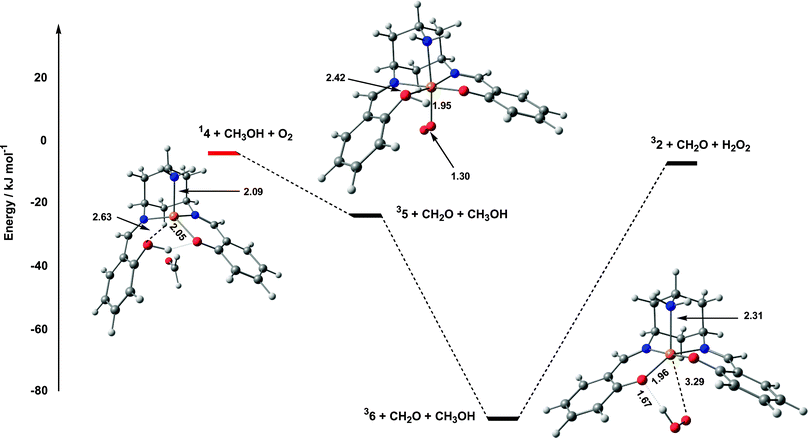 | ||
| Fig. 8 Potential energy surface for oxidation of 14 by O2. | ||
From 36, the catalytic cycle is closed by release of the hydroperoxyl radical, hydrogen-atom abstraction from a second molecule of CH3OH and coordination of the resultant methoxide radical to restore 32. We have not attempted to explore the sequence in which these events occur, largely because of the uncertain role of solvent, but the overall reaction is endothermic by 83.3 kJ mol−1.
Summary and comparison with galactose oxidase
The overall catalytic pathway is summarised in Fig. 9, where the oxidative and reductive components are brought together. The overall features of the potential energy surface are rather similar to those described by Himo and coworkers in their discussion of the galactose oxidase system itself,4 with a rate-limiting hydrogen-atom abstraction followed by exothermic release of aldehyde and reoxidation of the metal core. The most striking difference between our model system and galactose oxidase is the very high barrier to hydrogen-atom abstraction (100.0 vs. 46.4 kJ mol−1). However, in view of the fact that, following substrate binding, there is no spin density on the phenolate ring, it is perhaps surprising that hydrogen-atom abstraction occurs at all. The electron-transfer pathway proposed for galactose oxidase summarised in Scheme 1a illustrates the role of the tyrosyl radical in the process. In the model complex (Scheme 1b) where the radical character resides instead on the substrate oxygen, a significantly different cyclic redistribution of electrons occurs along the reaction pathway. The much higher barrier in the model system reflects this fundamental difference in mechanism.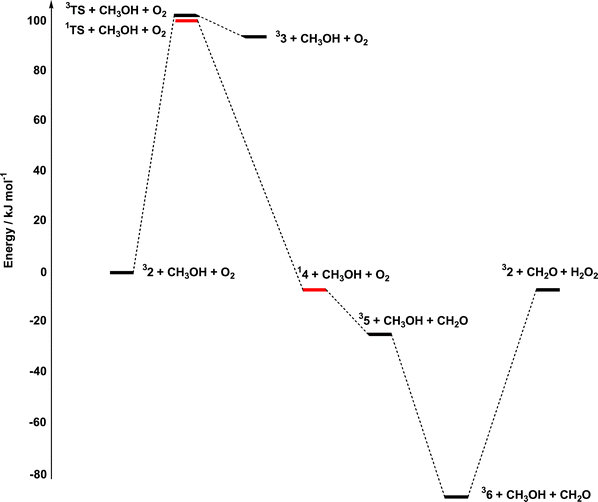 | ||
| Fig. 9 Summary of the catalytic pathway for aerobic oxidation of CH3OH. | ||
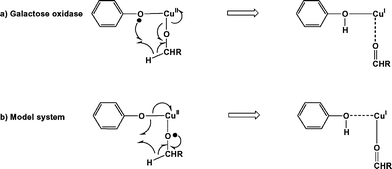 | ||
| Scheme 1 Hydrogen-atom abstraction and electron-transfer pathways in (a) galactose oxidase and (b) 32. | ||
The contrast between galactose oxidase and the model complex can be traced to the very different properties of the ligand sets involved. In galactose oxidase, the substrate displaces the fifth (exogenous) ligand, rather than filling a vacant coordination site. Moreover, proton transfer from the substrate leads to decoordination of the axial tyrosinase, giving a square planar complex with substrate in an equatorial site. In this geometry, the doubly-occupied dz2 orbital of CuII lies well below the tyrosine pi levels. In the model complex, in contrast, there is no readily displacable exogenous ligand, and, as noted previously, proton transfer leading to decoordination of a phenolate ring is prevented by the rigidity of the ligand framework. As a result, substrate binding generates an octahedral, rather than square planar, copper centre, and the presence of two axial ligands forces the Cu dz2–OR σ* orbital above the phenolate π* manifold. As a result, the active complex is better formulated as mixture of CuIII–OR and CuII–OR˙ resonance forms.
This detailed comparison of galactose oxidase and CuIIL clearly illustrates that, although the model complex is both a structural and functional mimic of galactose oxidase, the parallels between the two do not extend to the intimate details of mechanism. Thus, whilst nature has chosen to adopt the tyrosyl radical as a co-factor for catalytic alcohol oxidation, this is clearly not the only strategy available to the coordination chemist. The deeper understanding of the electronic factors involved in the process outlined in this paper will provide clear directions for the rational design of improved second-generation catalysts.
Acknowledgements
EZ acknowledges financial support from the Royal Society in the form of a visiting researcher grant.References
- S. J. Archibald, R. Bhalla, B. C. Gilbert, E. J. MacLean, A. K. Nairn, S. J. Teat and P. H. Walton, Dalton Trans., 2006 10.1039/b512296c.
- (a) N. Ito, S. E. V. Phillips, C. Stevens, Z. B. Ogel, M. J. McPherson, J. N. Keen, K. D. S. Yadav and P. F. Knowles, Nature (London), 1991, 350, 87 CrossRef CAS; (b) N. Ito, S. E. V. Phillips, C. Stevens, Z. B. Ogel, M. J. McPherson, J. N. Keen, K. D. S. Yadav and P. F. Knowles, Faraday Discuss., 1992, 93, 75 RSC; (c) N. Ito, S. E. V. Phillips, K. D. S. Yadav and P. F. Knowles, J. Mol. Biol., 1994, 238, 794 CrossRef CAS.
- (a) J. W. Whittaker, in Metalloenzymes Involving Amino Acid Residues and Related Radicals, ed. H. Sigel and A. Sigel, Marcel Dekker, New York, 1994, vol. 30, p. 315 Search PubMed; (b) P. F. Knowles and N. Ito, in Perspectives in Bio-inorganic Chemistry, Jai Press, London, 1994, vol. 2, p. 207 Search PubMed; (c) J. P. Klinman, Chem. Rev., 1996, 96, 2541 CrossRef CAS; (d) J. Stubbe and W. A. van der Donk, Chem. Rev., 1998, 98, 705 CrossRef CAS; (e) C. D. Borman, C. G. Saysell, A. Sokolowski, M. B. Twitchett, C. Wright and A. G. Sykes, Coord. Chem. Rev., 1999, 190–192, 771 CrossRef CAS; (f) J. W. Whittaker, Chem. Rev., 2003, 103, 2347 CrossRef CAS.
- F. Himo, L. A. Eriksson, F. Maseras and P. E. M. Siegbahn, J. Am. Chem. Soc., 2000, 122, 8031 CrossRef CAS.
- (a) U. Rothlisberger and P. Carloni, Int. J. Quantum Chem., 1999, 73, 209 CrossRef CAS; (b) U. Rothlisberger, P. Carloni, K. Doclo and M. Parrinello, J. Biol. Inorg. Chem., 2000, 5, 236 CrossRef CAS; (c) L. Guidoni, K. Spiegel, M. Zumstein and U. Rothlisberger, Angew. Chem., Int. Ed., 2004, 43, 3286 CrossRef CAS.
- A. M. Boulet, E. D. Walter, D. A. Schwartz, G. J. Gerfen, P. R. Callis and D. J. Singel, Chem. Phys. Lett., 2000, 331, 108 CrossRef CAS.
- M. Kaupp, T. Gress, R. Reviakine, O. L. Malkina and V. G. Malkin, J. Phys. Chem. B, 2003, 107, 331 CrossRef CAS.
- A. J. Baron, C. Stevens, C. Wilmot, K. D. Seneviratne, V. Blakeley, D. M. Dooley, S. E. V. Phillips, P. F. Knowles and M. J. McPherson, J. Biol. Chem., 1994, 269, 25095 CAS.
- (a) S. Itoh, K. Hirano, A. Furuta, M. Komatsu, Y. Ohshiro, A. Ishida, S. Takamuku, T. Kohzuma, N. Nakamura and S. Suzuki, Chem. Lett., 1993, 2099 CAS; (b) S. Itoh, S. Takayama, R. Arakawa, A. Furuta, M. Komatsu, A. Ishida, S. Takamuku and S. Fukuzumi, Inorg. Chem., 1997, 36, 1407 CrossRef CAS.
- (a) F. Himo, G. T. Babcock and L. A. Eriksson, Chem. Phys. Lett., 1999, 313, 374 CrossRef CAS; (b) M. A. Halcrow, Angew. Chem., Int. Ed., 2001, 40, 346 CrossRef CAS.
- (a) M. M. Whittaker, P. J. Kersten, N. Nakamura, J. Sanders-Loehr, E. S. Schweizer and J. W. Whittaker, J. Biol. Chem., 1996, 271, 681 CrossRef CAS; (b) M. M. Whittaker, P. J. Kersten, D. Cullen and J. W. Whittaker, J. Biol. Chem., 1999, 274, 36226 CrossRef CAS.
- (a) S. Itoh, M. Taki and S. Fukuzumi, Coord. Chem. Rev., 2000, 198, 3 CrossRef CAS and references therein; (b) B. A. Jazdzewski and W. B. Tolman, Coord. Chem. Rev., 2000, 200–202, 633 CrossRef CAS and references therein; (c) J.-L. Pierre, Chem. Soc. Rev., 2000, 29, 251 RSC and references therein.
- (a) M. Vaidyanathan and M. Palaniandavar, Proc. Indian Acad. Sci., Chem. Sci., 2000, 112, 223 Search PubMed; (b) M. Vaidyanathan, M. Palaniandavar and R. S. Gopalan, Inorg. Chim. Acta, 2001, 324, 241 CrossRef CAS; (c) T. Kruse, T. Weyhermüller and K. Wieghardt, Inorg. Chim. Acta, 2002, 331, 81 CrossRef CAS; (d) A. Neves, A. dos Anjos, A. J. Bortoluzzi, B. Szpoganicz, E. W. Schwingel and A. S. Mangrich, Inorg. Chim. Acta, 2003, 356, 41 CrossRef CAS.
- H.-J. Krüger, Angew. Chem., Int. Ed., 1999, 38, 627 CrossRef CAS.
- B. A. Jazdzewski, A. M. Reynolds, P. L. Holland, V. G. Young, S. Kaderli, A. D. Zuberbühler and W. B. Tolman, J. Biol. Inorg. Chem., 2003, 8, 381 Search PubMed.
- (a) J. Hockertz, S. Steenken, K. Wieghardt and P. Hildebrandt, J. Am. Chem. Soc., 1993, 115, 11222 CrossRef CAS; (b) A. Sokolowski, E. Bothe, E. Bill, T. Weyhermüller and K. Wieghardt, Chem. Commun., 1996, 1671 RSC; (c) B. Adam, E. Bill, E. Bothe, B. Goerdt, G. Haselhorst, K. Hildenbrand, A. Sokolowski, S. Steenken, T. Weyhermüller and K. Wieghardt, Chem. Eur. J., 1997, 3, 308 CrossRef CAS; (d) A. Sokolowski, B. Adam, T. Weyhermüller, A. Kikuchi, K. Hildenbrand, R. Schnepf, P. Hildebrandt, E. Bill and K. Wieghardt, Inorg. Chem., 1997, 36, 3702 CrossRef CAS; (e) A. Sokolowski, J. Müller, T. Weyhermüller, R. Schnepf, P. Hildebrandt, K. Hildenbrand, E. Bothe and K. Wieghardt, J. Am. Chem. Soc., 1997, 119, 8889 CrossRef CAS; (f) R. Schnepf, A. Sokolowski, J. Müller, V. Bachler, K. Wieghardt and P. Hildebrandt, J. Am. Chem. Soc., 1998, 120, 2352 CrossRef CAS; (g) M. D. Snodin, L. Ould-Moussa, U. Wallman, S. Lecomte, V. Bachler, E. Bill, H. Hummel, T. Weyhermüller, P. Hildebrandt and K. Wieghardt, Chem. Eur. J., 1999, 5, 2554 CrossRef CAS; (h) J. Müller, A. Kikuchi, E. Bill, T. Weyhermüller, P. Hildebrandt, L. Ould-Moussa and K. Wieghardt, Inorg. Chim. Acta, 2000, 297, 265 CrossRef CAS; (i) S. Itoh, H. Kumei, S. Nagatomo, T. Kitagawa and S. Fukuzumi, J. Am. Chem. Soc., 2001, 123, 2165 CrossRef CAS; (j) A. K. Nairn, R. Bhalla, S. P. Foxon, X. Liu, L. J. Yellowlees, B. C. Gilbert and P. H. Walton, J. Chem. Soc., Dalton Trans., 2002, 1253 RSC; (k) Y. Shimazaki, F. Tani, K. Fukui, Y. Naruta and O. Yamauchi, J. Am. Chem. Soc., 2003, 125, 10512 CrossRef CAS.
- (a) D. Zurita, I. Gautier-Luneau, S. Ménage, J.-L. Pierre and E. Saint-Aman, J. Biol. Inorg. Chem., 1997, 2, 46 CrossRef CAS; (b) D. Zurita, S. Ménage, J. L. Pierre and E. Saint-Aman, New J. Chem., 1997, 21, 1001 Search PubMed; (c) E. Saint-Aman, S. Ménage, J.-L. Pierre, E. Defrancq and G. Gellon, New J. Chem., 1998, 393 RSC; (d) F. Thomas, G. Gellon, I. Gautier-Luneau, E. Saint-Aman and J.-L. Pierre, Angew. Chem., Int. Ed., 2002, 41, 3047 CrossRef CAS; (e) A. Philibert, F. Thomas, C. Philouze, S. Hamman, E. Saint-Aman and J.-L. Pierre, Chem. Eur. J., 2003, 9, 3803 CrossRef CAS.
- (a) S. Itoh, M. Taki, S. Takayama, S. Nagatomo, T. Kitagawa, N. Sakurada, R. Arakawa and S. Fukuzumi, Angew. Chem., Int. Ed., 1999, 38, 2774 CrossRef CAS; (b) S. Itoh, M. Taki, H. Kumei, S. Takayama, S. Nagatomo, T. Kitagawa, N. Sakurada, R. Arakawa and S. Fukuzumi, Inorg. Chem., 2000, 39, 3708 CrossRef; (c) M. Taki, H. Kumei, S. Itoh and S. Fukuzumi, J. Inorg. Biochem., 2000, 78, 1 CrossRef CAS; (d) M. Taki, H. Kumei, S. Nagatomo, T. Kitagawa, S. Itoh and S. Fukuzumi, Inorg. Chim. Acta, 2000, 300–302, 622 CrossRef CAS.
- (a) Y. Shimazaki, S. Huth, A. Odani and O. Yamauchi, Angew. Chem., Int. Ed., 2000, 39, 1666 CrossRef CAS; (b) Y. Shimazaki, S. Huth, S. Hirota and O. Yamauchi, Bull. Chem. Soc. Jpn., 2000, 73, 1187 CrossRef CAS; (c) Y. Shimazaki, S. Huth, S. Hirota and O. Yamauchi, Inorg. Chim. Acta, 2002, 331, 168 CrossRef CAS.
- C. Ochs, F. E. Hahn and R. Fröhlich, Eur. J. Inorg. Chem., 2001, 2427 CrossRef CAS.
- M. Vaidyanathan, M. Palaniandavar and R. S. Gopalan, Indian J. Chem., Sect. A, 2003, 42, 2210.
- (a) J. A. Halfen, V. G. Young and W. B. Tolman, Angew. Chem., Int. Ed. Engl., 1996, 35, 1687 CrossRef CAS; (b) J. A. Halfen, B. A. Jazdzewski, S. Mahapatra, L. M. Berreau, E. C. Wilkinson, L. Que and W. B. Tolman, J. Am. Chem. Soc., 1997, 119, 8217 CrossRef CAS.
- (a) A. Sokolowski, H. Leutbecher, T. Weyhermüller, R. Schnepf, E. Bothe, E. Bill, P. Hildebrandt and K. Wieghardt, J. Biol. Inorg. Chem., 1997, 2, 444 CrossRef CAS; (b) J. Müller, T. Weyhermüller, E. Bill, P. Hildebrandt, L. Ould-Moussa, T. Glaser and K. Wieghardt, Angew. Chem., Int. Ed., 1998, 37, 616 CrossRef CAS; (c) E. Bill, J. Müller, T. Weyhermüller and K. Wieghardt, Inorg. Chem., 1999, 38, 5795 CrossRef CAS.
- (a) R. C. Pratt and T. D. P. Stack, Inorg. Chem., 2005, 44, 2367 CrossRef CAS; (b) Y. Wang and T. P. D. Stack, J. Am. Chem. Soc., 1996, 118, 13097 CrossRef CAS; (c) Y. Wang, J. L. DuBois, B. Hedman, K. O. Hodgson and T. D. P. Stack, Science, 1998, 279, 537 CrossRef CAS; (d) R. C. Pratt and T. D. P. Stack, J. Am. Chem. Soc., 2003, 125, 8716 CrossRef CAS; (e) R. J. M. K. Gebbink, M. Watanabe, R. C. Pratt and T. D. P. Stack, Chem. Commun., 2003, 630 RSC; (f) V. Mahadevan, R. J. M. K. Gebbink and T. D. P. Stack, Curr. Opin. Chem. Biol., 2000, 4, 228 CrossRef CAS.
- O. Sénèque, M. Campion, B. Douziech, M. Giorgi, Y. Le Mest and O. Reinaud, Dalton Trans., 2003, 4216 RSC.
- (a) P. Chaudhuri, M. Hess, U. Flörke and K. Wieghardt, Angew. Chem., Int. Ed., 1998, 37, 2217 CrossRef CAS; (b) P. Chaudhuri, M. Hess, T. Weyhermüller and K. Wieghardt, Angew. Chem., Int. Ed., 1999, 38, 1095 CrossRef CAS; (c) P. Chaudhuri, M. Hess, J. Müller, K. Hildenbrand, E. Bill, T. Weyhermüller and K. Wieghardt, J. Am. Chem. Soc., 1999, 121, 9599 CrossRef CAS; (d) T. K. Paine, T. Weyhermüller, K. Wieghardt and P. Chaudhuri, Dalton Trans., 2004, 2092 RSC.
- (a) M. A. Halcrow, L. M. L. Chia, X. Liu, E. J. L. McInnes, L. J. Yellowlees, F. E. Mabbs and J. E. Davies, Chem. Commun., 1998, 2465 RSC; (b) M. A. Halcrow, L. M. L. Chia, X. Liu, E. J. L. McInnes, L. J. Yellowlees, F. E. Mabbs, I. J. Scowen, M. McPartlin and J. E. Davies, J. Chem. Soc., Dalton Trans., 1999, 1753 RSC.
- L. Benisvy, A. J. Blake, D. Collison, E. S. Davies, C. D. Garner, E. J. L. McInnes, J. McMaster, G. Whittaker and C. Wilson, Chem. Commun., 2001, 1824 RSC.
- (a) P. Gamez, I. W. C. E. Arends, J. Reedijk and R. A. Sheldon, Chem. Commun., 2003, 2414 RSC; (b) A. Dijksman, I. W. C. E. Arends and R. A. Sheldon, Org. Biomol. Chem., 2003, 1, 3232 RSC and references therein.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Montgomery, Jr., T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, C. Gonzalez and J. A. Pople, Gaussian 03, Revision B.03, Gaussian, Inc., Pittsburgh, PA, 2003 Search PubMed.
- (a) F. Maseras and K. Morokuma, , J. Comput. Chem., 1995, 16, 1170 CrossRef CAS; (b) M. Svensson, S. Humbel, R. D. J. Froese, T. Matsubara, S. Sieber and K. Morokuma, J. Phys. Chem., 1996, 100, 19357 CrossRef CAS.
- A. K. Rappe, C. J. Casewit, K. S. Colwell, W. A. Goddard, III and W. M. Skiff, J. Am. Chem. Soc., 1992, 114, 10024 CrossRef CAS.
- (a) A. D. Becke, Phys. Rev. A, 1988, 38, 3098 CrossRef CAS; (b) A. D. Becke, J. Chem. Phys., 1993, 98, 1372 CrossRef CAS; (c) A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS; (d) S. H. Vosko, L. Wilk and M. Nusair, Can. J. Phys., 1980, 58, 1200 CrossRef CAS; (e) C. Lee, W. Yang and R. G. Parr, Phys. Rev. B, 1988, 37, 785 CrossRef CAS; (f) A. D. Becke, J. Chem. Phys., 1986, 84, 4524 CrossRef CAS; (g) J. P. Perdew, Phys. Rev. B, 1986, 33, 8822 CrossRef.
- (a) A. Ghosh, A and E. Steene, J. Biol. Inorg. Chem., 2001, 6, 739 Search PubMed; (b) A. Ghosh, B. J. Persson and P. R. Taylor, J. Biol. Inorg. Chem., 2003, 8, 507 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Optimised geometries and energies of all stationary points. See DOI: 10.1039/b512298h |
| This journal is © The Royal Society of Chemistry 2006 |