Tetrametallic clusters (Ir2Rh2) through an ancillary ortho-carborane-1,2-dichalcogenolato ligands
Guo-Xin
Jin
* and
Jian-Qiang
Wang
Laboratory of Molecular Catalysis and Innovative Material, Department of Chemistry, Fudan University, 200433, Shanghai, P. R. China. E-mail: gxjin@fudan.edu.cn; Fax: +86-21-65641740; Tel: +86-21-65643776
First published on 10th November 2005
Abstract
The tetrametallic cluster complexes {Cp*Ir[E2C2(B10H9)]}Rh2(cod){Cp*Ir[E2C2 (B10H10)]} (E = S (3a); Se (3b)) have been synthesized by reactions of the 16-electron half-sandwich iridium complexes [Cp*Ir{E2C2(B10H10)}] [Cp* = η5-C5Me5, E = S (1a), Se (1b)] with [Rh(cod)(µ-OEt)2] at room temperature in toluene solution. In the solid state, this tetrametallic cluster exhibits an irregular nearly planar metal skeleton with the two carborane dichalcogenolato ligands bridging the four metal centers from both sides of the tetrametallic plane. Even though all metal atoms coordinate bridging chalcogen atoms, they show different electronic and coordination environments. The molecular structures of 3a and 3b have been determined by X-ray crystallography.
Introduction
The chemistry of mixed-metal clusters has been of enduring interest because of the unique chemistry resulting from having two or more metals with different chemistry properties in close proximity and also because of their potential catalytic applications.1,2 Many dithiolene ligands have been employed for the preparation of heterometallic clusters.3 During the past decade, considerable attention has been devoted to metal complexes containing chelating 1,2-dicarba-closo-dodecaborane-1,2-dichalcogenolate ligands, to take advantage of their unique molecular structure.4 A number of mononuclear 16-electron Cp and Cp* half-sandwich complexes of Co, Rh and Ir have been described which contain a bidentate, chelating 1,2-dicarba-closo-dodecaborane-1,2-dichalcogenolato ligand, [(B10H10)C2E2]2− (E = S, Se) and a “pseudo-aromatic” metalladichalcogenolene five-membered ring.4 These compounds have been used as models to study the insertion of alkynes into one of the metal–sulfur bonds; this may lead to the formation of a metal-to-boron bond or substitution of the carborane cage in the positions of B(3)/B(6).5,6 In recently, we have developed a methodology to build up polynuclear frameworks, containing metal–metal bonds, by using the 16-electron mononuclear complexes of metals such as Cp*Ir[E2C2(B10H10)], Cp*Rh[E2C2(B10H10)] and Cp*Co[E2C2(B10H10)] (E = S, Se) (see Scheme 1 for selected examples A, B and C).5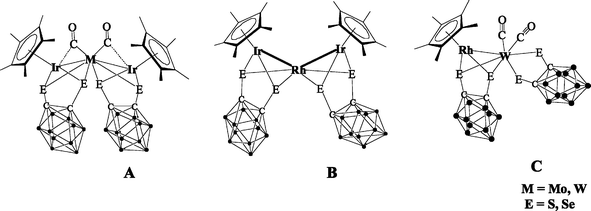 | ||
| Scheme 1 Examples of structurally characterized complexes. | ||
The incorporation of the 1,2-dithiolato or 1,2-diselenolato structural unit into the bulky closo-dicarbadodecaborane ligand protects the inner coordination sphere and stabilized the metal–metal bond, which has occasionally been observed at dithiolene ligands.7 Such properties aid in the construction of different types of binuclear, trinuclear and other unprecedented hetero-metallic clusters.
In previous work, we have already described, treatment of Cp*Ir[E2C2(B10H10)] with [Rh(cod)(µ-Cl)]2 resulted in the formation of a trimetallic cluster with the Ir2Rh core.5 For comparison, now the reactions of 1a, 1b with analogous complexes [Rh(cod)(µ-OEt)]2 have been investigated. Here, we present the results of these studied along with details of the synthesis and characterization of novel tetranuclear cluster complexes 3a, 3b.
Results and discussion
In our previous report, the dimer [Rh(cod)(µ-Cl)]2 was an acceptable material to produce the Ir2Rh trinuclear metallic clusters 2a, 2b.5 However, the work of Vinas et al. shows that the reaction of carborane complexes with other types of rhodium complexes, such as [Rh(cod)(acac)], would lead to some interesting reactions.8 Taking into account the similarity of [Rh(cod)(µ-OEt)]2 with [Rh(cod)(µ-Cl)]2, which has been long known to be an useful precursor to prepare a variety of rhodium complexes, we consider whether this similar reaction can give different results. The experimental procedure is similar to the reaction of [Rh(cod)(µ-Cl)]2. The 16-electron complexes 1a and 1b can be easily synthesized from the half-sandwich iridium dichloride complex [Cp*IrCl(µ-Cl)]2 with dilithium 1,2-dicarba-closo-dodecaborane (12)-1,2-dithiolate in THF solution (Scheme 3).91a and 1b were reacted with [Rh(cod)(µ-OEt)]2 in the molar ratio 2 : 1 in toluene, and the green solution changed to red gradually. After the products were separated by column chromatography, two products: {Cp*Ir[S2C2(B10H9)]}Rh(cod) (2a) and {Cp*Ir[S2C2(B10H9)]}Rh2(cod){Cp*Ir[S2C2(B10H10)]} (3a) were obtained in 18 and 30% yield, respectively (Scheme 2). In addition, analogous products {Cp*Ir[Se2C2(B10H9)]}Rh(cod) (2b) and {Cp*Ir[Se2C2(B10H9)]}Rh2(cod){Cp*Ir[Se2C2(B10H10)]} (3b) were also obtained by the same method in yields of 16 and 30% respectively. Complexes 3a and 3b can be obtained in higher yields when longer reaction times are applied.![Reaction of 1a and 1b with [Rh(cod)(µ-OEt)]2.](/image/article/2006/DT/b512027f/b512027f-s2.gif) | ||
| Scheme 2 Reaction of 1a and 1b with [Rh(cod)(µ-OEt)]2. | ||
 | ||
| Scheme 3 Synthesis of complexes 1a and 1b. | ||
The complexes 3a and 3b are neutral, diamagnetic and air-sensitive in solution. The IR spectra of the products 3a and 3b in the solid state exhibit intense B–H of carborane stretching at about 2573 (vs) and 2588 (vs) cm−1.
The molecule 2a is unsymmetrical, as confirmed by the 1H NMR spectrum with two singlets of Cp* rings at δ 2.11 and 2.19 ppm.5 The 1H NMR data for complexes 3a display two intense methyl singlets of pentamethylcyclopentandienyl ligands (2.07, 2.19 ppm) and a number of multiplets in the ranges 4.02–4.29 ppm (olefinic) and 1.58–1.83 ppm (aliphatic) which can be ascribed to the 1,5-cyclooctadiene ligand. Both molecules 2a and 3a contain an Ir–B bond, as indicated by the appearance of Ir–B resonances in the 11B NMR spectrum at about −20 ppm.10 Excellent elemental analyses were obtained for all complexes and consistent with the proposed structures.
Crystals of 2a, 3a, 2b and 3b suitable for X-ray diffraction study were obtained by slow diffusion of hexane into a concentrated solution of the complexes in dichloromethane at low temperature. The molecular structures of 2a and 2b have been described in our previous papers,5 so they were only used to compare with 3a and 3b here. Selected bond distances and angles of 3a and 3b are listed in Table 1.
| 3a | |||
| Ir(1)–S(1) | 2.332(3) | Ir(1)–S(2) | 2.347(3) |
| Ir(1)–Rh(2) | 2.5990(10) | C(1)–C(2) | 1.610(14) |
| Ir(2)–Rh(2) | 2.9814(10) | Rh(2)–S(3) | 2.304(3) |
| Rh(2)–S(4) | 2.295(3) | Ir(2)–Rh(1) | 2.7862(11) |
| Ir(2)–S(3) | 2.315(3) | Ir(2)–B(16) | 2.139(11) |
| Rh(1)–S(3) | 2.332(3) | Rh(1)–S(4) | 2.365(3) |
| Rh(1)–C(3) | 2.132(10) | C(3)–C(4) | 1.627(14) |
| S(1)–Ir(1)–S(2) | 57.69(7) | C(1)–S(1)–Ir(1) | 104.1(4) |
| C(1)–S(1)–Rh(2) | 103.3(3) | S(1)–Rh(2)–S(2) | 79.25(9) |
| Ir(1)–S(1)–Rh(2) | 66.76(7) | Ir(1)–S(2)–Rh(2) | 66.48(7) |
| Ir(1)–Rh(2)–Ir(2) | 168.99(3) | S(4)–Rh(2)–S(3) | 83.43(10) |
| S(3)–Rh(2)–Ir(2) | 49.95(7) | S(3)–Ir(2)–Rh(2) | 49.64(6) |
| S(3)–Ir(2)–Rh(1) | 53.45(7) | Rh(2)–S(4)–Rh(1) | 95.40(9) |
| Rh(1)–Ir(2)–Rh(2) | 73.32(3) | S(3)–Rh(1)–S(4) | 81.31(9) |
| C(3)–Rh(1)–S(4) | 75.0(3) | C(3)–Rh(1)–Ir(2) | 83.8(3) |
| C(4)–S(4)–Rh(1) | 81.6(3) | B(16)–Ir(2)–Rh(1) | 66.6(3) |
| 3b | |||
|---|---|---|---|
| Ir(1)–Se(1) | 2.4316(16) | Ir(1)–Se(2) | 2.4433(16) |
| Ir(1)–Rh(2) | 2.6281(12) | C(1)–C(2) | 1.614(19) |
| Ir(2)–Rh(2) | 3.0085(13) | Rh(2)–Se(3) | 2.4038(18) |
| Rh(2)–Se(4) | 2.3975(18) | Ir(2)–Rh(1) | 2.8258(13) |
| Ir(2)–Se(3) | 2.4435(16) | Ir(2)–B(16) | 2.151(15) |
| Rh(1)–Se(3) | 2.4578(18) | Rh(1)–Se(4) | 2.4777(18) |
| Rh(1)–C(3) | 2.138(15) | C(3)–C(4) | 1.648(19) |
| Se(1)–Ir(1)–Se(2) | 83.41(5) | C(1)–Se(1)–Ir(1) | 102.6(4) |
| C(1)–Se(1)–Rh(2) | 101.8(4) | Se(1)–Rh(2)–Se(2) | 81.42(6) |
| Ir(1)–Se(1)–Rh(2) | 64.63(4) | Ir(1)–Se(2)–Rh(2) | 64.39(4) |
| Ir(1)–Rh(2)–Ir(2) | 169.63(5) | Se(4)–Rh(2)–Se(3) | 83.96(6) |
| Se(3)–Rh(2)–Ir(2) | 52.23(4) | Se(3)–Ir(2)–Rh(2) | 51.04(4) |
| Se(3)–Ir(2)–Rh(1) | 55.03(4) | Rh(2)–Se(4)–Rh(1) | 94.15(6) |
| Rh(1)–Ir(2)–Rh(2) | 75.39(4) | Se(3)–Rh(1)–Se(4) | 81.18(6) |
| C(3)–Rh(1)–Se(4) | 76.2(4) | C(3)–Rh(1)–Ir(2) | 83.5(4) |
| C(4)–Se(4)–Rh(1) | 78.5(4) | B(16)–Ir(2)–Rh(1) | 66.2(4) |
The molecular structure of 3a is shown in Fig. 1. The metal atoms of the core adopt a quite irregular nearly planar Ir2Rh2 arrangement (Ir(1), Ir(2), Rh(1) and Rh(2) metals are coplanar with maximum deviation at 0.177 Å). To our knowledge, this type of Ir2Rh2 core cluster is rare, only some pyramidal shape metal carbonyl clusters of Ir2Rh2(CO)12 and its derivatives were reported previously.11 The two dithiolato ligands of carboranes are situated at the both sides of the tetrametallic plane with three of the sulfur atoms acting as µ2-bridging ligands and the other sulfide simultaneously connecting three metals. The Ir(2), Rh(1) and Rh(2) metals exhibit six-coordinate environments and Ir(1) is five coordinate if considering the metal–metal bonds, and their coordination spheres are clearly different (Fig. 2). Ir(2) coordinates a different set of ligands bonding to Ir(1), boron and two metal bonds with Rh(1) and Rh(2), except for the similar Cp* bridging thiolates ligands. By use of the thiolate ligands, Rh(2) connects the Ir(1) and Ir(2) atoms.
 | ||
| Fig. 1 Molecular structure of the tetranuclear complex 3a. | ||
 | ||
| Fig. 2 Central skeleton of 3a showing the different metal coordination spheres. | ||
Compared with structures of 2a and 3a, the difference lies in the [(cod)Rh] fragment in 3a. We can presume that in the whole reaction the [(cod)Rh] fragment exists in some reactive form and inserts into the original S(3)–C(3) bond, accompanying the cleavage of S(3)–C(3) bond, and 3a is formed. The detailed mechanism of the insertion reaction of [(cod)Rh] into S–C bonds is still not clear, and further investigations of these aspects are in progress.
The structure of 3a shows three short intermetallic separations: Ir(1)–Rh(2) 2.5990(10), Rh(2)–Ir(2) 2.9814(10), Ir(2)–Rh(1) 2.7862(11) Å. All these lie in the expected range for a metal–metal bond. Compared with the analogous fragment in 2a (2.685(3) Å), the Ir(1)–Rh(2) metal bond become much shorter. Slightly longer Rh–Ir separations were found in many cases, such as [Cptt2Zr(µ-S)2Ir(CO)2Rh(cod)2] (2.8205(10) Å)12 (with a µ-S supported metal–metal bond), and the heterodinuclear cations RhIr(CO)3(µ-dppm)2]+ (2.7722(7) Å)13 and [RhIr(CH3)(CO)3(µ-dppm)2]+ (2.743(1) Å).14 Much longer metal–metal distances were observed in the cluster [(Cp*Ir)2(µ3-S)2Rh(cod)][RhCl2(cod)] (2.906(1) and 2.913(9) Å).15 The presence of a shorter d8–d8 Ir(1)–Rh(2) interaction in 3a is also supported by the Ir(1)–S–Rh(2) angles (66.76(7) and 66.48(7)°), which are smaller than the analogous Rh–S–Ir angles in 2a (69.59(18) and 69.8(2)°). The Ir–S distances (2.332(3)–2.347(3) Å) and Rh–S distances (2.295(3)–2.365(3) Å) in 3a are in the normal range of Ir–S and Rh–S single bonds, but the Ir–S distances are significantly longer than those in the “pseudo-aromatic” iridadithiolene ring in Cp*Ir[S2C2(B10H10)] (1a) (2.252(5) and 2.251(6) Å).4 Due to the coordination of the S atoms of carborane to Rh, the pseudo-aromatic iridadithiolene heterocyclic system (Ir(1)–S(1)–C(1)–C(2)–S(2)) is destroyed and bent with a dihedral angle of 135.3° along the S(1)⋯S(2) vector.
The complex 3b crystallizes in the form of dark-red block in the monoclinic space group P21/n with four molecular in the unit cell. Its molecular structure is similar to 3a. Fig. 3 depicts the molecular structure of 3b. Complex 3b also contain four metal atoms, which form an irregular nearly planar Ir2Rh2 core (The Ir(1), Ir(2), Rh(1) and Rh(2) metals are coplanar with maximum deviation at 0.167 Å) similarly with 3a. There are three metal bonds: Ir(1)–Rh(2) 2.6281(12), Rh(2)–Ir(2) 3.0085(13), Ir(2)–Rh(1) 2.8258(13) Å. The Ir(1)–Se–Rh(2) angles (64.63(4) and 64.39(4)°) are smaller than the analogous Ir(1)–S–Rh(2) angles in 3a (66.76(7) and 66.48(7)°). The planar pseudo-aromatic system of the iridadiselene heterocycle 1b is also no longer present in 3b, the dihedral angle at the Se⋯Se vector in the IrSe2C2 ring is 135.8°. The Ir–Se bond distances (2.4316(16)–2.4435(16) Å) are similar to those in normal selenolate complexes, but are significantly longer than those in the “pseudo-aromatic” iridadiselene ring in Cp*Ir[Se2C2(B10H10)] (1b) (2.3753(9) and 2.3656(9) Å).9
 | ||
| Fig. 3 Molecular structure of the tetranuclear complex 3b. | ||
Conclusion
In this work, we carried out the reactions of 16-electron coordinatively unsaturated iridium dichalcogenolato complexes 1a, 1b with [Rh(cod)(µ-OEt)2]. Two novel tetranuclear cluster complexes were prepared and fully characterized. The resultant clusters 3a, 3b feature an Ir2Rh2 core containing three Ir–Rh metal bonds.Experimental
General information
All manipulations were performed under an atmosphere of nitrogen using standard Schlenk techniques. Solvents were dried by refluxing over sodium/benzophenone ketyl (toluene, hexane, dichloromethane) and distilled just before use. Cp*Ir[S2C2(B10H10)] (1a),4,9 Cp*Ir[Se2C2(B10H10)] (1b)9 and [Rh(cod)(µ-OEt2)]16 were prepared by methods reported previously. IR spectra were recorded on a Nicolet AVATAR-360IR spectrometer, whereas 1H (500 MHz) and 11B (160 MHz) NMR spectra were obtained on a Bruker DMX-500 spectrometer in CDCl3 solution. Elemental analyses were performed on an Elementar vario EI Analyzer.![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) ), 4.29 (m, 2H, CH). 11B NMR (25 °C, BF3·Et2O): δ
−3.2, −6.7, −9.0, −11.1, −13.6, −15.8, −20.2; IR (KBr disk): 2573, 2588 cm−1 (νB–H).
), 4.43 (m, 2H, CH); 11B NMR (25 °C, BF3·Et2O): δ
−2.9, −6.1, −8.8, −12.5, −14.7, −19.8; IR (KBr disk): 2577, 2585 cm−1 (νB–H).
), 4.29 (m, 2H, CH). 11B NMR (25 °C, BF3·Et2O): δ
−3.2, −6.7, −9.0, −11.1, −13.6, −15.8, −20.2; IR (KBr disk): 2573, 2588 cm−1 (νB–H).
), 4.43 (m, 2H, CH); 11B NMR (25 °C, BF3·Et2O): δ
−2.9, −6.1, −8.8, −12.5, −14.7, −19.8; IR (KBr disk): 2577, 2585 cm−1 (νB–H).
Crystallographic analysis
Dark-red crystals of 3a and 3b were grown by slow diffusion from dichloromethane–hexane solution. The data crystals of both compounds were mounted by gluing onto the end of a thin glass fiber. X-Ray intensity data were collected on the CCD-Bruker SMART APEX system. All the determinations of unit cell and intensity data were performed with graphite-monochromated Mo-Kα radiation (λ = 0.71073 Å). All the data were collected at room temperature using the ω scan technique. These structures were solved by direct methods, using Fourier techniques, and refined on F2 by a full-matrix least-squares method. All non-hydrogen atoms were refined with anisotropic thermal parameters. All the hydrogen atoms were included but not refined. All the calculations were carried out the SHELXTL program.17 Crystal data, data collection parameters, and the results of the analyses of compounds 3a, 3b are listed in Table 2.| 3a | 3b | |
|---|---|---|
| Formula | C32H61B20S4Ir2Rh2S4·CH2Cl2 | C32H61B20S4Ir2Rh2Se4·CH2Cl2 |
| M | 1465.39 | 1652.99 |
| Crystal system | Monoclinic | Monoclinic |
| Space group | P21/n | P21/n |
| a/Å | 11.066(4) | 11.104(3) |
| b/Å | 26.140(8) | 26.517(6) |
| c/Å | 18.062(6) | 18.003(4) |
| β/° | 98.633(5) | 98.155(4) |
| V/Å3 | 5165(3) | 5247(2) |
| Z | 4 | 4 |
| D c/g cm−3 | 1.884 | 2.092 |
| µ(Mo-Kα)/mm−1 | 6.057 | 8.578 |
| Collected reflections | 21604 | 22009 |
| Unique | 9083 | 9264 |
| Parameters | 597 | 595 |
| Goodness of fit | 1.020 | 1.016 |
| R 1 (I > 2σ(I)) | 0.0499 | 0.0549 |
| wR 2 (I > 2σ(I)) | 0.1174 | 0.1424 |
| Max., min. residual density/e Å−3 | 1.917, −0.986 | 1.399, −1.083 |
CCDC reference numbers 281883 and 281884.
For crystallographic data in CIF or other electronic format see DOI: 10.1039/b512027f
Acknowledgements
This work was supported by the National Science Foundation of China for Distinguished Young Scholars (29925101, 20421303) and by the National Basic Research Program of China (2005CB623802), and is gratefully acknowledged.References
- P. Braunstein and J. Rose, in Comprehensive Organometallic Chemistry, ed. E. W. Abel, F. G. A. Stone and G. Wilkinson, Pergamon, Oxford, UK, 1995, vol. 10, ch. 7 Search PubMed.
- Selected review: M. Herberhold and G.-X. Jin, Angew. Chem., Int. Ed. Engl., 1994, 33, 964–966 Search PubMed; M. C. Comstock and J. R. Shapley, Coord. Chem. Rev., 1995, 143, 501–533 CrossRef; L. H. Gade, Angew. Chem., Int. Ed., 2000, 39, 2658–2678 CrossRef CAS.
- M. Fourmigue, Coord. Chem. Rev., 1998, 178–180, 823–864 CrossRef CAS.
- M. Herberhold, G.-X. Jin, H. Yan, W. Milius and B. Wrackmeyer, J. Organomet. Chem., 1999, 587, 252–257 CrossRef CAS; J. Y. Bae, Y. I. Park, J. Ko, K. I. Park, S. I. Cho and S. O. Kang, Inorg. Chim. Acta, 1999, 289, 141–148 CrossRef CAS; J.-H. Won, D.-H. Kim, B. Y. Kim, S.-J. Kim, C. Lee, S. Cho, J. Ko and S. O. Kang, Organometallics, 2002, 21, 1443–1453 CrossRef CAS; X.-F. Hou, X.-C. Wang, J.-Q. Wang and G.-X. Jin, J. Organomet. Chem., 2004, 689, 2228–2235 CrossRef CAS.
- G.-X. Jin, J.-Q. Wang, Z. Zhang, L. H. Weng and M. Herberhold, Angew. Chem., Int. Ed., 2005, 44, 259–262 CrossRef CAS; J.-Q. Wang, L. H. Weng and G.-X. Jin, J. Organomet. Chem., 2005, 690, 249–252 CrossRef CAS; J.-Q. Wang, L. H. Weng and G. X. Jin, Rev. Inorg. Chem., 2005, 25, 55–66; J.-Q. Wang, X. Hou, L. H. Weng and G.-X. Jin, Organometallics, 2005, 24, 826–830 CrossRef CAS; S. Cai, J.-Q. Wang and G.-X. Jin, Organometallics, 2005, 24, 4226–4231 CrossRef CAS; J.-Q. Wang, S. Y. Cai, L. H. Weng, M. Herberhold and G.-X. Jin, Chem.–Eur. J., 2005, 11, 7343–7351; W. Li, L.-H. Weng and G.-X. Jin, Inorg. Chem. Commun., 2004, 1174–1177 CrossRef CAS; S. Y. Cai, Y. Lin and G.-X. Jin, Dalton Trans., 2006 10.1039/b512027f.
- M. Herberhold, H. Yan, W. Milius and B. Wrackmeyer, Angew. Chem., Int. Ed., 1999, 38, 3689–3691 CrossRef; M. Herberhold, H. Yan, W. Milius and B. Wrackmeyer, Chem.–Eur. J., 2000, 6, 3026–3032 CrossRef CAS; M. Herberhold, H. Yan, W. Milius and B. Wrackmeyer, J. Chem. Soc., Dalton Trans., 2001, 1782–1789 RSC.
- M. Nihei, T. Nankawa, M. Kurihara and H. Nishihara, Angew. Chem., Int. Ed., 1999, 38, 1098–1100 CrossRef CAS.
- F. Teixidor, M. A. Flores, C. Vinas, R. Sillanpaa and R. Kivekas, J. Am. Chem. Soc., 2000, 122, 1963–1973 CrossRef CAS.
- M. Herberhold, G.-X. Jin, H. Yan, W. Milius and B. Wrackmeyer, Eur. J. Inorg. Chem., 1999, 873–875 Search PubMed.
- J.-Y. Bae, Y.-J. Lee, S.-J. Kim, J. Ko, S. Cho and S. O. Kang, Organometallics, 2000, 19, 1514–1521 CrossRef CAS; Y.-J. Lee, J.-D. Lee, S.-J. Kim, J. Ko, I.-H. Suh, M. Cheong and S. O. Kang, Organometallics, 2004, 23, 135–143 CrossRef CAS.
- L. J. Farrugia, A. G. Orpen and F. G. A. Stone, Polyhedron, 1983, 2, 171–173 CrossRef CAS; B. Giacomo, S. Gianfranco, R. Renzo, R. Raymond, G. Fabrizia and B. Dario, Helv. Chim. Acta, 1993, 76, 2913–2925 CrossRef CAS; J. R. Galsworthy, A. D. Hattersley, C. E. Housecroft, A. L. Rheingold and A. Waller, J. Chem. Soc., Dalton Trans., 1995, 549–557 RSC.
- M. A. F. Hernandez-Gruel, J. J. Perez-Torrente, M. A. Ciriano, F. J. Lanoz and L. A. Oro, Angew. Chem., Int. Ed., 1999, 38, 2769–2771 CrossRef CAS.
- R. McDonald and M. Cowie, Inorg. Chem., 1990, 29, 1564–1571 CrossRef CAS.
- F. H. Antwi-Nsiah, O. Oke and M. Cowie, Organometallics, 1996, 15, 1042–1054 CrossRef CAS.
- Z. Tang, Y. Nomura, Y. Ishii, Y. Mizobe and M. Hidai, Organometallics, 1997, 16, 151–154 CrossRef CAS.
- G. Pannetier, P. Fougeroux, R. Bonnaire and N. Platzer, J. Less-Common Met., 1971, 24, 83–92 CrossRef CAS; R. Uson, L. A. Oro and J. A. Cabeza, Inorg. Synth., 1985, 23, 126–130 CAS; L. M. Green and D. W. Meek, Organometallics, 1989, 8, 659–666 CrossRef.
- SHELXTL 6.10, Bruker Analytical Instrumentation, Madison, WI, USA, 2000 Search PubMed.
| This journal is © The Royal Society of Chemistry 2006 |