Hydride transfer reactivity of tetrakis(trimethylphosphine)(hydrido)(nitrosyl)molybdenum(0)†
Yin Zhao, Helmut W. Schmalle, Thomas Fox, Olivier Blacque and Heinz Berke*
Department of Inorganic Chemistry, University of Zürich, Winterthurerstrasse 190, 8057, Zürich, Switzerland. E-mail: yzhao@aci.unizh.ch; schmalle@aci.unizh.ch; fox@aci.unizh.ch; oblacque@aci.unizh.ch; hberke@aci.unizh.ch
First published on 23rd November 2005
Abstract
The tetrakis(trimethylphosphine) molybdenum nitrosyl hydrido complex trans-Mo(PMe3)4(H)(NO) (2) and the related deuteride complex trans-Mo(PMe3)4(D)(NO) (2a) were prepared from trans-Mo(PMe3)4(Cl)(NO) (1). From 2H T1 min measurements and solid-state 2H NMR the bond ionicities of 2a could be determined and were found to be 80.0% and 75.3%, respectively, indicating a very polar Mo–D bond. The enhanced hydridicity of 2 is reflected in its very high propensity to undergo hydride transfer reactions. 2 was thus reacted with acetone, acetophenone, and benzophenone to afford the corresponding alkoxide complexes trans-Mo(NO)(PMe3)4(OCHR′R″) (R′ = R″ = Me (3); R′ = Me, R″ = Ph (4); R′ = R″ = Ph (5)). The reaction of 2 with CO2 led to the formation of the formato-O-complex Mo(NO)(OCHO)(PMe3)4 (6). The reaction of 2 with HOSO2CF3 produced the anion coordinated complex Mo(NO)(PMe3)4(OSO2CF3) (7), and the reaction with [H(Et2O)2][BArF4] with an excess of PMe3 produced the pentakis(trimethylphosphine) coordinated compound [Mo(NO)(PMe3)5][BArF4] (8). Imine insertions into the Mo–H bond of 2 were also accomplished. PhCH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NPh (N-benzylideneaniline) and C10H7CHNPh (N-1-naphthylideneaniline) afforded the amido compounds Mo(NO)(PMe3)4[NR′(CH2R″)] (R′ = R″ = Ph (9), R′ = Ph, R″ = naphthyl (11)). 9 could not be obtained in pure form, however, its structure was assigned by spectroscopic means. At room temperature 11 reacted further to lose one PMe3 forming 12 (Mo(NO)PMe3)3[N(Ph)CH2C10H6)]) with agostic stabilization. In a subsequent step oxidative addition of the agostic naphthyl C–H bond to the molybdenum centre occurred. Then hydrogen migration took place giving the chelate amine complex Mo(NO)(PMe3)3[NH(Ph)(CH2C10H6)] (15). The insertion reaction of 2 with C10H7NCHPh led to formation of the agostic compound Mo(NO)(PMe3)3[N(CH2Ph)(C10H7)] (10). Based on the knowledge of facile formation of agostic compounds the catalytic hydrogenation of C10H7NCHPh and PhNCHC10H7 with 2 (5 mol%) was tested. The best conversion rates were obtained in the presence of an excess of PMe3, which were 18.4% and 100% for C10H7NCHPh and PhNCHC10H7, respectively.
NPh (N-benzylideneaniline) and C10H7CHNPh (N-1-naphthylideneaniline) afforded the amido compounds Mo(NO)(PMe3)4[NR′(CH2R″)] (R′ = R″ = Ph (9), R′ = Ph, R″ = naphthyl (11)). 9 could not be obtained in pure form, however, its structure was assigned by spectroscopic means. At room temperature 11 reacted further to lose one PMe3 forming 12 (Mo(NO)PMe3)3[N(Ph)CH2C10H6)]) with agostic stabilization. In a subsequent step oxidative addition of the agostic naphthyl C–H bond to the molybdenum centre occurred. Then hydrogen migration took place giving the chelate amine complex Mo(NO)(PMe3)3[NH(Ph)(CH2C10H6)] (15). The insertion reaction of 2 with C10H7NCHPh led to formation of the agostic compound Mo(NO)(PMe3)3[N(CH2Ph)(C10H7)] (10). Based on the knowledge of facile formation of agostic compounds the catalytic hydrogenation of C10H7NCHPh and PhNCHC10H7 with 2 (5 mol%) was tested. The best conversion rates were obtained in the presence of an excess of PMe3, which were 18.4% and 100% for C10H7NCHPh and PhNCHC10H7, respectively.
Introduction
The search for activated transition metal hydrides is of continuing interest in particular with regard to hydrogenations. Earlier, it has been shown that the strength of LnM–H bonds depends on the electronegativity of the metal centre.1a Low electronegativities cause weaker bonds to hydrogen and in addition a hydridic polarization. On the basis of a ligand tuning nitrosyl substitution may be used to effect low orbital electronegativities1b at various metal centres thus simulating early transition metals.2–10 It has also been demonstrated with trans-W(CO)2(H)(NO)(PR3)22,11–13 and mer-W(CO)(H)(NO)(PMe3)314 that the hydridic character of the hydride is phosphine-dependent. Thus an increased number of phosphine ligands enhances the hydridic character of the hydride, which has also been observed in a Re(CO)n(H)(PR3)5−n (n = 1–3) series.5,15 Based on the fact that molybdenum has generally weaker bonds to H than tungsten but similar electronegativity, it can be inferred that in a Mo(NO)(CO)4−n(H)(PR3)n (n = 2–4) series of complexes the Mo–H bond is expected to display higher reactivity than the analogous tungsten series.The synthesis of mer-Mo(CO)(PMe3)3(H)(NO) and the reactivity of this hydride was studied earlier in our group.16 As a natural extension to this the Mo(PMe3)4(H)(NO) complex was prepared and studied for reactivity.
Results and discussion
Synthesis and characterization of trans-Mo(PMe3)4(H)(NO) (2)
Even though Liang et al.16 and Hillhouse et al.17 could prepare mer-Mo(CO)(PMe3)3(H)(NO) and mer-Mo(CO)(PMePh2)3(H)(NO) as complexes related to trans-Mo(PMe3)4(H)(NO) (2) using trans-Mo(AlCl4)(CO)4(NO) as a starting material, attempts to obtain 2 with a similar route failed. The substitution of the “fourth” CO ligand of trans-Mo(AlCl4)(CO)4(NO) could not be accomplished. Carmona et al.18 reported the preparation of trans-Mo(PMe3)4(Cl)(NO) using 1% sodium amalgam reducing MoCl3(NO)(PMe3)3 in the presence of PMe3. MoCl3(NO)(PMe3)3 was obtained from the reaction of MoCl3(PMe3)3 with NO gas, a reaction difficult to control. This route was modified as shown in Scheme 1 using Mo(NO)Cl3(CH3CN)219 and 5 equivalents of PMe3. At room temperature the complex Mo(NO)Cl3(PMe3)3 was formed, which was not isolated and immediately subjected to reduction with sodium amalgam. The purification of trans-Mo(PMe3)4(Cl)(NO) was carried out as described in the literature.18 Compound trans-Mo(PMe3)4(Cl)(NO) (1) was obtained as yellow crystals in ca. 85% yield. The IR spectrum of 1 in solution shows a ν(NO) band in THF at 1544 cm−1 and at 1522 cm−1 in the solid state. In the 1H NMR spectrum (benzene-d6) at room temperature a broadened singlet resonance is found at 1.30 ppm for the Me groups. The broadening is interpreted in terms of an unresolved very small coupling to the HMe, CH2 protons. The 31P NMR spectrum reveals a singlet at −2.3 ppm and in the 13C NMR spectrum one signal of higher order is detected at 19.3 ppm in agreement with the high symmetry of 1. | ||
| Scheme 1 | ||
2 was obtained by the treatment of trans-Mo(PMe3)4(Cl)(NO) with 5 equivalents of NaBH4 and 10 equivalents of PMe3 in THF at 50 °C for 5 days (Scheme 1). After extraction with pentane, subsequent sublimation of the impurities at 60 °C in vacuo, and recrystallization from pentane 2 was isolated in 87% yield. Hydride 2 has been fully characterized by NMR, IR, Raman and MS spectroscopies and elemental analysis. The 1H NMR spectrum of 2 in benzene-d6 reveals a quintet at −2.51 ppm (2JHP = 33.0 Hz), for the hydrido ligand coupled to the four chemically equivalent phosphorus nuclei of the PMe3 ligands. The 31P{1H} NMR spectrum in toluene shows a singlet at 5.1 ppm.
The IR spectrum of 2 in the solid-state revealed a band at 1521 cm−1 assigned to overlapping ν(NO) and ν(Mo–H) vibrations. In hexane at room temperature the IR spectrum of 2 shows one ν(NO) band at 1562 cm−1 and at −25 °C a moderately strong additional band at 1521 cm−1. The latter was attributed to the ν(Mo–H) vibration. To assure this assignment the IR spectrum of the corresponding deuteride trans-Mo(PMe3)4(D)(NO) (2a) was studied in the solid state and in hexane. 2a can be obtained by a procedure analogous to the preparation of hydride 2 using NaBD4 instead of NaBH4. In the solid state IR spectrum one finds the expected ν(NO) absorption of 2a at 1498 cm−1, which is shifted by 23 cm−1 to lower wavenumbers with respect to 2 indicating coupling of ν(NO) and ν(Mo–H) in 2. The ν(Mo–D) vibration is observed at 1063 cm−1 in accord with the isotope shift. The isotopic ratio of ν(Mo–H)/ν(Mo–D) ratio is 1.43, which is close to the theoretical value of √2.20 In the IR spectrum of 2a in hexane one finds the ν(NO) vibration at 1548 cm−1, which is shifted by −14 cm−1 with respect to 2. The Raman spectrum of 2 exhibits in the solid-state a weak band at 1526 cm−1 which is attributed to the ν(NO) vibration. A ν(Mo–H) band expected at 1508 cm−1 could not be detected. The Raman spectrum of 2a reveals a ν(Mo–D) band at 1066 cm−1 and a ν(NO) vibration at 1507 cm−1; the latter is shifted to low wavenumbers by 19 cm−1 with respect to ν(NO) of 2 confirming again vibrational coupling.
Cooling a saturated pentane solution of 2 to −30 °C gave crystals suitable for an X-ray diffraction study. The X-ray data collection and processing parameters are compiled in Table 7. Selected bond distances and angles are given in Table 1. Fig. 1 shows the structure of 2, which displays pseudo-octahedral coordination of the molybdenum centre.
| Mo–N | 1.827(3) | Mo–P(1) | 2.4566(10) |
| Mo–H | 2.06(4) | Mo–P(2) | 2.4629(10) |
| N–O | 1.218(4) | Mo–P(3) | 2.4597(10) |
| Mo–P(4) | 2.4375(10) | ||
| N–Mo–H | 175.7(11) | P(4)–Mo–P(2) | 147.96(3) |
| O–N–Mo | 176.2(3) | P(4)–Mo–P(1) | 91.89(3) |
| P(1)–Mo–H | 92.1(11) | P(4)–Mo–P(3) | 91.01(3) |
| P(2)–Mo–H | 70.8(11) | P(1)–Mo–P(3) | 169.25(3) |
| P(3)–Mo–H | 98.6(11) | N–Mo–P(1) | 83.61(11) |
| P(4)–Mo–H | 77.2(11) | N–Mo–P(2) | 109.31(11) |
| P(1)–Mo–P(2) | 91.38(3) | N–Mo–P(3) | 85.66(11) |
| P(3)–Mo–P(2) | 91.65(3) | N–Mo–P(4) | 102.73(11) |
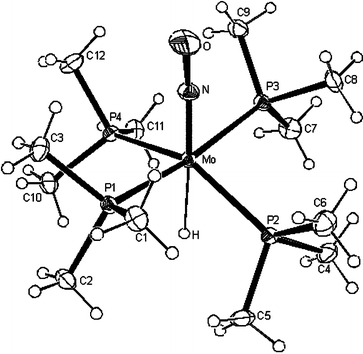 | ||
| Fig. 1 ORTEP plot of the structure of 2. Displacement ellipsoids are drawn with 30% probability. | ||
The hydride is located trans to the nitrosyl ligand. Two trans phosphine ligands P(1) and P(3) were found pointing toward the hydride ligand, whilst the other two phosphine ligands P(2) and P(4) are bent up toward the nitrosyl group. The H atom was localized in the difference Fourier maps and could be refined with an isotropic displacement parameter of 0.021(10) Å2. Thus, it is believed that the Mo bound H position can be taken with confidence. It is noteworthy that the Mo–H bond is quite long (2.06(4) Å) in comparison with that of mer-Mo(H)(CO)(NO)(PMe3)3 (1.84(3) Å).16
Ionicity of the Mo–H bond of trans-Mo(PMe3)4(H)(NO) (2)
LnM–H bonds can be described as polar covalent linkages, in which a charge distribution is imposed on an essentially covalent bond between the transition metal and the hydrogen atom. Bond ionicities have been used to determine the hydridic character of M–H bonds.21 The bond ionicity of the Mo–H bond of 2 was studied by deuterium quadrupole coupling constant (DQCC) measurements.21 The direction of the polarization of such bonds can principally not be obtained by such experiments, however based on electronegativity it seems reasonable to assume that it is hydridic. It is also expected that the bond ionicities i remain the same in the case of an isotopic replacement.In the 2H NMR spectra (in toluene-h8) the resonance for 2a is observed at −2.60 ppm (2JPD = 5 Hz). The 31P {1H} NMR spectra (in toluene-h8) shows a 1 : 1 : 1 triplet at 4.23 ppm and JDP is 5 Hz.
The DQCC values were first attempted to be determined from 2H NMR T1 min measurements in toluene and then from a static solid-state 2H NMR spectrum.21,22 The plot of lnT1vs. 1/T is shown in Fig. 2, which allowed for the calculation of T1 min = 55 ms at 188 K and subsequently a DQCC value of 45.5 kHz. The ionicity i of the Mo–D bond is 80.0%.
 | ||
| Fig. 2 Variable-temperature 2H T1 data (T1 in ms) of 2a in toluene-d8. | ||
The bond ionicitiy i of 2a could be determined from a static solid-state 2H NMR spectrum. A Δ value of the “Pake Doublet” of 42 kHz was derived from a solid-state 2H NMR spectrum, which allowed for the calculation of a DQCC value of 56 kHz, and a bond ionicity i of 75.3%. The deviating bond ionicities in solution and solid-state is presumably due to the non-zero asymmetry parameter in the solid state.23
The smallest experimental DQCC value was found for LiD (33 kHz), corresponding to a bond ionicity of 86%.24 The large i value for 2a, which is close to that of LiD, demonstrates a high ionicity of the Mo–D bond in 2a. The i value is greater than that of its congener W(D)(NO)(PMe3)4 (i = 77.6%),25 which mirrors the fact that the electronegativity of Mo is smaller than that of W. Compared with the bond ionicity of mer-Mo(H)(CO)(NO)(PMe3)3 (78.5%),26 the stronger bond polarity of 2 is expected to give rise to enhanced reactivity.
Insertion reactions of trans-Mo(PMe3)4(H)(NO) (2) with ketones and CO2
Insertion reactions of 2 were attempted with acetone, acetophenone and bezophenone at room temperature affording the corresponding alkoxide complexes 3, 4 and 5 (Scheme 2).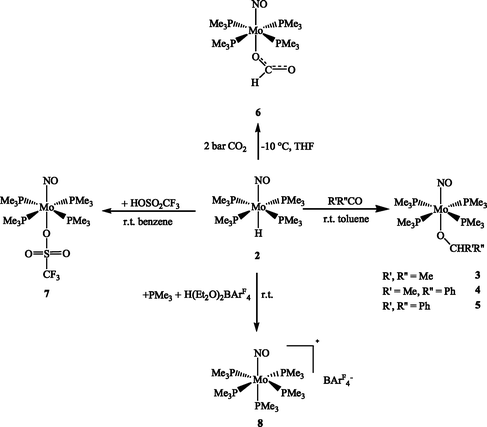 | ||
| Scheme 2 | ||
Monitored by NMR spectroscopy in toluene-d8 solution at room temperature, the reaction of 2 with 1 equivalent of acetone provided the isopropoxide 3 within 30 min. After a longer reaction time signals of an as yet unidentified product appeared. 3 was isolated as an analytically pure yellow solid in almost quantitative yield. Similarly the reaction of 2 with 1 equivalent of acetophenone or benzophenone in toluene took at room temperature about 4 or 10 hours for completion. The yellow alkoxides 4 and 5 were obtained in 85% and almost quantitative yield, respectively.
The IR specta of 3, 4 and 5 revealed in the solid-state bands at 1500 cm−1, 1508 cm−1 and 1502 cm−1, corresponding to their ν(NO) bands. In the 1H NMR spectra of 3, 4 and 5 (benzene-d6) the characteristic resonance of the newly formed CH moieties were observed at 3.70 ppm (sept., 3JHH = 6 Hz), 4.51 ppm (quart., 3JPH = 6 Hz) and 5.35 ppm (s, 3JHH = 6 Hz), respectively. In the 13C {1H} NMR spectra these CH carbon atoms gave rise to characteristic multiplets or a singlet at 68.9 ppm (3), 77.4 ppm (4) and 87.9 ppm (5). The 31P {1H} NMR spectra of 3, 4 and 5 showed singlets at −5.3 ppm, −5.3 ppm and −5.6 ppm, respectively. All complexes gave the correct elemental analyses.
Suitable crystals for an X-ray diffraction study were obtained from 5 by recrystallization from pentane at −30 °C. The structure was determined by X-ray diffraction and is shown in Fig. 3. Selected bond lengths and angles are listed in Table 2. The X-ray data collection and processing parameters are compiled in Table 7.
| Mo(1)–N(1) | 1.7803(16) | N(1)–O(1) | 1.227(2) |
| Mo(1)–O(2) | 2.1025(14) | O(2)–C(1) | 1.375(2) |
| Mo(1)–P(1) | 2.5083(5) | C(1)–C(2) | 1.536(3) |
| Mo(1)–P(2) | 2.4979(5) | C(1)–C(8) | 1.542(3) |
| Mo(1)–P(3) | 2.4840(5) | C(1)–H(1) | 0.9800 |
| Mo(1)–P(4) | 2.4872(6) | ||
| N(1)–Mo(1)–O(2) | 172.09(7) | O(1)–N(1)–Mo(1) | 176.75(18) |
| N(1)–Mo(1)–P(3) | 95.37(5) | C(1)–O(2)–Mo(1) | 135.93(14) |
| O(2)–Mo(1)–P(3) | 79.27(5) | O(2)–C(1)–C(2) | 109.96(17) |
| N(1)–Mo(1)–P(4) | 84.07(6) | O(2)–C(1)–C(8) | 110.54(17) |
| O(2)–Mo(1)–P(4) | 101.60(5) | C(2)–C(1)–C(8) | 110.30(15) |
| P(3)–Mo(1)–P(4) | 89.78(2) | O(2)–C(1)–H(1) | 108.7 |
| N(1)–Mo(1)–P(2) | 82.20(6) | C(2)–C(1)–H(1) | 108.7 |
| O(2)–Mo(1)–P(2) | 91.96(5) | C(8)–C(1)–H(1) | 108.7 |
| P(3)–Mo(1)–P(2) | 90.37(2) | O(2)–Mo(1)–P(1) | 89.25(5) |
| P(4)–Mo(1)–P(2) | 166.229(19) | P(3)–Mo(1)–P(1) | 168.288(18) |
| N(1)–Mo(1)–P(1) | 96.28(5) | P(4)–Mo(1)–P(1) | 90.22(2) |
| P(2)–Mo(1)–P(1) | 92.396(19) | ||
 | ||
| Fig. 3 ORTEP plot of the structure of 5. Displacement ellipsoids are drawn with 30% probability. | ||
CO2 insertion into the metal–hydride bond to generate a metal formato-O-complex has been extensively studied, in particular with regard to the hydrogenation of CO2 to formic acid.27
The reaction of 2 with 2 bar of CO2 afforded at −10 °C the formato-O-complex Mo(η1-OCHO)(PMe3)4(NO) (6) within 1 h (Scheme 2), which can be isolated as analytically pure brown crystals in 95% yield from diethyl ether at −30 °C. Full characterization was achieved by IR, NMR, elemental analysis and mass spectroscopy, which confirmed the η1-formato structure. The IR spectrum of 6 revealed two characteristic bands: an intense ν(NO) band at 1533 cm−1 and a ν(OCO) band at 1626 cm−1.14,16 In the 1H NMR spectrum in THF-d8 the formato group of 6 displayed a singlet at 8.06 ppm and in the 13C {1H} NMR spectrum the resonance of the respective carbon atom was observed as a singlet at 166.1 ppm.
Reactions of trans-Mo(PMe3)4(H)(NO) (2) with acids
Coordinatively unsaturated cationic species, as well as cationic complexes of weakly coordinating anions and solvent molecules are particularly useful in Lewis acidic catalysis and organometallic synthesis. The Mo–H bond of 2 was shown to possess a hydridic H atom, which may in turn point at the existence of a stabilized 16e− [Mo(PMe3)4(NO)]+ cation. Removal of the hydride anion from hydride complexes is one of the routes generating such electrophilic cationic species with weakly coordinating or non-coordinating anions. Therefore the reactions of 2 with the strong acids HOSO2CF3 and [H(Et2O)2][BArF4] were studied.The reaction of 2 with 1 equivalent of HOSO2CF3 was monitored by NMR spectroscopy in benzene-d6. It was completed immediately and H2 was released. The triflate complex 7 was obtained in quantitative yield (Scheme 2). Complex 7 has been fully characterized by elemental analyses, IR, NMR spectroscopy and mass spectrometry, which revealed a pseudo-octahedral structure with a coordinated triflate anion. The IR spectrum in the solid state shows a band at 1551 cm−1 corresponding to the ν(NO) vibration. In the 1H NMR spectrum of 7 (benzene-d6) reveals a singlet resonance for the PMe3 group at 1.24 ppm. The 31P {1H} NMR spectrum displays a singlet at −3.2 ppm corresponding to the four chemically equivalent phosphorus atoms. The triflate ligand tends to be labile in coordination compounds, for that reason it was tested whether an intermediate 16e− species would exist, persistent enough to allow further reactions. 7 did however not react under hydrogenation conditions with H2 and acetophenone, even under a pressure of 80 atm H2. The OSO2CF3− anion seems to be too strongly coordinated to the Mo centre forming a relatively stable 18 electron compound.
The reaction of 2 with 1 equivalent of [H(Et2O)2][BArF4] at room temperature was monitored by NMR spectroscopy in diethyl ether-d10. The reaction was completed within 3 hours (Scheme 2). The 1H NMR spectrum shows a singlet at 1.52 ppm and a doublet at 1.28 ppm in a 4 : 1 ratio. Accordingly, the 31P {1H} NMR spectrum revealed a doublet at −11.6 ppm and a quintet at −35.9 ppm with a 4 : 1 ratio, which have the same coupling constant (26 Hz). This indicated the formation of an octahedral molecule with five PMe3 ligands, four PMe3 ligands are in one planar arrangement, while the other PMe3 group is located trans to the nitrosyl ligand. Suitable crystals for X-ray diffraction analysis were obtained by pentane diffusion into the diethyl ether solution of the reaction mixture. The isolated yield of 8 was approximately 30%. The X-ray data collection and processing parameters are compiled in Table 7. A structural model is displayed in Fig. 4. The Mo centre possesses a pseudo-octahedral geometry; the bond length of Mo–P4 (trans to NO) is a little longer than the other Mo–P bonds (see Table 3). This is supposed to be due to the trans influence of the NO group.
 | ||
| Fig. 4 ORTEP plot of the structure of 8. Only the cation is shown. Displacement ellipsoids are drawn with 30% probability. | ||
| Mo(1)–N(1) | 1.778(6) | Mo(1)–P(3) | 2.519(2) |
| N(1)–O(1) | 1.187(8) | Mo(1)–P(5) | 2.506(3) |
| Mo(1)–P(1) | 2.543(2) | Mo(1)–P(4) | 2.682(2) |
| Mo(1)–P(2) | 2.499(3) | ||
| N(1)–Mo(1)–P(1) | 83.1(2) | P(1)–Mo(1)–P(5) | 88.43(9) |
| N(1)–Mo(1)–P(2) | 89.4(2) | P(2)–Mo(1)–P(3) | 93.66(11) |
| N(1)–Mo(1)–P(3) | 86.6(2) | P(2)–Mo(1)–P(4) | 86.60(9) |
| N(1)–Mo(1)–P(4) | 175.8(2) | P(2)–Mo(1)–P(5) | 176.31(13) |
| N(1)–Mo(1)–P(5) | 93.6(2) | P(3)–Mo(1)–P(4) | 92.57(7) |
| P(1)–Mo(1)–P(2) | 89.73(8) | P(3)–Mo(1)–P(5) | 88.72(10) |
| P(1)–Mo(1)–P(3) | 169.14(7) | P(4)–Mo(1)–P(5) | 90.48(9) |
| P(1)–Mo(1)–P(4) | 97.93(7) | O(1)–N(1)–Mo(1) | 179.7(7) |
When the reaction was carried out in the presence of 5 equivalents of PMe3, 8 was obtained almost quantitatively and the reaction was complete within 10 minutes at room temperature. During this period of time 8 partly precipitated from solution. By this route 8 was obtained as orange crystals by diffusion of pentane into the reaction mixture. The solid state IR spectrum of 8 showed a band at 1593 cm−1 corresponding to the ν(NO) vibration. The elemental analysis is consistent with the composition of the complex.
Insertion reactions of trans-Mo(PMe3)4(H)(NO) (2) with imines
The insertion of imines into transition metal–hydride bonds is assumed to be the crucial step in various catalytic hydrogenations of imines generating amines,28–34 but it is difficult to isolate this step.16 Recently insertion reactions of the hydride trans-Mo(dmpe)2(H)(NO) with imines were reported. Disubstitued aromatic imines were shown to insert into its Mo–H bond.35 Therefore, the insertion capability of 2 was studied with several imines Ph(Me)CNPh, Ph2CNH, PhCHNPh, PhCHNC10H7 and C10H7CHNPh. The C-disubstituted imines showed no reaction at room temperature and even at 60 °C. The other imines inserted into the Mo–H bond to form amido complexes, but in most cases these were not stable and underwent further transformations.Treatment of 2 with an equimolar amount of N-benzylideneaniline in toluene-d8 at room temperature afforded after 3 days the amido complex 9 (Scheme 3). In the 1H NMR spectrum a characteristic singlet at 4.42 ppm was observed for the newly formed NCH2 group. In the 31P {1H} NMR spectrum of 9 a very broad signal at −9.0 ppm was found at room temperature, which became sharp and shifted to −7.1 ppm when the sample was cooled to −50 °C. This presumably indicates reversible PMe3 dissociation at room temperature.
 | ||
| Scheme 3 | ||
The reaction of 2 with N-benzylidene-1-naphthylamine was monitored by 1H NMR spectroscopy in toluene-d8 at room temperature. After 3 days the reaction is almost complete. The C–H agostic tris(trimethylphosphine) complex 10 was isolated in 59% yield (Scheme 3). The structure of 10 was assigned based on NMR spectroscopy. The 31P {1H} NMR spectrum of 10 at room temperature displays a triplet at 16.7 ppm (2JPP = 20 Hz) and a broadened signal at −5.4 ppm, indicating the meridional disposition of the three phosphine ligands. When the 31P {1H} NMR spectrum is recorded at −70 °C, the broadened signal splits into two doublet of doublet patterns at 1.2 and −8.4 ppm with 2JPP-cis = 20 Hz, 2JPP-trans = 190 Hz couplings, respectively. In the 1H NMR spectrum there is a singlet at 4.43 ppm corresponding to the resonance of the newly formed NCH2 group and a broadened signal appearing at 2.97 ppm. From the 13C–1H correlation spectrum (Fig. S.1, ESI†) it was derived that this signal possesses correlation with a 13C resonance having a chemical shift of 117.0 ppm in the aromatic range. The drastic upfield shift of this proton resonance supports its agostic nature.36 Further evidence for the agostic interaction comes from 1H {31P} NMR spectroscopy. In this spectrum the resonance at 2.97 ppm is sharp and split into a doublet due to coupling with the neighboringing aromatic proton (2JHH = 5 Hz). The broadening is interpreted in terms of an additional coupling to phosphorus nuclei through the agostic bond.
From the gated 13C NMR spectrum of 10 (Fig. S.2, ESI†), the 1JCH of the agostic proton is determined to be 125 Hz, significantly lower than usual aromatic 1JCH couplings (155–165 Hz). The coupling constant is reduced by about 30–35 Hz, which is a typical value for an agostic interaction. The ν(CH) bond of the agostic C–H bond could not be detected by IR or Raman spectroscopy. The ν(NO) absorption appears at 1557 cm−1 in the solid state IR spectrum, and at 1558 cm−1 in the solid state Raman spectrum.
The reaction of 2 with N-1-naphthylideneaniline, an isomer of N-benzylidene-1-naphthylamine, has been monitored by NMR spectroscopy in toluene-d8 at room temperature. After 4 days the reaction was almost complete and showed the amido complex 11 as the only product. Beside signals for the aromatic protons of the amido moiety appearing in the range 6.10–9.12 ppm and a singlet for the methyl protons of the PMe3 ligands at 1.12 ppm, the 1H NMR spectrum of the reaction mixture in toluene-d8 revealed the characteristic resonance of the NCH2 group as a singlet at 4.89 ppm. In the 31P {1H} NMR spectrum a very broad signal at −9.0 ppm was observed at room temperature. The signal appeared at −7.6 ppm and became sharp at −30 °C suggesting that there is ligand dissociation as in 9. 11 however could not be isolated, because upon crystallization it lost a PMe3 ligand to form the agostic complex 12 (Scheme 4). 12 was obtained as red crystals in 45% yield by diffusion of pentane into a saturated toluene solution. Similar to 10, the agostic ν(CH) band of 12 could not be observed in the IR or Raman spectra. In the solid-state IR spectrum a band at 1578 cm−1 is assigned to the ν(NO) vibration.
 | ||
| Scheme 4 | ||
The 31P {1H} NMR spectrum of 12 (toluene-d8) displayed at room temperature very broadened signals, which became sharp at low temperature. In the 31P {1H} NMR spectrum at −60 °C a doublet and a triplet resonance appeared at −8.0 and 33.1 ppm (2JPP = 17 Hz) typical for a meridonal phosphine substitution pattern. In the 1H NMR spectrum the agostic proton appeared at 3.29 ppm and was also identified in a 13C–1H correlation spectrum (Fig. S.3, ESI†). The corresponding “agostic” 1H NMR signal shows correlation with a 13C NMR resonance in the aromatic region (91.5 ppm). A 1JCH of 131 Hz is determined for the agostic proton. The agostic interaction causes the coupling constant to be reduced by about 25–30 Hz.
The molecular structure of 12 was determined by an X-ray diffraction analysis. Suitable crystals were obtained by diffusion of pentane into a concentrated toluene solution of 12. The ORTEP plot is shown in Fig. 5. Selected bond lengths and bond angles are listed in Table 4. The X-ray data collection and processing parameters are compiled in Table 7. The Mo centre possesses a pseudo-octahedral geometry: the amido group is located trans to the nitrosyl ligand and the 5-phenyl proton (H15) in involved in the agostic interaction with the molybdenum centre. The agostic H is trans to a phosphorus atom, forming an angle of 166° (P2–Mo–H15). The distance between H15 and the Mo centre is 2.11 Å, and of C15 and Mo is 2.68 Å, the angle of the agostic hydrogen C15–H15–Mo is 117°. All these structural data are typical for agostic C–H bonding.37
| Mo(1)–N(1) | 1.783(3) | N(2)–C(10) | 1.386(5) |
| Mo(1)–N(2) | 2.193(3) | N(2)–C(161) | 1.448(14) |
| Mo(1)–P(1) | 2.5044(11) | N(2)–C(162) | 1.456(9) |
| Mo(1)–P(2) | 2.4134(10) | C(161)–C(17) | 1.585(19) |
| Mo(1)–P(3) | 2.5036(11) | C(162)–C(17) | 1.487(11) |
| N(1)–O(1) | 1.232(4) | ||
| N(1)–Mo(1)–N(2) | 166.98(12) | P(2)–Mo(1)–P(3) | 93.89(4) |
| N(1)–Mo(1)–P(1) | 91.95(10) | C(10)–N(2)–Mo(1) | 100.9(2) |
| N(1)–Mo(1)–P(2) | 92.33(10) | C(161)–N(2)–Mo(1) | 130.9(7) |
| N(1)–Mo(1)–P(3) | 84.45(10) | C(162)–N(2)–Mo(1) | 127.6(4) |
| N(2)–Mo(1)–P(1) | 90.60(8) | C(10)–N(2)–C(161) | 110.6(14) |
| N(2)–Mo(1)–P(2) | 100.31(8) | C(10)–N(2)–C(162) | 121.9(7) |
| N(2)–Mo(1)–P(3) | 91.51(8) | N(2)–C(161)–C(17) | 113.9(13) |
| P(1)–Mo(1)–P(2) | 92.64(4) | N(2)–C(162)–C(17) | 119.6(6) |
| P(1)–Mo(1)–P(3) | 172.67(4) | ||
 | ||
| Fig. 5 ORTEP plot of the structure of 12. Displacement ellipsoids are drawn with 10% probability. | ||
When the reaction of 2 was carried out at 60 °C with 1 equivalent of N-1-naphthylideneaniline the amino complex 15 was obtained within 2 weeks. The 31P {1H} NMR spectrum of 15 displayed a doublet and a triplet at −10.1 ppm and −19.5 ppm (2JPP = 23 Hz), corresponding to a meridional arrangement of the three phosphine ligands. The 1H NMR spectrum of 15 (benzene-d6) showed besides resonances for the aromatic protons in the range 9.03 to 6.42 ppm and those for the PMe3 ligand in the range 1.02 to 0.98 ppm, a characteristic doublet at 4.32 ppm (2JHH = 5 Hz) attributed to the NCH2 group. In the 13C {1H} NMR spectrum the resonance of the respective carbon atom appears as a singlet at 46.3 ppm. The 1H NMR spectrum of 15 displays another broadened signal at 3.3 ppm, which can be assigned to the NH group. This assignment was further supported by a NOE experiment. When the signal at 3.3 ppm was irradiated, apparently positive NOE signals were detected at 6.42 ppm and 4.32 ppm, which were attributed to the resonance for the o-Ph and the NCH2 protons.
Complex 15 could only be isolated with some impurities, thus preventing its full spectroscopic characterization. However a single crystal was picked out, which allowed its structure to be established by X-ray diffraction analysis. The ORTEP plot is shown in Fig. 6. Selected bond lengths and bond angles are listed in Table 5. The X-ray data collection and processing parameters are compiled in Table 7.
| Mo(1)–N(1) | 1.7819(18) | N(1)–O(1) | 1.224(2) |
| Mo(1)–C(2) | 2.112(2) | N(2)–C(11) | 1.315(3) |
| Mo(1)–N(2) | 2.2551(17) | N(2)–C(12) | 1.420(3) |
| Mo(1)–P(1) | 2.4835(6) | N(2)–H(2) | 0.9300 |
| Mo(1)–P(2) | 2.5674(6) | C(1)–C(11) | 1.416(3) |
| Mo(1)–P(3) | 2.4945(6) | C(1)–C(2) | 1.427(3) |
| N(1)–Mo(1)–C(2) | 95.02(8) | N(2)–C(11)–C(1) | 118.5(2) |
| N(1)–Mo(1)–N(2) | 168.82(7) | C(11)–N(2)–C(12) | 118.46(19) |
| N(1)–Mo(1)–P(1) | 90.13(6) | C(11)–N(2)–Mo(1) | 115.16(14) |
| N(1)–Mo(1)–P(2) | 92.09(6) | C(12)–N(2)–Mo(1) | 126.32(14) |
| N(1)–Mo(1)–P(3) | 91.11(6) | C(11)–N(2)–H(2) | 90.8 |
| N(2)–Mo(1)–P(1) | 87.40(5) | C(12)–N(2)–H(2) | 90.8 |
| N(2)–Mo(1)–P(2) | 99.07(5) | Mo(1)–N(2)–H(2) | 90.8 |
| N(2)–Mo(1)–P(3) | 89.37(5) | C(11)–C(1)–C(2) | 113.99(19) |
| P(1)–Mo(1)–P(2) | 99.52(2) | C(11)–C(1)–C(9) | 123.1(2) |
| P(1)–Mo(1)–P(3) | 169.48(2) | C(2)–C(1)–C(9) | 122.9(2) |
| C(2)–Mo(1)–N(2) | 73.94(7) | C(1)–C(2)–C(3) | 115.40(19) |
| C(2)–Mo(1)–P(1) | 85.67(6) | C(1)–C(2)–Mo(1) | 118.18(15) |
| C(2)–Mo(1)–P(2) | 171.20(6) | C(3)–C(2)–Mo(1) | 126.33(16) |
| C(2)–Mo(1)–P(3) | 83.81(6) | C(13)–C(12)–N(2) | 119.1(2) |
| P(3)–Mo(1)–P(2) | 90.88(2) | C(17)–C(12)–N(2) | 121.8(2) |
| O(1)–N(1)–Mo(1) | 176.07(17) | ||
 | ||
| Fig. 6 ORTEP plot of the structure of 15. Displacement ellipsoids are drawn with 30% probability. | ||
Scheme 4 presents a plausible pathway for the formation of 15. The first step is imine insertion into the Mo–H bond to form the amido complex 11, which subsequently loses one PMe3 ligand and establishes an equilibrium with the agostic complex 12. When the system was heated to 60 °C, other equilibria are assumed to come into play leading to the agostic species 13. Oxidative addition of the agostic C–H bond produces 14 and H transfer to the amide ligand eventually gives 15. The intermediates 13 and 14 were not observed. However, an analogous seven-coordinate hydride species has been isolated from the reaction of a related tungsten hydride W(NO)(H)(PMe3)4 with N-benzylidene-1-naphthylamine.28 This might point to the fact that in the molybdenum hydride case, the related seven-coordinated hydride complex is less stable and is immediately transformed to 15.
Investigations on the homogeneous hydrogenation of imines
The transition-metal catalyzed hydrogenation of imines is more difficult to achieve than that of olefins and ketones.37 There are only a few imine hydrogenation systems, based on Rh(I),38–46 Ir(I),45,47–56 Ru(II)57–65 and titanocene catalysts,27,28,66,67 since the catalytic generation of amines is quite often circumvented by the hydrogenation of enamines. H2 activation by transition metal complexes is well understood, but relatively little is known about the activation of imines and the required H-atom transfer steps. Several factors effect the hydrogenation of imines.33 First, there is a smaller thermodynamic gain from the reduction of CN bonds (approx. −60 kJ mol−1) relative to CC bonds (approx. −130 kJ mol−1). Secondly, the typical end-on η1 binding mode of the azomethine groups contrasts with the side-on η2 binding of olefins, which results in less effective orbital overlaps with the metal centre. Thirdly there is competitive coordination with the products of the hydrogenation (amines) causing ‘catalyst self poisoning’.68,69The reactions of complexes 10 and 12 with H2 have been investigated applying various conditions. 10 and 12 can be viewed as a 16 electron species stabilized by an agostic interaction. The relative stability of these coordinatively unsaturated species challenged us to investigate their potential for the catalytic hydrogenation of respective imines.
Stoichiometric hydrogenation of N-benzylidene-1-naphthylamine and N-1-naphthylideneaniline
The reaction of 10 with H2 (1.5 bar), has been monitored by NMR spectroscopy in toluene-d8 at room temperature and also at −30 °C with the same results for both experiments. After one day, the 1H NMR spectrum of the reaction mixture revealed a doublet at 4.08 ppm (3JHH = 5 Hz) and a broadened signal at 4.52 ppm, which corresponded to NCH2 and NH resonances of the formed N-[(1-naphthyl)methyl]aniline (C10H7NHCH2Ph). The presence of amine was also proven by MS. The 31P {1H} NMR spectrum of the reaction mixture showed besides the resonance of 10 also singlets at 32.2 and −61.3 ppm, which indicated that under these conditions 10 partly decomposed to release PMe3 and unidentifiable products. After 2 days, the resonances of 10 disappeared in the 1H and 31P {1H} NMR spectra. Only the resonances of a decomposed species (32.2 ppm) and of free PMe3 (−61.3 ppm) remained in the 31P {1H} NMR spectrum.The insertion reaction of 2 with N-benzylidene-1-naphthylamine to yield 10 and 1 equivalent of PMe3 took 3 days. This reaction mixture was treated with 1.5 bar H2, which is related to the reaction of 10 with H2 in the presence of 1 equivalent of PMe3 and was then monitored by NMR spectroscopy in toluene-d8 at room temperature. After one day, the 1H NMR spectrum showed the resonances of the amine C10H7NHCH2Ph and a characteristic quintet of the hydride 2 at −2.50 ppm (3JPH = 33 Hz). The 31P {1H} NMR spectrum of the reaction mixture displayed besides the resonances of 2 and 10 also resonances of decomposition products and PMe3. After still longer reaction time (>4 day), the resonances of 2 increased and those of 10 disappeared in the 1H and 31P {1H} NMR spectra.
The latter reaction has also been monitored by NMR spectroscopy at −30 °C. The result was different from the reaction at room temperature. In the 1H and 31P {1H} NMR spectra only resonances of the amine product (C10H7NHCH2Ph) and of the decomposition species were observed.
Based on the fact that only the reaction of 10 with H2 in the presence of additional PMe3 at room temperature afforded the amine and 2, a possible reaction pathway could be established. According to Scheme 5 the agostic complex 10 reacts with H2 to form either the dihydride or the dihydrogen intermediate 16. Heterolysis with H+ transfer37 leads to the amine species 17 from which the amine is supposed to be replaced by PMe3 giving 2. 16 and 17 could not be detected in the NMR spectra. In particular 17 is suspected to be so short-lived that it rapidly decomposes to the mentioned unidentifiable species, when PMe3 is not present for its stabilization.
 | ||
| Scheme 5 | ||
The reactions of 12 with H2 (1.5 bar) in the presence of and without additional PMe3 have also been studied by NMR spectroscopy in toluene-d8 at room temperature. They showed the same results. After one day the reaction mixtures revealed in their 1H NMR spectrum a doublet at 4.25 (3JHH = 5 Hz) besides a characteristic quintet of hydride 2 at −2.50 ppm (3JPH = 33 Hz) and a broadened signal at 3.33 ppm corresponding to the resonance of the NCH2 and NH protons of the benzylnaphth-1-ylamine (PhNHCH2C10H7). Its presence was additionally proven by GC-MS. In the 31P {1H} NMR spectrum the resonance of 2 was observed at 5.0 ppm.
Similar to 10 the reaction of the agostic species 12 with H2 was supposed to proceed via the H2 complex 18 and the amine species 19 (Scheme 5). The substitution of the amine ligand of 19 for PMe3 is supposed to be more facile than in 17 revealing 2 as the main NMR detectable product.
Catalytic hydrogenation of N-benzylidene-1-naphthylamine and N-1-naphthylideneaniline
Based on the fact that the reactions of 10 or 12 with H2 produced the corresponding amine and the hydride complex 2, the optimization of these hydrogenations of N-benzylidene-1-naphthylamine and of N-1-naphthylideneaniline in terms of catalysis were of great interest. These reactions have therefore been tested under various conditions. Table 6 shows results of the hydrogenation pursued by GC.| Substrate | Catalyst | Pressure/bar | Temperature/°C | Conversion (%) |
|---|---|---|---|---|
| a 5 mol%.b Mix 5 mol% of 2 and C10H7NCHPh at r.t. for 3 days.c 5 mol% of 2 and 25 mol% of PMe3.d 5 mol%.e Mix 5 mol% of 2 and PhNCHC10H7 at r.t. for 3 days.f 5 mol% of 2 and 25 mol% of PMe3.g 5 mol% of 2 and 100 mol% of PMe3. | ||||
| C10H7NCHPh | 2a | 60 | r.t. | 7.1 |
| 10a | 60 | 60 | 6.4 | |
| 2a | 60 | 60 | 13.8 | |
| 10 + PMe3b | 60 | 60 | 7.2 | |
| 2a | 120 | r.t. | 5.3 | |
| 2a | 120 | 60 | 15.5 | |
| 2 + PMe3c | 120 | 60 | 18.4 | |
| PhNCHC10H7 | 2d | 60 | r.t | 5.1 |
| 12d | 60 | 60 | 5.1 | |
| 2d | 60 | 60 | 16.2 | |
| 12 + PMe3e | 60 | 60 | 14.4 | |
| 2d | 120 | r.t. | 4.2 | |
| 2d | 120 | 60 | 18.3 | |
| 2 + PMe3f | 120 | 60 | 85.6 | |
| 2 + PMe3g | 120 | 60 | 100 | |
From Table 6 one can derive that the catalytic hydrogenation reactions of both PhCHNC10H7 and C10H7CHNPh are slow at room temperature. Highest conversion rates are 15.5% for C10H7NCHPh and 18.3% for PhNCHC10H7. At 60 °C the catalyses showed higher conversions.
In the proposed catalytic cycle of Scheme 5, PMe3 was believed to stabilize the intermediates 16, 17, 18 and 19. Therefore if additional PMe3 is added into the reaction system, higher catalyst stabilities were found. Therefore experiments were carried out in the presence of excess of PMe3, which produced satisfactory results. 5 equivalents of PMe3 H2 (pressure of 120 bar and a temperature of 60 °C) brought about conversions of 18.6% and 85.6% for C10H7NCHPh and PhNCHC10H7, respectively. With 20 equivalents of PMe3, 120 bar H2 pressure and 60 °C even 100% conversion of PhNCHC10H7 was reached.
Conclusions
The tetrakis(phosphine)-substituted molybdenum hydride 2 displays a Mo–H bond with a high bond ionicity value of 80.0%. Presumably based on this, 2 shows a very high propensity for hydride transfer to ketones, CO2 and imines. Due to facile loss of PMe3 catalysis of imine hydrogenation could become reality. The catalytic hydrogenation of N-benzylidene-1-naphthylamine (C10H7NCHPh) and N-1-naphthylideneaniline (PhNCHC10H7) with 2 (5 mol%) have thus been investigated under various conditions. 100% conversion was observed for PhNCHC10H7 in the presence of an excess of PMe3.Experimental
General procedures
All reactions and manipulations with air-sensitive compounds were performed under an atmosphere of dry nitrogen using conventional Schlenk techniques or a glovebox. Solvents were dried by standard methods and freshly distilled under nitrogen before use (e.g., pentane, hexane, diethyl ether, benzene, toluene, THF were purified by reflux over sodium/benzophenone, dichloromethane and acetonitrile were refluxed over P2O5). The deuterated solvents were also dried over appropriate drying agents and vacuum transferred before use. NMR spectra were recorded on the following spectrometers. Varian Gemini-300 instrument; 1H at 300.1 MHz, 13C at 75.4 MHz, 31P at 121.5 MHz, 19F at 282.2 MHz. Varian Gemini-200 instrument: 1H at 200.0 MHz, 13C at 50.3 MHz, 31P at 80.9 MHz, 19F at 188.2 MHz. Bruker DRX-500 instrument: 1H at 500.2 MHz, 2H at 76.7 MHz, 13C at 125.8 MHz, 31P at 202.5 MHz, 11B at 160.5 MHz. δ(1H), δ(13C) relative to SiMe4, δ(31P) relative to 85% H3PO4, δ(19F) relative to trifluorotoluene and δ(11B) rel to BF3·OEt2. IR spectra: Biorad FTS-45 and FTS-3500 instrument. Raman spectra: Renishaw Labram Raman microscope. Mass spectra: Finnigan-MAT-8400 spectrometer. Elemental analyses: Leco CHN(S)-932 instrument. GC-MS: Varian CP-3800 instrument; MS: Saturn 2000. The compounds Mo(NO)Cl3(CH3CN)2,19,70N-1-naphthylideneaniline,71N-benzenylidene-1-naphthylamine,71 [H(Et2O)][BArF4],72trans-Mo(CO)4(NO)(ClAlCl3)73 were prepared according to literature procedures. Other reagents were obtained from commercial supplies.Syntheses
At room temperature. A mixture of 0.032 g (0.074 mmol) of 2 and 0.016 g (0.069 mmol) of N-1-naphthylideneaniline was dissolved in ca. 0.7 ml of toluene-d8 in an NMR tube. The reaction was monitored by NMR at room temperature. After 4 days the reaction was complete (1H and 31P NMR monitoring). The solvent was removed in vacuo, the residue was dissolved in ca. 0.1 ml toluene and diffusion of pentane into the toluene solution afforded 12 as red crystals. Yield: 0.013 g (45%). IR (cm−1, ATR): 1578 (s) (NO). Raman (cm−1): 1582 (NO). 1H NMR (toluene-d8, 500.2 MHz, −60 °C): δ 8.11 (d, 2JHH = 8 Hz, 1H, 8-naphthyl), 7.61 (d, 2JHH = 8 Hz, 1H, 5-naphthyl), 7.53 (d, 2JHH = 8 Hz, 1H, 2-naphthyl), 7.47 (d, 2JHH = 8 Hz, 1H, 4-naphthyl), 7.39 (t, 2JHH = 8 Hz, 1H, 7-naphthyl), 7.29 (t, 2JHH = 8 Hz, 1H, 6-naphthyl), 7.23 (d, 2JHH = 8 Hz, 1H, 3-Ph), 7.21 (t, 2JHH = 8 Hz, 1H, 3-naphthyl), 7.00 (m, 1H, 5-Ph), 6.58 (t, 2JHH = 8 Hz, 1H, 4-Ph), 5.87 (d, 2JHH = 8 Hz, 1H, 6-Ph), 4.74 (s, 2H, NCH2), 3.29 (br, 1H, 2-Ph), 0.98 (s, 18H, PMe3cis-agostic), 0.94 (m, 9H, PMe3trans-agostic). 13C {1H} NMR (toluene-d8, 125.8 MHz, −60 °C): δ 157.8 (s, i-Ph), 136.0 (s, 1-naphthyl), 135.6 (s, 3-Ph), 134.1 (s, 9-naphthyl), 131.7 (s, 10-naphthyl), 131.1 (s, 5-Ph), 129.4 (s, 5-naphthyl), 126.4 (s, 4-naphthyl), 125.7 (s, 3-naphthyl), 125.3 (s, 7-naphthyl), 125.2 (s, 6-naphthyl), 124.2 (s, 2-naphthyl), 122.4 (s, 8-naphthyl), 113.4 (s, 4-Ph), 108.9 (s, 6-Ph), 91.5 (s, br, 2-Ph), 51.4 (s, NCH2), 20.9 (m, PMe3trans-agostic), 17.6 (m, PMe3cis-agostic). 31P {1H} NMR (toluene-d8, 202.5 MHz, −60 °C): δ 33.1 (t, 2JPP = 17 Hz, trans-agostic), −8.0 (d, 2JPP = 17 Hz, PMe3cis-agostic). Anal. Calcd for C26H41MoN2OP3: C 53.25, H 7.05, N 4.78. found: C 53.63, H 7.14, N 4.87%.
At 60 °C. A 0.014 g (0.032 mmol) sample of 2 was dissolved in ca. 0.7 ml of toluene-d8 in an NMR tube then 0.008 g (0.034 mmol) of N-1-naphthylideneaniline was added. The resulting solution was heated to 60 °C. After 2 days the reaction was complete (1H and 31P NMR monitoring). The solvent was removed in vacuo and the remaining residue was extracted with pentane. The combined solutions were concentrated and cooled to −30 °C overnight, which gave a mixture of a brown solid and green-brown crystals. Single crystals suitable for a X-ray diffraction study could be picked out from the mixture, which was demonstrated to be 15. An analytically pure product could not be obtained.
Catalytic hydrogenations of imines with 2 under H2 pressure
These reactions were carried out as follows: a mixture of the catalyst 2 and PhCHNC10H7 or C10H7CHNPh was dissolved according to Table 6 in ca. 2 ml of toluene in a steel autoclave. The reaction vessel was then pressurized with 60 or 120 bar H2. The contents were stirred and heated for given temperature and periods of times. The mixtures were filtered over silica gel. The conversions were determined by GC.X-Ray crystal structure analyses of 2, 5, 8, 12 and 15
All crystals were grown and prepared under a nitrogen atmosphere in a glove box in order to avoid deterioration of these compounds by air and moisture. Before the crystals were sent out for selection, they were embedded in polybutene oil in the glove box. Good quality single crystals for the X-ray diffraction studies have been chosen under polarized light using a polarizing microscope. The selected single crystal was mounted on top of a glass fibre and fixed on a goniometer head, then it was transferred immediately to the Stoe IPDS diffractometer where the crystal was cooled to 183(2) K, using an Oxford Cryogenic System. Centreing of the crystal was performed with the support of a video camera, which is incorporated in the Stoe IPDS diffractometer and controlled by the computer terminal.X-Ray diffraction data were collected at 183(2) K using an imaging plate detector system (Stoe IPDS) with graphite monochromated Mo-Kα radiation. A total of 200, 218, 200, 200 and 300 images were exposed at constant times of 7.00, 1.00, 2.00, 2.20 and 1.50 min per image for 2, 5, 8, 12, 15 respectively. The crystal-to-image distances were set to 50.0 mm (60.0 mm for 8 and 15) (θmax = 30.29°, 30.34°, and 30.41° for 2, 5, and 12; θmax = 28.09° and 28.09° for 8 and 15). ϕ-oscillation (2) or rotation scan modes (5, 8, 12, 15) were selected for the ϕ increments of 1.3°, 1.1°, 1.2°, 1.4° and 0.7° per exposure for 2, 5, 8, 12, 15 respectively. Total exposure times for the five compounds were 37, 19, 21, 17 and 28 hours in the order of the complexes as given above. The intensities were integrated after using a dynamic peak profile analysis and estimated mosaic spread (EMS) check73 was performed to prevent overlapping intensities. 8000 reflections (7998 for 8) were selected out of the whole limiting sphere for the cell parameter refinements. A total of 16665, 44839, 33425, 23862 and 44150 reflections were collected, of which 6197, 9353, 14600, 8795 and 6985 reflections were unique (Rint = 5.93%, 4.43%, 6.28%, 7.44% and 5.05%); data reduction and numerical absorption correction used 7, 14, 9, 7 and 16 indexed crystal faces.74 All five structures were solved by the Patterson method using an improved version of SHELXS-9775 and refined with SHELXL-97.76 Compound 2 crystallizes in the non-centrosymetric achiral77 space group Cc (no. 9). Many structures published wrongly in space group Cc were detected by Baur et al.78 and Marsh.79,80 Compound 8 crystallizes in the non-centrosymmentric achiral space group Pn (no. 7, cell choice 2 of space group Pc), which is also a candidate for overlooked higher symmetry (e.g. space groups P2/n or P21/n). The program PLATON81 was used to find centrosymmetric symmetry for structure 2 (e.g. space group C2/c, no. 14) and structure 8 (e.g. P2/n, or P21/n, no. 13 or no. 14), however, no higher symmetry was found for both structures. The absolute structures of 2 and 8 and the chiral structure of 12 (space group P21) were determined by using Flack's X-parameter refinement.82,83 The absolute structure parameters X for compounds 2, 8 and 12 are given in Table 7. No twinning by merohedry was noted for these non-centrosymmetric structures.
CCDC reference numbers 281546–281550.
For crystallographic data in CIF or other electronic format see DOI: 10.1039/b511797f
| 2 | 5 | 8 | 12 | 15 | |
|---|---|---|---|---|---|
| a R1 = ∑(Fo − Fc)/ΣFo; I>2σ(I); wR2 = {∑w(Fo2 − Fc2)2/∑w(Fo2)2}1/2. | |||||
| Empirical formula | C12H37MoNOP4 | C25H47MoNO2P4 | C47H57BF24MoNOP5 | C26H41MoN2OP3 | C26H41MoN2OP3 |
| Formula weight/g mol−1 | 431.25 | 613.46 | 1369.54 | 586.46 | 586.46 |
| T/K | 183(2) | 183(2) K | 183(2) | 183(2) | 183(2) |
| Crystal system | Monoclinic | Monoclinic | Monoclinic | Monoclinic | Orthorhombic |
| Space group | Cc | P21/c | Pn | P21 | Pbca |
| a/Å | 9.5261(7) | 10.0261(9) | 13.1223(18) | 10.1601(7) | 13.4047(9) |
| b/Å | 15.6484(15) | 16.7255(10) | 13.2643(12) | 13.9020(9) | 14.3370(13) |
| c/Å | 14.6607(11) | 18.7690(18). | 18.473(2) | 10.9422(8) | 30.111(2) |
| α/° | 90 | 90 | 90 | 90 | 90 |
| β/° | 98.876(9) | 94.046(11) | 108.728(15) | 107.548(8) | 90 |
| γ/° | 90 | 90 | 90 | 90 | 90 |
| V/Å3 | 2159.3(3) | 3139.6(5) | 3045.2(6) | 1473.62(18) | 5786.9(8) |
| Z, dcalcd/Mg m−3 | 4, 1.327 | 4, 1.298 | 2, 1.494 | 2, 1.322 | 8, 1.346 |
| μ/mm−1) | 0.899 | 0.642 | 0.455 | 0.628 | 0.640 |
| F(000) | 904 | 1288 | 1384 | 612 | 2448 |
| Reflections collected/unique | 16665/6197 [R(int) = 0.0593] | 44839/9353 [R(int) = 0.0443] | 33425/14600 [R(int) = 0.0628] | 23862/8795 [R(int) = 0.0744] | 44150/6985 [R(int) = 0.0505] |
| Goodness-of-fit on F2 | 0.882 | 1.000 | 0.869 | 0.671 | 0.876 |
| Final R indices [I > 2σ(I)]a | R1 = 0.0337, wR2 = 0.0709 | R1 = 0.0305, wR2 = 0.0787 | R1 = 0.0611, wR2 = 0.1474 | R1 = 0.0348, wR2 = 0.0687 | R1 = 0.0300, wR2 = 0.0759 |
| R indices (all data) | R1 = 0.0469, wR2 = 0.0737 | R1 = 0.0408, wR2 = 0.0809 | R1 = 0.1017, wR2 = 0.1637 | R1 = 0.0622, wR2 = 0.0712 | R1 = 0.0410, wR2 = 0.0780 |
| Absolute struct. param. | 0.01(4) | −0.01(4) | 0.03(3) | ||
Acknowledgements
Financial support from the Swiss National Science Foundation (SNSF) and University of Zürich are gratefully acknowledged.References
- (a) H. Jacobsen and H. Berke, bookpubl., ed. R. Poli, Elsevier, Amsterdam, Holland, 2001, p. 89 Search PubMed; (b) H. Berke and P. Burger, Comments Inorg. Chem., 1994, 16, 279 CrossRef CAS.
- A. A. H. van der Zeijden, C. Sontag, H. W. Bosch, V. Shklover, H. Berke, D. Nanz and W. von Philipsborn, Helv. Chim. Acta, 1991, 74, 1194 CrossRef CAS.
- A. A. H. van der Zeijden, H. W. Bosch and H. Berke, Organometallics, 1992, 11, 2051 CrossRef CAS.
- A. A. H. van der Zeijden and H. Berke, Helv. Chim. Acta, 1992, 75, 513 CrossRef CAS.
- D. Nietlispach, D. Veghini and H. Berke, Helv. Chim. Acta, 1994, 77, 2197 CrossRef CAS.
- F. Furno, T. Fox, H. W. Schmalle and H. Berke, Organometallics, 2000, 19, 3620 CrossRef CAS.
- D. Nietlispach, H. W. Bosch and H. Berke, Chem. Ber., 1994, 127, 2403 CrossRef CAS.
- A. Messmer, H. Jacobsen and H. Berke, Chem. Eur. J., 1999, 5, 3341 CrossRef CAS.
- D. G. Gusev, A. Llamazares, G. Artus, H. Jacobsen and H. Berke, Organometallics, 1999, 18, 75 CrossRef CAS.
- H. Jacobsen, K. Heinze, A. Llamazares, H. W. Schmalle, G. Artus and H. Berke, J. Chem. Soc., Dalton Trans., 1999, 1717 RSC.
- A. A. H. van der Zeijden, V. Shklover and H. Berke, Inorg. Chem., 1991, 30, 4393 CrossRef CAS.
- A. A. H. van der Zeijden, H. W. Bosch and H. Berke, Organometallics, 1992, 11, 563 CrossRef CAS.
- F. Bäth, Ph. D. Thesis, University of Zürich, 1998.
- J. Höck, H. Jacobsen, H. W. Schmalle, G. R. J. Artus, T. Fox, J. I. Amor, F. Bäth and H. Berke, Organometallics, 2001, 20, 1533 CrossRef.
- D. G. Gusev, D. Nietlispach, I. L. Eremenko and H. Berke, Inorg. Chem., 1993, 32, 3628 CrossRef CAS.
- F. P. Liang, H. Jacobsen, H. W. Schmalle, T. Fox and H. Berke, Organometallics, 2000, 19, 1950 CrossRef CAS.
- T. Y. Cheng, J. S. Southern and G. L. Hillhouse, Organometallics, 1997, 16, 2335 CrossRef CAS.
- E. Carmona, E. Gutierrezpuebla, A. Monge, P. J. Perez and L. J. Sanchez, Inorg. Chem., 1989, 28, 2120 CrossRef CAS.
- R. Taube and K. Seyferth, Z. Anorg. Allg. Chem., 1977, 437, 213 CrossRef CAS.
- K. Nakamoto, Infrared and Raman Spectra of Inorganic and Coordination Compounds (Part A), John Wiley & Sons, Inc., New York, 1997 Search PubMed.
- D. Nietlispach, V. I. Bakhmutov and H. Berke, J. Am. Chem. Soc., 1993, 115, 9191 CrossRef CAS; V. I. Bakhmutov, in Recent Advances in Hydride Chemistry, ed. M. Peruzzini and R. Poli, Elsevier, Amsterdam, 2001, ch. 13 Search PubMed.
- C. A. Fyre, CFC Press, Guelph, 1983.
- K. Guo, W. L. Jarrett and L. G. Butler, Inorg. Chem., 1987, 26, 3001 CrossRef CAS.
- L. Wharton, W. Klemperer and L. P. Gold, J. Chem. Phys., 1962, 37, 2149 CrossRef CAS.
- Z. L. Chen, Ph. D. Thesis, University of Zürich, 2004.
- F. P. Liang, H. W. Schmalle, T. Fox and H. Berke, Organometallics, 2003, 22, 3382 CrossRef CAS.
- P. G. Jessop, F. Joo and C. C. Tai, Coord. Chem. Rev., 2004, 248, 2452; W. Leitner, Angew. Chem., Int. Ed. Engl., 1995, 2207 CrossRef CAS.
- C. A. Willoughby and S. L. Buchwald, J. Am. Chem. Soc., 1994, 116, 11703 CrossRef CAS; C. A. Willoughby and S. L. Buchwald, J. Am. Chem. Soc., 1994, 116, 8952 CrossRef CAS.
- Y. Obora, T. Ohta, C. L. Stern and T. J. Marks, J. Am. Chem. Soc., 1997, 119, 3745 CrossRef CAS.
- S. Kobayashi and H. Ishitani, Chem. Rev., 1999, 99, 1069 CrossRef CAS.
- M. Ringwald, R. Sturmer and H. H. Brintzinger, J. Am. Chem. Soc., 1999, 121, 1524 CrossRef CAS.
- B. R. James, Catal. Today, 1997, 37, 209 CrossRef CAS.
- P. Schnider, G. Koch, R. Pretot, G. Z. Wang, F. M. Bohnen, C. Kruger and A. Pfaltz, Chem.-Eur. J., 1997, 3, 887 CrossRef CAS.
- H. U. Blaser and F. Spindler, Top. Catal., 1997, 4, 275 CrossRef.
- F. P. Liang, H. W. Schmalle and H. Berke, Inorg. Chem., 2004, 43, 993 CrossRef CAS.
- M. Brookhart and M. L. H. Green, J. Organomet. Chem., 1983, 250, 395 CrossRef CAS.
- G. J. Kubas, Metal Dihydrogen and σ-Bond Complexes, Kluwer Academic/Plenum Publishers, New York, 2001 Search PubMed.
- H. U. Blaser and F. Spindler, in Comprehensive Asymmetric Catalysis I, ed. E. N. Jacksen and A. H. Y. Pfaltz, Springer, Berlin, Heidelberg, 1999, p. 247 Search PubMed.
- Z. X. Zhou, B. R. James and H. Alper, Organometallics, 1995, 14, 4209 CrossRef CAS.
- W. R. Cullen, M. D. Fryzuk, B. R. James, J. P. Kutney, G. J. Kang, G. Herb, I. S. Thorburn and R. Spogliarich, J. Mol. Catal., 1990, 62, 243 CrossRef CAS.
- M. D. Fryzuk and W. E. Piers, Organometallics, 1990, 9, 986 CrossRef CAS.
- J. Bakos, I. Toth, B. Heil, G. Szalontai, L. Parkanyi and V. Fulop, J. Organomet. Chem., 1989, 370, 263 CrossRef CAS.
- G. J. Kang, W. R. Cullen, M. D. Fryzuk, B. R. James and J. P. Kutney, J. Chem. Soc., Chem. Commun., 1988, 1466 RSC.
- H. Moser, G. Rihs and H. Sauter, Z. Naturforsch., Teil B, 1982, 37, 451.
- M. A. Andrews and H. D. Kaesz, J. Am. Chem. Soc., 1977, 99, 6763 CrossRef CAS.
- F. Spindler, B. Pugin, H. P. Jalett, U. Pittelkow and H. U. Blaser, Chem. Ind., 1996, 68, 153 CAS.
- A. G. Becalski, W. R. Cullen, M. D. Fryzuk, B. R. James, G. J. Kang and S. J. Rettig, Inorg. Chem., 1991, 30, 5002 CrossRef CAS.
- K. Tani, J. Onouchi, T. Yamagata and Y. Kataoka, Chem. Lett., 1995, 955 CAS.
- T. Morimoto, N. Nakajima and K. Achiwa, Chem. Pharm. Bull., 1994, 42, 1951 CAS.
- H. Brunner and C. Huber, Chem. Ber./Recl., 1992, 125, 2085 CrossRef CAS.
- M. J. Burk and J. E. Feaster, J. Am. Chem. Soc., 1992, 114, 6266 CrossRef CAS.
- C. Lensink and J. G. Devries, Tetrahedron: Asymmetry, 1992, 3, 235 CrossRef CAS.
- J. Bakos, A. Orosz, B. Heil, M. Laghmari, P. Lhoste and D. Sinou, J. Chem. Soc., Chem. Commun., 1991, 1684 RSC.
- Y. N. C. Chan and J. A. Osborn, J. Am. Chem. Soc., 1990, 112, 9400 CrossRef.
- Y. N. C. Chan, D. Meyer and J. A. Osborn, J. Chem. Soc., Chem. Commun., 1990, 869 RSC.
- F. Spindler, B. Pugin and H. U. Blaser, Angew. Chem., Int. Ed. Engl., 1990, 29, 558 CrossRef.
- H. A. Brune, J. Unsin, R. Hemmer and M. Reichhardt, J. Organomet. Chem., 1989, 369, 335 CrossRef CAS.
- N. Uematsu, A. Fujii, S. Hashiguchi, T. Ikariya and R. Noyori, J. Am. Chem. Soc., 1996, 118, 4916 CrossRef CAS.
- A. Fujii, S. Hashiguchi, N. Uematsu, T. Ikariya and R. Noyori, J. Am. Chem. Soc., 1996, 118, 2521 CrossRef CAS.
- S. Hashiguchi, A. Fujii, J. Takehara, T. Ikariya and R. Noyori, J. Am. Chem. Soc., 1995, 117, 7562 CrossRef CAS.
- D. E. Fogg and B. R. James, Inorg. Chem., 1995, 34, 2557 CrossRef CAS.
- D. E. Fogg, B. R. James and M. Kilner, Inorg. Chim. Acta, 1994, 222, 85 CrossRef CAS.
- P. Krasik and H. Alper, Tetrahedron: Asymmetry, 1992, 3, 1283 CrossRef CAS.
- R. Noyori, Chem. Soc. Rev., 1989, 18, 187 RSC.
- B. R. James, A. Pacheco, S. J. Rettig, I. S. Thorburn, R. G. Ball and J. A. Ibers, J. Mol. Catal., 1987, 41, 147 CrossRef CAS.
- I. S. Thorburn, S. J. Rettig and B. R. James, J. Organomet. Chem., 1985, 296, 103 CrossRef CAS.
- A. Viso, N. E. Lee and S. L. Buchwald, J. Am. Chem. Soc., 1994, 116, 9373 CrossRef CAS.
- C. A. Willoughby and S. L. Buchwald, J. Am. Chem. Soc., 1992, 114, 7562 CrossRef CAS.
- P. Marcazzan, B. O. Patrick and B. R. James, Organometallics, 2003, 22, 1177 CrossRef CAS.
- K. Seyferth, R. Taube, L. Bencze and L. Marko, J. Organomet. Chem., 1977, 137, 275 CrossRef CAS.
- F. P. Liang, Ph.D. Thesis, University of Zürich, 2001.
- M. Brookhart, B. Grant and A. F. Volpe, Organometallics, 1992, 11, 3920 CrossRef CAS.
- K. Seyferth and R. Taube, J. Organomet. Chem., 1982, 229, 275 CrossRef CAS.
- Stoe IPDS software for data collection, cell refinement and data reduction. Version 2.92 ed., Stoe & Cie, Darmstadt, Germany, 1997–1999 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A, 1990, 46, 467 CrossRef.
- G. M. Sheldrick, SHELXL-97, Uinversity of Göttingen, Germany, 1997 Search PubMed.
- H. D. Flack, Helv. Chim. Acta, 2003, 86, 905 CrossRef CAS.
- W. H. Baur and D. Kassner, Acta Crystallogr., Sect. B, 1992, 48, 356 CrossRef.
- R. E. Marsh, Acta Crystallogr., Sect. B, 1997, 53, 317 CrossRef.
- R. E. Marsh, Acta Crystallogr., Sect. B, 2004, 60, 252 CrossRef.
- A. L. Spek, J. Appl. Crystallogr., 2003, 36, 7 CrossRef CAS.
- H. D. Flack, Acta Crystallogr., Sect. A, 1983, 39, 876 CrossRef.
- G. Bernardinelli and H. D. Flack, Acta Crystallogr., Sect. A, 1985, 41, 500 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: 2D and 1D NMR spectra of 10 and 12 . See DOI: 10.1039/b511797f |
| This journal is © The Royal Society of Chemistry 2006 |