The synthesis, characterisation and reactivity of 2-phosphanylethylcyclopentadienyl complexes of cobalt, rhodium and iridium†
Ann C.
McConnell
a,
Peter J.
Pogorzelec
b,
Alexandra M. Z.
Slawin
a,
Gary L.
Williams
c,
Paul I. P.
Elliott
c,
Anthony
Haynes
c,
Andrew C.
Marr
a and
David J.
Cole-Hamilton
*a
aEaStCHEM, School of Chemistry, University of St. Andrews, St. Andrews, Fife, UK KY16 9ST. E-mail: djc@st-and.ac.uk; Fax: +44-1334-463808; Tel: +44-1334-463805
bCatalyst Evaluation and Optimisation Service, EaStCHEM, School of Chemistry, University of St. Andrews, St. Andrews, Fife, UK KY16 9ST
cDepartment of Chemistry, University of Sheffield, Dainton Building, Brook Hill, Sheffield, UK S3 7HF. E-mail: a.haynes@sheffield.ac.uk; Fax: +44-114 222 9346; Tel: +44-114 2229326
First published on 21st November 2005
Abstract
2-Phosphanylethylcyclopentadienyl lithium compounds, Li[C5R′4(CH2)2PR2] (R = Et, R′ = H or Me, R = Ph, R′ = Me), have been prepared from the reaction of spirohydrocarbons C5R′4(C2H4) with LiPR2. C5Et4HSiMe2CH2PMe2, was prepared from reaction of Li[C5Et4] with Me2SiCl2 followed by Me2PCH2Li. The lithium salts were reacted with [RhCl(CO)2]2, [IrCl(CO)3] or [Co2(CO)8] to give [M(C5R′4(CH2)2PR2)(CO)] (M = Rh, R = Et, R′ = H or Me, R = Ph, R′ = Me; M = Ir or Co, R = Et, R′ = Me), which have been fully characterised, in many cases crystallographically as monomers with coordination of the phosphorus atom and the cyclopentadienyl ring. The values of νCO for these complexes are usually lower than those for the analogous complexes without the bridge between the cyclopentadienyl ring and the phosphine, the exception being [Rh(Cp′(CH2)2PEt2)(CO)] (Cp′ = C5Me4), the most electron rich of the complexes. [Rh(C5Et4SiMe2CH2PMe2)(CO)] may be a dimer. [Co2(CO)8] reacts with C5H5(CH2)2PEt2 or C5Et4HSiMe2CH2PMe2 (L) to give binuclear complexes of the form [Co2(CO)6L2] with almost linear PCoCoP skeletons. [Rh(Cp′(CH2)2PEt2)(CO)] and [Rh(Cp′(CH2)2PPh2)(CO)] are active for methanol carbonylation at 150 °C and 27 bar CO, with the rate using [Rh(Cp′(CH2)2PPh2)(CO)] (0.81 mol dm−3 h−1) being higher than that for [RhI2(CO)2]− (0.64 mol dm−3 h−1). The most electron rich complex, [Rh(Cp′(CH2)2PEt2)(CO)] (0.38 mol dm−3 h−1) gave a comparable rate to [Cp*Rh(PEt3)(CO)] (0.30 mol dm−3 h−1), which was unstable towards oxidation of the phosphine. [Rh(Cp′(CH2)2PEt2)I2], which is inactive for methanol carbonylation, was isolated after the methanol carbonylation reaction using [Rh(Cp′(CH2)2PEt2)(CO)]. Neither of [M(Cp′(CH2)2PEt2)(CO)] (M = Co or Ir) was active for methanol carbonylation under these conditions, nor under many other conditions investigated, except that [Ir(Cp′(CH2)2PEt2)(CO)] showed some activity at higher temperature (190 °C), probably as a result of degradation to [IrI2(CO)2]−. [M(Cp′(CH2)2PEt2)(CO)] react with MeI to give [M(Cp′(CH2)2PEt2)(C(O)Me)I] (M = Co or Rh) or [Ir(Cp′(CH2)2PEt2)Me(CO)]I. The rates of oxidative addition of MeI to [Rh(C5H4(CH2)2PEt2)(CO)] and [Rh(Cp′(CH2)2PPh2)(CO)] are 62 and 1770 times faster than to [Cp*Rh(CO)2]. Methyl migration is slower, however. High pressure NMR studies show that [Co(Cp′(CH2)2PEt2)(CO)] and [Cp*Rh(PEt3)(CO)] are unstable towards phosphine oxidation and/or quaternisation under methanol carbonylation conditions, but that [Rh(Cp′(CH2)2PEt2)(CO)] does not exhibit phosphine degradation, eventually producing inactive [Rh(Cp′(CH2)2PEt2)I2] at least under conditions of poor gas mixing. The observation of [Rh(Cp′(CH2)2PEt2)(C(O)Me)I] under methanol carbonylation conditions suggests that the rhodium centre has become so electron rich that reductive elimination of ethanoyl iodide has become rate determining for methanol carbonylation. In addition to the high electron density at rhodium.
Introduction
The carbonylation of methanol to ethanoic acid is one of the most successful industrial applications of homogeneous catalysis.1–3 The first process to be developed was based on cobalt, but required very forcing conditions (600–700 bar, 250 °C).4 In the mid 1960's Monsanto developed a rhodium based process in which the active species is [RhI2(CO)2]−.5–8 This operates under much milder conditions (30 bar, 180 °C). The most recent developments have involved [IrI2(CO)2]− promoted by e.g. [RuI2(CO)3].9 In all cases, the mechanism involves oxidative addition of MeI, formed from a reaction of methanol with HI, meaning that highly electron rich metal centres are required. In the case of the rhodium based system, this oxidative addition to [RhI2(CO)2]− is the rate determining step, despite the estimate of Forster that oxidative addition of MeI to the anionic [RhI2(CO)(EPh3)]− is faster by a factor of 105 than to the neutral [RhI(CO)(EPh3)2] (E = P, As, Sb).10 The other steps in the catalytic cycle are fast and the short lifetime of many of the intermediates ensures the very high selectivity (>99%) to the desired ethanoic acid.In order to try to increase the rate of the reaction even further, we11–14 and others1 have investigated using highly electron donating ligands to increase the electron density on the metal and hence increase the rate of the oxidative addition step. This strategy has proven effective in that the rate has been increased but, especially when using tertiary phosphines, the catalysts are rather unstable. In the case of PEt3,11 where the active species is [RhI(CO)(PEt3)2], the rate of methanol carbonylation at 150 °C in the presence of 17% added water is 1.5 times that using [RhI2(CO)2]−, but the rate drops to the same as that using [RhI2(CO)2]−, over a period of 15 min. The rate of oxidative addition of MeI to [RhI(CO)(PEt3)2] at room temperature is 57 times that to [RhI2(CO)2]−, whilst the migration of the methyl group onto CO is 38 times faster for [RhMeI3(CO)2]− than for [RhMeI2(CO)(PEt3)2]. Despite this, oxidative addition of MeI is rate determining in both cases. The deactivation of the PEt3 containing catalyst is caused by loss of the PEt3 groups, which are mainly oxidised to Et3PO, with only a small amount of quaternisation to [Et3MeP]I and [Et3PH]I being observed. High activity with better stability has been observed using bidentate ligands involving at least one P donor,15–18 but the best results are obtained if the ligand is unsymmetrical.16,17 Phosphine oxidation is still a concern over longer reaction times. There has been one report of a non-phosphine based ligand which gives high rate and good stability. This involves the multifunctinal thiophene-2,5-(carboxylatomethylenebenzotriazole) ligand.19 Because of the problems associated with phosphine ligands, we attempted to increase the electron density on the metal by using highly electron donating pentamethylcyclopentadienyl (Cp*) ligands. [Cp*Rh(CO)2] proved to be highly active for the carbonylation of methyl ethanoate to ethanoic anhydride,13 but the most promising results were obtained using [Cp*Co(CO)2] in the presence of PEt3.14 At 100 bar and 120 °C, this system gave an initial rate of methanol carbonylation of 44 mol dm−3 h−1, 1.5 times the rate using the same concentration of [RhI2(CO)2]− under the same conditions and faster than obtained in the commercial rhodium based systems at 180 °C (ca. 20 mol dm−3 h−1). This catalytic system was again not stable and after 200 s a period of inactivity (400 s) was followed by a prolonged reaction with a rate of 2.5 mol dm−3 h−1. Although the actual catalytically active species were not identified, it was proposed that the fast step was catalysed by [Cp*Co(CO)(PEt3)] and the slower step by [Cp*Co(CO)I]−. Unusually, the PEt3 appeared to transfer between cobalt atoms without being quaternised as [CoI(CO)2(PEt3)2] was isolated from the final solution of a catalytic reaction at low water content and crystallographically characterised.
Assuming that the initial highly active cobalt based catalyst was [Cp*Co(CO)(PEt3)], we reasoned that its stability might be improved if we connected the phosphine to the Cp* ligand, since the chelate effect should ensure a higher stability constant for phosphine coordination. In this paper, we describe the synthesis of such ligands, their coordination to Co, Rh and Ir, studies of the rate of reaction of some of these complexes with MeI and the use of the resulting complexes in methanol carbonylation. A preliminary report of some of these results has appeared.20
Experimental
Microanalyses were by the University of St. Andrews microanalytical service. Infrared spectra were obtained using a Nicolet Protege 460 Fourier Transform Spectrometer with CsI optics. The infrared spectrometer was interfaced to a personal computer via the OMNIC operating system. GCMS analysis was carried out using a Hewlett Packard GC system with a 5973 mass selective detector fitted with a 5% phenyl methyl siloxane capilliary column. Carbon, proton and phosphorus NMR spectra were recorded on a Bruker AM 300 NMR spectrometer, a Bruker Avance 300 or a Varian 300 NMR spectrometer. The two-dimensional spectra were recorded on a Bruker Avance 300. Broad band decoupling was used for 13C spectra and 31P spectra. 1H and 13C NMR spectra were referenced internally to deuteriated solvents and are reported relative to TMS. 31P chemical shifts are to high frequency of external H3PO4 (85%).All manipulations were performed in a nitrogen atmosphere, unless otherwise stated, using standard Schlenk line and catheter tubing techniques. Nitrogen/argon was passed through a glass column containing Cr(II) absorbed onto silica, prior to use. All gases were purchased from BOC gases. All solvents were freshly distilled and dried; petroleum ether (bp 40–60 °C), tetrahydrofuran and diethyl ether were distilled over sodium diphenylketyl. Dichloromethane was distilled over calcium hydride.
The metal complexes [RhCl(CO)2]2, [Co2(CO)8], [IrCl(CO)(PPh3)2] and [IrCl(CO)3]n were all purchased from Strem. Diethylphosphine and diphenylphosphine were both purchased from Strem. Cyclopentadiene, tetramethylcyclopentadiene, n-butyllithium, dichlorodimethylsilane, methyl iodide, acetic acid and methyl acetate were all purchased from Aldrich. Deuteriated solvents were purchased from Cambridge Isotope Laboratories, degassed by repeated freeze–pump–thaw cycles under high vacuum or by deoxygenation with dry argon/nitrogen prior to use. [Rh(C5H4(CH2)2PPh2)(CO)] was prepared by a published method.21
1,2,3,4-Tetramethyl-5-(2-chloroethyl)cyclopentadiene22
1,2,3,4-Tetramethylcyclopentadiene (30.5 g, 0.25 mol) was dissolved in diethyl ether (700 cm3) and cooled to 0 °C. n-BuLi (160 cm3, 1.6 mol dm−3, 0.26 mol in hexane) was added dropwise over 2 h and the solution was stirred at room temperature for 18 h. 1-Bromo-2-chloroethane (36.0 g, 0.25 mol) was added to the mixture all at once and the resulting mixture stirred for 30 days at room temperature (reaction was followed by GCMS). Water (100 cm3) was added to the reaction mixture and the phases were separated. The organic layer was washed once with saturated aqueous NaCl solution (50 cm3), dried over MgSO4, filtered and the solvent removed in vacuo. The product was a pale yellow oil (32.0 g) which according to GCMS contained approximately 50% of geminal isomers and 50% of non-geminal isomers.1,2,3,4,-Tetramethylbicyclo[2,4]hepta-1,3-diene22
1,2,3,4-Tetramethyl-5-(2-chloroethyl)cyclopentadiene (32.0 g) was dissolved in THF (300 cm3). The solution was cooled to −60 °C and n-BuLi (115 cm3, 1.6 mol dm−3, 0.18 mol in hexane) was added dropwise with stirring. The reaction mixture was brought to room temperature and stirred for 40 h. The THF was removed in vacuo and the residue dissolved in diethyl ether (200 cm3). Water (100 cm3) was added and the two phases were then separated. The organic layer was washed with water (100 cm3). The combined water layers were extracted with diethyl ether (50 cm3). The combined organic layers were washed with saturated NaCl (50 cm3), dried over NaSO4, filtered and the solvent removed in vacuo to give a pale yellow oil (27.0 g). This mixture of spirohydrocarbon and geminal isomers was purified using column chromatography (silica gel 60, mobile solvent petroleum) to give the pure spirohydrocarbon product as a pale yellow oil (14.8 g, 40%). 1H NMR (300 MHz, C6D6) δ 1.95 (s, 6H, CH3), 1.6 (s, 6H, CH3) and 1.1 (s, 4H, CH2). 13C NMR (75.45 MHz, C6D6) δ 134.37 (s, CCH3), 134.06 (s, CCH3), 38.05 (C ring), 12.11 (s, CH3), 11.22 (s, CH2) and 9.31 (s, CH3).1,2,3,4-Tetramethyl-5-(2-P,P-diethylphosphinoethyl)cyclopentadienyl lithium
LiPEt2 (1.15 g, 12.0 mmol) was dissolved in THF (50 cm3) and stirred with the spirohydrocarbon (2.0 g, 13.5 mmol) at room temperature for 3 days. The THF was removed in vacuo and the residue washed with petroleum (50 cm3 × 2) to leave a white solid (2.55 g, 87% yield). Most of the lithium salts were highly air sensitive and were characterised spectroscopically. 1H NMR (300 MHz, d8-THF) δ 2.27 (s(b), 2H, CH2 (10)), 1.76 (s(b), 12H, CH3 (6,7,8,9)), 1.40 (m(b), 4H, CH2CH3), 1.33 (m, 2H, CH2 (11)) and 1.06 (dt, 3JHH = 7.42 Hz, 3JHP = 13.83 Hz, 6H, CH2CH3). 13C NMR (75.4 MHz, d8-THF) δ 113.06 (d, 3JCP 11.51 Hz, ring C (5)), 106.81 (s, ring C (1,4)), 105.85 (s, ring C (2,3)), 30.33 (d, 1JCP = 14.37 Hz, CH2 (11)), 22.97 (d, 2JCP = 13.26 Hz, CH2 (10)), 19.86 (d, 1JCP = 13.81 Hz, CH2CH3), 11.22 (s, CH3 (6,7,8,9)) and 10.45 (d, 2JCP = 13.27 Hz, PCH2CH3). 31P NMR (121.4 Hz, d8-THF) δ −21.93 (b). For numbering scheme see Scheme 1.![Synthesis of Cp′H(CH2)2PR2 (R = Et or Ph) and metal complexes of the related anion. (i) BuLi, THF; (ii) Br(CH2)2Cl, Et2O, 30 d, geminal isomers removed by chromatography; (iii) LiPR2, THF, −60 °C, 3 d; (iv) aqueous work-up; (v) BuLi; (vi) M = Co, [Co2(CO)8], 170 °C; M = Rh, [RhCl(CO)2]2; M = Ir, [IrCl(CO)3]n or [IrCl(CO)(PPh3)2].](/image/article/2006/DT/b512054c/b512054c-s1.gif) | ||
| Scheme 1 Synthesis of Cp′H(CH2)2PR2 (R = Et or Ph) and metal complexes of the related anion. (i) BuLi, THF; (ii) Br(CH2)2Cl, Et2O, 30 d, geminal isomers removed by chromatography; (iii) LiPR2, THF, −60 °C, 3 d; (iv) aqueous work-up; (v) BuLi; (vi) M = Co, [Co2(CO)8], 170 °C; M = Rh, [RhCl(CO)2]2; M = Ir, [IrCl(CO)3]n or [IrCl(CO)(PPh3)2]. | ||
1,2,3,4-Tetramethyl-5-(2-P,P-diphenylphosphinoethyl)cyclopentadienyl lithium22
LiPPh2 (2.3 g, 12.0 mmol) was dissolved in THF (50 cm3) and stirred with the spirohydrocarbon (50) (2.0 g, 13.5 mmol) at room temperature for 3 days. The THF was removed in vacuo and the residue washed with petroleum (50 cm3 × 2) to leave a white solid (3.7 g, 90% yield). 1H NMR (300 MHz, d8-THF) δ 7.39 (m (b), 4H, PhH ortho), 7.25 (m(b), 6H, PhH meta and para), 2.40 (s (vb), 2H, CH2 (10)), 2.04 (s (vb), 2H, CH2 (11)) and 1.73 (s(vb), 12H, CH3 (6,7,8,9)). 13C NMR (75.4 MHz, d8-THF) δ 141.40 (d, 2JCP = 14.70 Hz, Ph (C) meta), 133.80 (d, 1JCP = 17.30 Hz, Ph (C) ortho), 128.90 (m, Ph (C) para), 112.50 (d, 3JCP = 14.96 Hz, ring C (5)), 106.80 (s, ring C (1,4)), 106.10 (s, ring C (2,3)), 32.26 (s, CH2 (11)), 23.55 (d, 1JCP = 17.48 Hz, CH2 (10)) and 11.23 (s, CH3 (6,7,8,9)). 31P {1H} NMR (121.43 Hz, d8-THF) δ −15.10 (b).(2-P,P-Diethylphosphinoethyl)cyclopentadienyl lithium23
LiPEt2 (0.77 g, 6.5 mmol) was dissolved in THF (40 cm3) and refluxed with bicyclo[2,4]hepta-1,3-diene (0.65 g, 7.03 mmol) for 1 h. The solvent was removed in vacuo and the residue washed with petroleum (50 cm3 × 2) to leave a white solid (0.98 g, 80% yield).1H NMR (300 MHz, d8-THF) δ 5.52 (d (b), 2H, C5H4), 5.45 (d (b), 2H, C5H4), 2.61 (m, 2H, CH2 (7)), 1.65 (m, 2H, CH2 (6)), 1.35 (m, 4H, CH2 (8,10)) and 1.10 (m, 6H, CH3 (10,11)). 13C NMR (75.43 MHz, d8-THF) δ 102.28 (d, ring C), 31.11 (d, JCP = 9.21 Hz, CH2 (6)), 27.29 (d, JCP = 13.81 Hz, CH2 (7)), 19.43 (d, JCP = 11.5 Hz, CH2 (8,9)) and 10.16 (d, 2JCP = 12.66 Hz, CH3 (9,11)). 31P NMR (121.4 MHz, d8-THF) δ −25.05.

Preparation of 1,2,3,4-tetra(ethyl)cyclopentadiene24
An aqueous solution (50%) of NaOH (1050 g, 26.25 mol) was cooled to 10 °C. Aliquat 336 (32.0 g, 79.0 mmol) and freshly cracked cyclopentadiene (51.0 g, 0.77 mol) were added to this cooled solution with constant stirring using a mechanical stirrer. The reaction mixture was stirred for 15 min. Ethyl bromide (344 g, 3.19 mol) was added over 1 h at 10 °C. The mixture was stirred for 1 h at room temperature then heated to 35 °C for a further 6 h. Stirring was stopped and the mixture allowed to separate into two phases. The water layer was drawn off and fresh NaOH solution (50%, 1050 g, 26.25 mol)) added. The mixture was stirred for a further 5 h at room temperature. Stirring was stopped and the organic phase separated from the aqueous phase. The product was distilled (11 mbar, 90 °C) to give 1,2,3,4-tetra(ethyl)cyclopentadiene as a pale yellow oil (60 g, 44% yield). 1H NMR (300 MHz, CD2Cl2) δ 2.75 (s, 2H, CH2), 2.34 (q, 3JHH = 7.58 Hz, 4H, CH2), 2.23 (q, 3JHH = 7.58 Hz, 4H, CH2), 1.06 (t, 3JHH = 7.54 Hz, 6H, CH3) and 1.01 (t, 3JHH = 7.54 Hz, 6H, CH3). 13C NMR (75.4 MHz, CD2Cl2) δ 141.80 (s, ring C), 139.55 (s, ring C), 42.30 (s, CH2 (ring)), 22.12 (s, CH2), 19.54 (s, CH2) and 15.81 (s, CH3).1-Chloromethyl(dimethyl)silyl-2,3,4,5-tetraethylcyclopentadiene
A solution of n-BuLi in hexane (35 cm3, 1.6 mol dm−3, 56 mmol) was added dropwise to a solution of tetra(ethyl)cyclopentadiene (10.0 g, 56 mmol) in THF (300 cm3) at 0 °C. The solution was stirred for 2 h before the addition of Me2SiCl2 (15.11 g, 56.0 mmol) at −90 °C. The mixture was stirred overnight at room temperature. The THF was removed in vacuo and the resulting oily solid extracted with petroleum (50 cm3 × 2). The solvent was removed in vacuo from the filtrate and the yellow oil was distilled under reduced pressure at 80 °C to give a pale yellow oil (13.1 g, 88% yield). 1H NMR (300 MHz, C6D6) δ 3.42 (s (b), 1H, CH), 2.41 (m (b), 4H, CH2), 2.26 (q, 3JHH = 7.72 Hz, 4H, CH2), 0.99 (t, 3JHH = 7.72 Hz, 12H, CH3) and 0.19 (s, 6H, SiCH3). 13C NMR (75.4 MHz, C6D6) δ 143.71 (s, ring), 139.90 (s, ring), 56.30 (s, CH), 21.92 (s, CH2), 19.68 (s, CH2), 15.83 (s, CH3) and 1.03 (s, SiCH3).1-Dimethylphosphinomethyl(dimethyl)silyl-2,3,4,5-tetraethylcyclopentadiene
(C5Et5H)SiMe2Cl (5.9 g, 0.020 mol) was dissolved in THF (100 cm3) and cooled to −78 °C. This was treated dropwise with a solution of LiCH2PMe2 (1.80 g, 0.020 mol) in THF (50 cm3). The solution was stirred for 24 h at room temperature. The THF was removed in vacuo and petroleum ether (100 cm3) was added to the residue. After filtration the petroleum was removed in vacuo to give a sticky oily product (4.5 g, 66% yield). 1H NMR (300 MHz, C6D6) δ 3.36 (s (b), 1H, CH), 2.41 (m, 8H, CH2), 1.18 (t, 3JHH = 6.80 Hz, 12H, CH3), 0.99 (d, 2JHP = 4.00 Hz, 6H, PCH3), 0.47 (d, 2JHP = 3 Hz, 2H, CH2P) and 0.22 (s, 6H, SiCH3). 13C NMR (75.4 MHz, C6D6) δ 141.59 (s, C ring), 140.95 (s, C ring), 49.67 (s, CH), 22.26 (s, CH2), 19.75 (s, CH2), 18.51 (d, JCP = 18.85 Hz, PCH2) 17.10 (d, JCP = 28.1 Hz, PCH3), 15.30 (s, CH3), 15.1 (s, CH3) and −0.70 (s, SiCH3). 31P NMR (121.40 MHz, C6D6) δ −53.90.Carbonyl(κ2-1-diethylphosphinoethyl-2,3,4,5-tetramethylcyclopentadienyl)rhodium(I)
A solution of [RhCl(CO)2]2 (0.2 g, 0.52 mmol) in THF (50 cm3) was treated dropwise with a solution of Cp′(CH2)2PEt2Li (Cp′ = C5Me4, 0.27 g, 1.12 mmol) in THF (10 cm3). Immediately on addition of the ligand to the dimer solution, a colour change from pale yellow to dark red was noted. The mixture was refluxed for 8 h. The solvent was removed in vacuo and petroleum added (50 cm3). After filtration to leave a white solid (LiCl), the red solution was evaporated in vacuo to approximately half volume (25 cm3) and cooled to −78 °C to yield red crystals (0.17 g, 65% yield). 1H NMR (299.9 MHz, C6D6) δ 2.37 (dt, 3JHP = 9.61 Hz, 3JHH = 7.07 Hz, 2H, CH2 (11)), 2.18 (d, JHP = 2.95 Hz, 6H, CH3, (7,8)), 2.13 (d, JHRh = 0.7 Hz, 6H, CH3 (6,9)), 2.03 (dt, 2JHP = 26.98 Hz, 3JHH 6.95 Hz, CH2 (10)), 1.36 (m, 4H, CH2CH3) and 1.04 (dt, 3JHP = 17.53 Hz, 3JHH = 7.73 Hz, 6H, CH2CH3). 13C NMR (75.4 MHz, C6D6) δ 195.94 (dd, 1JCRh = 87.48 Hz, 2JCP = 20.72 Hz, CO (16)), 107.96 (m, ring(5)), 99.96 (m, ring (1,4)), 97.44 (s, ring (2,3)), 46.17 (d, 1JCP = 24.17 Hz, CH2 (11)), 23.44 (d, 1JCP = 25.32 Hz, CH2CH3), 20.36 (d, 2JCP = 2.30 Hz, CH2 (10)), 11.90 (s, CH3 (7,8)), 11.59 (s, CH3 (6,9)) and 8.93 (s, CH2CH3). 31P NMR (121.4 MHz, CD2Cl2) δ 76.30 (d, JPRh = 189.21 Hz, PEt2). For numbering scheme see Scheme 1. IR (petroleum ether) λmax 1898 cm−1. Found C 52.69, H 7.75%; C16H26OPRh requires C 52.18%, H 7.12%.Carbonyl(κ2-1-diphenylphosphinoethyl-2,3,4,5-tetramethylcyclopentadienyl)rhodium(I)
A solution of [RhCl(CO)2]2 (0.2 g, 0.52 mmol) in THF (50 cm3) was treated dropwise with a solution of Cp′(CH2)2PPh2Li (0.38 g, 1.12 mmol) in THF (10 cm3). Immediately on addition of the ligand to the dimer solution, a colour change from pale yellow to dark red was noted. The mixture was refluxed for 8 h. The solvent was removed in vacuo and petroleum added (50 cm3). After filtration to leave a white solid (LiCl), the red solution was evaporated in vacuo to approximately half volume (25 cm3) and cooled to −78 °C to yield red crystals (0.17 g, 45% yield). 1H NMR (300 MHz, CD2Cl2) δ 7.67 (m, 4H, PhH), 7.50 (m, 6H, PhH), 3.45 (dt, 3JHH = 6.94 Hz, 3JHP = 10.97 Hz, 2H, CH2 (11)), 2.23 (dt, 3JHH = 6.92 Hz, 3JHP = 29.10 Hz, CH2 (10)), 1.88 (d, JHP = 5.29 Hz, CH3 (7,8)) and 1.59 (s, 6H, CH3 (6,9)). 13C NMR (75.4 MHz, CD2Cl2) δ 135.40 (s, PhC), 133.80 (s, C(Ph)), 132.03 (s, PhC), 129.67 (d, JCP = 10.4 Hz, PhC), 114.20 (m, ring (3,2)), 109.40 (m, ring (1,4)), 84.10 (m, ring (5)), 46.50 (d, JCP = 31.2 Hz, CH2 (11)), 19.49 (s, CH2 (10)), 9.68 (s, CH3 (7,8)) and 9.22 (s, CH3 (6,9)). 31P NMR (121.4 MHz, CD2Cl2) δ 52.12 (d, JPRh = 174.99 Hz, PRh). For numbering scheme, see Scheme 1. IR (petroleum ether) λmax 1933 cm−1. Found: C 62.49%, H 5.53%; C24H26OPRh requires C 62.08%, H 5.64.Carbonyl(κ2-diethylphosphinoethylcyclopentadienyl)rhodium(I)
A solution of [RhCl(CO)2]2 (0.20 g, 0.52 mmol) in THF (50 cm3) was treated dropwise with a solution of Cp(CH2)2PEt2Li (0.21 g, 1.12 mmol) in THF (10 cm3). Immediately on addition of the ligand to the dimer solution, a colour change from pale yellow to dark red was noted. The mixture was refluxed for 8 h. The solvent was removed in vacuo and petroleum added (50 cm3). After filtration to leave a white solid (LiCl), the red solution was evaporated in vacuo to approximately half volume (25 cm3) and cooled to −78 °C to yield red crystals (0.14 g, 70% yield). 1H NMR (299.9 MHz, C6D6) δ 5.59 (m, 2H, H (ring)), 5.46 (m, 2H, H (ring)), 2.04 (m, 2H, CH2 (7)), 1.81 (dt, 2JHP = 28.26 Hz, 3JHH = 7.30 Hz, CH2 (6)), 1.35 (m, 4H, CH2 (8,10)) and 1.04 (dt, 3JHP = 17.84 Hz, 3JHH = 7.53 Hz, CH3 (9,11)). 13C NMR (75.4 MHz, C6D6) δ 195.10 (dd, JCRh = 88.63 Hz, 2JCP = 20.72 Hz, CO), 112.0 (s, C (5)), 87.22 (m, C (2,3)), 85.89 (m, C (1,4)), 46.23 (d, JCP = 25.32 Hz, CH2 (7)), 24.69 (s, CH2 (6)), 24.23 (dd, 1JCP = 27.62 Hz, 2JCRh = 2.30 Hz, CH2 (8,10)) and 9.57 (s, CH3 (9,11)). 31P NMR (121.4 MHz, C6D6) δ 80.29 (d, 1JPRh = 191.31 Hz, PEt2). For numbering scheme, see the free ligand. IR (petroleum ether) λmax1938 cm −1. Found C 46.94%, H 6.35%; C12H19OPRh requires C 46.17%, H 5.81%.Carbonyl(κ2-1-dimethylphosphinomethyl(dimethyl)silyl-2,3,4,5-tetraethylcyclopentadienyl)rhodium(I)
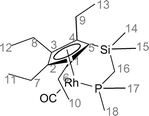
A solution of [RhCl(CO)2]2 (0.2 g, 0.52 mmol) in THF (50 cm3) was treated dropwise with a solution of C5Et4SiMe2CH2PMe2Li (0.35 g,1.12 mmol) in THF (10 cm3). Immediately on addition of the ligand to the dimer solution, a colour change from pale yellow to dark red was noted. The solution was stirred at room temperature for 1 h and the solvent was removed in vacuo. Petroleum was added to the oily solid (50 cm3). After filtration to leave a white solid (LiCl) the red/brown solution was evaporated in vacuo to approximately half volume (25 cm3) and cooled to −78 °C, −40 °C and −2 °C. This failed to yield crystals. The solution was reduced in vacuo to give a red/brown oil (0.21 g, 62% yield).1H NMR (300 MHz, C6D6) δ 2.62 (dq, 3JHH = 7.33 Hz, 2JHH = 14.70 Hz, 2H, CH2 (6a,9a)), 2.45 (dq, 3JHH = 7.33 Hz, 2JHH = 14.70 Hz, 2H, CH2 (6b,9b)), 2.39 (qt (b), 3JHH = 7.33 Hz, J = 1.33 Hz, 4H, CH2 (7,8)), 1.58 (dd, 2JHP = 13.39 Hz, 3JHRh = 1.07 Hz, 2H, CH2 (16)), 1.35 (t, 3JHH = 7.33 Hz, 6H, CH3 (10,13)), 1.32 (dd, 2JHP = 10.18 Hz, 3JHRh = 1.59 Hz, 6H, CH3 (17,18)), 1.19 (t, 3JHH = 7.33 Hz, 6H, CH3 (11,12)) and 0.40 (s, 6H, CH3 (14,15)). 13C NMR (75.4 MHz, C6D6) δ 196.40 (dd, JCRh = 86.3 Hz, JCP = 25.1 Hz, CO), 36.25 (d, JCP = 13.11 Hz, CH2 (16)), 23.70 (d, JCP = 26.88 Hz, CH3 (17,18)), 21.22 (s, CH3 (10,13)), 20.80 (s, CH3 (11,12)), 19.82 (s, CH2 (6,9)), 18.28 (s, CH2 (7,8)) and 1.10 (s, CH3 (14,15)). 31P NMR (121.4 MHz, C6 D6) δ 15.22 (d, JPRh = 185.37 Hz). IR (petroleum ether) λmax 2029, 1967 and 1928 cm−1.
Carbonyl(κ2-1-diethylphosphinoethyl-2,3,4,5-tetramethylcyclopentadienyl)iridium(I)
A solution of [Ir(PPh3)2(CO)Cl] (0.79 g, 1.0 mmol) in THF (30 cm3) was treated dropwise with a solution of Cp′(CH2)2PEt2Li (0.34 g, 1.4 mmol) in THF (20 cm3). The mixture was refluxed for 3 h. The solvent was removed in vacuo and petroleum added (50 cm3). After filtration to leave a white solid (LiCl), the yellow solution was evaporated in vacuo to approximately half volume (25 cm3) and cooled to 2 °C for several days to yield yellow crystals (0.22 g, 49% yield). 1H NMR (300 MHz, C6D6) δ 2.48 (dt, 3JHP = 9.73 Hz, 3JHH = 7.29 Hz, 2H, CH2 (11)), 2.18 (d, JHP = 2.40 Hz, 6H, CH3 ring methyl (7,8)), 2.16 (s(b), 6H, CH3 ring methyl (6,9)), 1.80 (dt, 3JHP = 24.19 Hz, 3JHH = 7.29 Hz, 2H, CH2 (10)), 1.48 (m, CH2CH3) and 1.04 (dt, 3JHP = 17.79 Hz, 3JHH = 7.55 Hz, 6H, CH2CH3). 13C NMR (75.4 MHz, C6D6) δ 181.10 (d, 2JCP = 11.51 Hz, CO (16)), 104.65 (d, 3JCP = 9.21 Hz, ring C (5)), 94.52 (d, 2JCP = 6.91 Hz, ring C (1,4)), 93.04 (s, ring C (2,3)), 48.49 (d, 1JCP = 31.08 Hz, CH2 (11)), 24.40 (d, 1JCP = 34.53 Hz, CH2CH3), 19.66 (s, CH2 (10)), 11.87 (s, ring C), 11.41 (s, ring C) and 9.47 (s, CH2CH3). 31P NMR (121.4 MHz, C6D6) δ 37.67. For numbering scheme, see Scheme 1. IR (petroleum ether) λmax 1910 cm −1.Hexacarbonylbis(2-(2′,3′,4′,5′-tetramethylcyclopentadienylethyl)diphenylphosphine)dicobalt(0)
A solution of [Co2(CO)8] (0.15 g, 0.44 mmol) and 1,3-cyclohexadiene (0.05 g, 0.66 mmol) in methylene chloride (25 cm3) was treated dropwise with a solution of Cp′H(CH2)2PPh2 (0.3 g, 0.89 mmol) in THF (25 cm3). The mixture was refluxed for 4 h. The solvent was removed in vacuo and petroleum added (50 cm3) to the red oil. After filtration, the red solution was cooled to 2 °C for several days to yield red/brown crystals (0.14 g, 35% yield). 1H NMR‡ (300 MHz, CD2Cl2) δ 7.55 (m, 10H, PhH), 2.67 (s (b), 1H, CH), 2,43 (m (b), 2H, CH2 (10)), 1.92 (m (b), 2H, CH2 (11)) and 1.67 (m (b), 12H, CH3). 13C NMR‡ (Bruker, CD2Cl2) δ 202.75 (t, J = 9.77 Hz, CO), 139.95 (s, ring C), 137.25 (s, ring C), 134.21 (s, ring C), 132.29 (m, PhC), 130.82 (m, PhC), 129.14 (m, PhC), 56.12 (d, 1JCP = 15.86 Hz, CH2 (11)), 52.09 (s, CH), 22.50 (s, CH2 (10)), 11.80 (s, CH3) and 11.20 (s, CH3). 31P NMR (121.4 MHz, CD2Cl2) δ 61.17 (s, PPh), 59.87 (m, PPh) and 59.57 (m, PPh). IR (CH2Cl2) λmax 1973 and 1951 cm−1.Hexacarbonylbis(2,3,4,5-tetramethyl(dimethyl)silylmethyl)(dimethyl)phosphinedicobalt(0)
A solution of [Co2(CO)8] (0.27 g, 0.81 mmol) and 1,3-cyclohexadiene (0.090 g, 1.2 mmol) in THF (25 cm3) was treated dropwise with a solution of C5Et4HSiMe2CH2PMe2 (0.5 g, 1.6 mmol) in THF (25 cm3). The mixture was refluxed for 5 h. The solvent was removed in vacuo and petroleum added (50 cm3) to the red oil. After filtration, the red solution was cooled to 2 °C for several days to yield red/brown crystals (0.30 g, 41% yield). 1H NMR (300 MHz, C6D6) δ 2.95 (s (b), 1H, CH), 2.36 (m, 4H, CH2), 2.07 (m, 4H, CH2), 1.05 (b, 12H, CH3), 0.88 (b, 6H, PCH3), 0.77 (b, 4H, CH2P) and 0.28 (s, SiCH3). 13C NMR (75.4 MHz, C6D6) δ 203.92 (m, CO), 142.27 (s, C ring), 140.73 (s, C ring), 49.86 (s, CH), 22.66 (d, JCP = 27.90 HZ, PMe2), 22.09 (s, CH2), 19.63 (s, CH2), 18.29 (b, PCH2), 16.25 (s, CH3), 16.02 (s, CH3) and 0.46 (s, SiCH3). 31P NMR (121.4 MHz, C6H6) δ 32.26. IR (petroleum ether) λmax 1969 and 1951 cm−1.Hexacarbonylbis(2-(2′,3′,4′,5′-tetramethylcyclopentadienylethyl)diethylphosphine)dicobalt(0)
A solution of [Co2(CO)8] (0.30 g, 0.89 mmol) and 1,3-cyclohexadiene (0.14 g, 1.8 mmol) in THF (25 cm3) was treated dropwise with a solution of Cp′H(CH2)2PEt2 (0.43 g, 1.8 mmol) in THF (25 cm3). This solution was transferred to an autoclave using standard Schlenk techniques under carbon monoxide. The autoclave was pressurized to approximately 20 bar of carbon monoxide and heated to 100 °C for 60 min. The solvent was removed in vacuo and petroleum added (50 cm3) to the red oil. After filtration, the red solution was cooled to 2 °C. No crystals were isolated. 31P NMR (crude product solution) (121.4 MHz, C6D6) δ 58.20, 57.60 and 55.76. IR (petroleum ether) λmax 2078, 2009, 1993 and 1951 cm−1.Carbonyl(κ2-1-diethylphosphinoethyl-2,3,4,5-tetramethylcyclopentadienyl)cobalt(I)
A solution of [Co2(CO)8] (0.36 g, 1.0 mmol) in THF (15 cm3) was treated dropwise with a solution of Cp′(CH2)2PEt2Li (0.26 g, 1.1 mmol) in THF (15 cm3). Using standard Schlenk techniques this solution was transferred to an autoclave under nitrogen, sealed and heated to approximately 160 °C for 90 min. The solvent was removed in vacuo and petroleum added (50 cm3) to the red oil. After filtration, the red solution was cooled to −78 °C to yield dark red/brown crystals (0.17 g, 52% yield). 1H NMR (300 MHz, C6D6) δ 2.31 (dt, 3JHP = 8.96, 3JHH = 7.93 Hz, 2H, CH2 (11)), 2.12 (d, JHP = 2.10 Hz, 6H, CH3 (7,8)), 2.09 (s, CH3 (6,9), 1.82 (dt, 2JHP = 22.27 Hz, 3JHH = 7.93 Hz, 2H, CH2 (10)), 1.39 (m, 4H, CH2CH3), 1.07 (dt, 3JHP = 16.64 Hz, 3JHH = 7.42 Hz, 6H, CH2CH3). 13C NMR (75.4 MHz, C6D6) δ 207.86 (s, CO (16)), 105.80 (d, JCP = 9.20 Hz, ring C), 94.84 (d, JCP = 4.59 Hz, ring C), 91.35 (s, ring C), 43.89 (d, 1JCP = 25.32 Hz, CH2 (11)), 22.99 (d, 1JCP = 24.17 Hz, CH2CH3), 20.10 (d, 2JCP = 6.91 Hz, CH2 (10)), 12.26 (s, ring methyl), 12.12 (s, ring methyl), 9.14 (s, CH2CH3). 31P NMR (121.4 MHz, C6D6) δ 87.60 (b). For numbering, see Scheme 1. IR (petroleum ether) λmax 1910 cm−1. Found C 59.18%, H 8.59%; C16H26CoOP requires C 59.26%, H 8.08%.Dichloro(κ2-1-diethylphosphinoethyl-2,3,4,5-tetramethylcyclopentadienyl)rhodium(I)
[Rh(Cp′(CH2)2PEt2)(CO)] (0.050 g, 0.14 mmol) was dissolved in CD2Cl2 (5 cm3) and stirred at room temperature for 24 h. The product was recrystallised from benzene to give bright orange crystals (0.050 g, 87% yield). 1H NMR (299.9 MHz, CD2Cl2) δ 2.89 (dt, 3JHH = 7.5 Hz, 3JHP = 10.45 Hz, 2H, CH2 (11)), 2.31 (dt, 3JHH 7.5 Hz, 3JHP = 28.86 Hz, 2H, CH2 (10)), 2.22 (m, 2H, CH2CH3 (Hb), 1.93 (m, 2H, CH2CH3 (Ha), 1.77 (d, JHP = 7.5 Hz, CH3 (7,8)), 1.60 (s, 3H, CH3 (6,9)) and (dt, 3JHH = 7.16 Hz, 3JHP = 15.5 Hz, 6H, CH2CH3). 13C NMR (75.4 MHz, CD2Cl2) δ 114.20 (t, J = 8.15 Hz, C ring), 111.28 (dd, JCP = 8.2 Hz, JCRh = 4.08 Hz, C ring), 86.39 (dd, JCP = 7.34 Hz, JCRh = 3.26 Hz, C ring), 41.66 (dd, 1JCP = 27.6 Hz, 2JCRh = 2.87 Hz, CH2 (11)), 20.10 (s, CH2 (10)), 18.39 (d, 2JCP = 26.3 Hz, CH2CH3), 9.40 (s, CH3 (6,9)), 9.31 (d, JCP = 3.12 Hz, CH3 (7,8)) and 7.64 (d, 2JCP = 5.20 Hz, CH2CH3). 31P NMR (121.4 MHz, CD2Cl2) δ 72.80 (d, JPRh = 146.50 Hz). For numbering see Scheme 1. Found C 44.62%, H 5.21%; C15H26Cl2Rh requires C 43.82%, H 6.37%.Dichloro(κ2-1-diphenylphosphinoethyl-2,3,4,5-tetramethylcyclopentadienyl)rhodium(I)
[Rh(Cp′(CH2)2PPh2)(CO)] (0.050 g, 0.11 mmol) was dissolved in CD2Cl2 (5 cm3) and stirred at room temperature for 72 h. The product was recrystallised from benzene to give bright orange crystals (0.030 g, 69% yield). 1H NMR (300 MHz, CD2Cl2) δ 7.84 (m, 4H, PhH), 7.43 (m, 6H, PhH), 3.50 (dt, 3JHH = 7.24 Hz, 3JHP = 10.61 Hz, 2H, CH2 (11)), 2.30 (dt, 3JHH = 7.24 Hz, 1JHP = 29.45 Hz, 2H, CH2 (10)), 1.79 (d, JHP = 5.79 Hz, 6H, CH3 (7,8)) and 1.44 (s, 6H, CH3 (6,9)). 13C NMR (299.98 MHz, CD2Cl2) δ 133.36 (d, JCP = 9.77 Hz, PhC), 131.61 (d, JCP = 2.44 Hz, PhC), 129.14 (d, JCP = 10.99 Hz, PhC), 45.61 (d, JCP = 31.74 Hz, CH2 (11)), 18.87 (s, CH2 (10)), 9.56 (s, CH3 (7,8)) and 9.49 (s, CH3 (6,9)). 31P NMR (121.4 Hz, CD2Cl2) δ 51.45 (d, JPRh = 157.14 Hz, PPh). For numbering, see Scheme 1).(κ2-1-Diethylphosphinoethyl-2,3,4,5-tetramethylcyclopentadienyl)diiodorhodium(I)
[Rh(Cp′(CH2)2PEt2)I2] was formed from reaction of [Rh(Cp′(CH2)2PEt2)(CO)] with methyl iodide, methyl ethanoate, water and ethanoic acid (under 30 bar CO and 150 °C) under catalytic conditions. The exact details of the experiment are given later. 1H NMR (300 MHz, C6D6) δ 2.47 (m, J = 7.72 Hz, 2H, CH2CH3 (Ha), 2.05 (dt, 3JHP = 10.61 Hz, 3JHH = 7.26 Hz, 2H, CH2 (11)), 1.98 (d, JHP = 5.30 Hz, 6H, CH3 (7,8)), 1.49 (s, 6H, CH3 (6,9)), 1.25 (dt,2JHP = 27.0 Hz, 3JHH = 7.20 Hz, 2H, CH2 (10)), 1.55–1.12 (m, 2H, CH2CH3 (Hb) and 0.85 (dt, 2JHP = 15.92 Hz, 3JHH = 7.72 Hz, 6H, CH2CH3 (13,15)). 13C NMR (75.4 MHz, C6D6) δ 43.17 (d, 2JCP = 26.48 Hz, CH2 (11)), 23.22 (d, JCP = 27.63 Hz, CH2CH3), 18.52 (s, CH2 (10)), 11.47 (s, CH3 (8,9)), 10.72 (s, CH3 (6,7)) and 8.80 (d, 2JCP = 5.76 Hz, CH2CH3). 31P NMR (121.4 MHz, C6D6) δ 63.71 (d, 1JPRh 148.29 Hz, PEt2). For numbering, see Scheme 1.(κ2-1-Diethylphosphinoethyl-2,3,4,5-tetramethylcyclopentadienyl)ethanoyliodorhodium(III)
A solution of [Rh(Cp′(CH2)2PEt2)(CO)] (0.10 g, 0.27 mmol) in THF (15 cm3) was treated with an excess of methyl iodide (2 cm3, 32.1 mmol) and stirred for 4 h. The solvent was removed in vacuo and the red solid product was recrystallised from petroleum ether (25 cm3) (0.10 g, 75% yield). 1H NMR (299.98 MHz, C6D6)§δ 3.18 (s, 3H, C(O)CH3), 2.24–1.14§ (m, 8H, CH2 (10, 11, CH2CH3)), 2.09 (d, JHP = 5.63 Hz, 3H, CH3 (ring methyl)), 1.74 (d, JHP = 0.92 Hz, 3H, CH3 (ring methyl)), 1.43 (dd, JHP = 2.7 Hz, JHRh = 1.5 Hz, 3H, CH3 (ring methyl)), 1.34 (dd, JHP = 1.2 Hz, JHRh = 0.60 Hz, 3H, CH3 (ring methyl)), 1.02 (dt, 3JHH = 8.27 Hz, 3JHP = 9.30 Hz, CH2CH3) and 0.56 (dt, 3JHH = 8.06 Hz, 3JHP = 9.30 Hz, CH2CH3). 13C NMR (75.4 MHz, C6D6) δ 121.30 (d, JCP = 11.90 Hz, C (ring)), 110.75 (d, JCP = 12.1 Hz, C (ring)), 105.66 (s, C (ring)), 95.36 (s, C (ring)), 84.60 (d, JCP = 7.18 Hz, C (ring)), 54.20 (s, C(O)CH3), 43.97 (d, 1JCP = 27.63 Hz, CH2 (11)), 23.39 (d, JCP = 26.48 Hz, CH2CH3), 19.25 (s, CH3 (10)), 16.17 (d, 2JCP = 24.17 Hz, CH2CH3), 9.83 (s, CH3 (ring methyl)), 9.37 (s, CH3, (ring methyl)), 9.08 (s, CH3 (ring methyl)), 8.73 (s, ring methyl) and 7.95 (s, CH2CH3). 31P NMR (121.4 Hz, C6H6) δ 68.57 (d, JPRh = 167.58 Hz). IR (petroleum ether) λmax 1629 cm−1. For numbering, see Scheme 1). Found C 40.71%, H 5.02%; C17H29IOPRh requires C 40.02%, H 5.73%.Carbonyl(κ2-1-diethylphosphinoethylcyclopentadienyl)methylrhodium(III) tetrafluoroborate
A sample of [Rh(C5H4(CH2)2PEt2)(CO)] (20 mg) was dissolved in neat MeI which, after 10 min, was removed with a stream of N2. The resulting sample of [Rh(C5H4(CH2)2PEt2)(C(O)Me)I] was redissolved in CH2Cl2 and treatment with a small excess of AgBF4 to give a yellow precipitate of AgI which was removed by filtration. The resulting solution displayed a single νCO band in the IR spectrum at 2059 cm−1 assigned to [Rh(C5H4(CH2)2PEt2)Me(CO)]BF4. The solvent was removed and NMR spectra of the product were recorded in CD2Cl2. 1H NMR δ 0.95 (dd, 3H, Rh–CH3, J = 2.3, 3.5 Hz), 1.04–1.22 (m, 6H, CH2CH3), 1.94–2.24, 2.39–2.72, 2.91–3.17 (each m, 4H, 2H, 2H resp., CH2) 5.55, 5.60, 5.80, 6.05 (each m, 1H, ring CH). 31P NMR δ 72.2 (d, JRhP = 129.5 Hz).Carbonyl(κ2-1-diethylphosphinoethyl-2,3,4,5-tetramethylcyclopentadienyl)methyliridium(III) iodide
A solution of [Ir(Cp′(CH2)2PEt2)(CO)] (0.10 g, 0.22 mmol) in THF (15 cm3) was treated with an excess of methyl iodide (2 cm3, 32.1 mmol) and stirred for 4 h. The solvent was removed in vacuo and the pale yellow product was recrystallised from an ethanol (10 cm3)/petroleum ether (2 cm3) solution (0.070 g, 53% yield). 1H NMR (CD2Cl2, 300 MHz) δ 3.39 (m, 2H, CH2 (11)), 2.59 (m, 2H, CH2 (10)), 2.43 (s, 3H, CH3 (ring methyl)), 2.27 (d, JHP = 3.07 Hz, CH3 (ring methyl)), 2.25 (m, 2H, CH2CH3), 2.02 (m, 2H, CH2CH3), 1.92 (s, 3H, CH3 (ring methyl)), 1.85 (d, JHP = 3.07 Hz, CH3 (ring methyl)), 1.19 (dt, 3JHH = 7.68 Hz, 3JHP = 12.08 Hz, 3H, CH2CH3), 1.12 (dt, 3JHH = 7.68 Hz, 3JHP = 11.52 Hz, 3H, CH2CH3) and 0.62 (d, 3JHP = 3.84 Hz, 3H, Ir–CH3). 31P NMR (CD2Cl2, 121.44 MHz) δ 32.07. For numbering, see Scheme 1. IR (CD2Cl2) λmax 2038 cm−1.(κ2-1-Diethylphosphinoethyl-2,3,4,5-tetramethylcyclopentadienyl)ethanoyliodocobalt(III)
A solution of [Co(Cp′(CH2)2PEt2)(CO)] (0.10 g, 0.31 mmol) in THF (15 cm3) was treated with an excess of methyl iodide (2 cm3, 32.1 mmol) and stirred for 4 h. The solvent was removed in vacuo and the red/brown solid product was recrystallised from petroleum ether (25 cm3) (0.090 g, 66% yield). 1H NMR (C6D6, 300 MHz) δ 3.41 (s, 3H, C(O)CH3), 2.28 (m, 2H, CH2 (10)), 2.00 (m, 2H, CH2CH3), 1.89 (d, JHP = 3.07 Hz, ring methyl), 1.72 (s, ring methyl), 1.44 (m, 2H, CH2 (11)), 1.23 (d, 3H, JHP = 1.54 Hz, ring methyl), 1.09 (m, 2H, CH2CH3), 0.96 (s, 3H, ring methyl), 0.64 (dt, 3JHH = 7.68 Hz, 3JHP = 15.62 Hz, CH2CH3) and 0.56 (dt, 3JHH = 7.43 Hz, 3JHP = 14.34 Hz, CH2CH3). 13C NMR (C6D6, 75.4 MHz) δ 114.50 (d, JCP = 11.00 Hz, ring C), 100.90 (d, JCP = 7.31 Hz, ring C), 98.17 (d, JCP = 5.50 Hz, ring C), 93.94 (d, JCP = 2.20 Hz, ring C), 78.99 (s, ring C), 53.30 (d, JCP = 7.16 Hz, C(O)CH3), 39.30 (d, JCP = 26.76 Hz, CH2 (11)), 21.90 (d, JCP = 25.64 Hz, CH2CH3), 17.87 (d, JCP = 3.66 Hz, CH2 (10)), 13.50 (d, JCP = 23.60 Hz, CH2CH3), 12.23 (s, ring methyl), 9.88 (s, ring methyl), 9.13 (s, ring methyl), 7.91 (s, ring methyl), 7.81 (d, JCP = 2.34 Hz, CH2CH3) and 7.78 (d, JCP = 2.48 Hz, CH2CH3). For numbering, see Scheme 1. 31P NMR (C6D6, 121.44 MHz) δ 69.10.Kinetic measurements
Reaction monitoring for kinetic experiments was achieved using a Perkin-Elmer GX FTIR spectrometer (2 cm−1 resolution) controlled by Spectrum TimeBase software. Pseudo-first order conditions were employed, with at least a 10-fold excess of MeI, relative to the Rh complex. A solution containing the required concentration of MeI in CH2Cl2 was prepared in a 5 cm3 graduated flask. A portion of this solution was used to record a background spectrum. Another portion (typically 500 µl) was added to the solid Rh complex in a sample vial to give a reaction solution containing typically 5–10 mmol dm−3 [Rh]. A portion of the reaction solution was quickly transferred to the IR cell and data collection was started. The IR cell (0.5 mm pathlength, CaF2 windows) was maintained at constant temperature by a thermostated jacket. Spectra (2200–1600 cm−1) were scanned and saved at regular time intervals under computer control. For the faster oxidative addition reaction of [Rh(Cp′(CH2)2PPh2)(CO)], a rapid mixing stopped-flow infrared accessory (Hi-Tech FMA-10) was used in combination with a Nicolet Magna 560 FTIR instrument fitted with an MCT detector and using OMNIC Series software. Absorbance vs. time data for the appropriate νCO bands were extracted and analyzed off-line using Kaleidagraph curve-fitting software. For each experiment, the decay of the appropriate νCO band was fitted to an exponential curve, with correlation coefficient ≥0.999, to give a pseudo-first order rate constant. Each kinetic run was repeated at least twice to check reproducibility, the kobs data given being averaged values with component measurements deviating from each other by ≤5%.High pressure NMR studies
High pressure NMR studies were carried out in a sapphire tube fitted with a titanium head. The tube was loaded using standard Schlenk line and catheter tubing techniques.In a typical procedure, [Rh(Cp′(CH2)2PEt2)(CO)] (0.02 g, 0.05 mmol) was dissolved in a stock solution of D2O : ethanoic acid : methyl ethanoate : methyl iodide (1 : 6 : 2 : 1) (2 cm3). The resulting solution was transferred to the previously degassed sapphire cell and pressurized with approximately 15 bar of carbon monoxide. The cell was allowed to stand for 15 h before the first spectrum was collected at room temperature. The temperature was increased in increments of 10 °C and equilibrated at each temperature for 30 min before a spectrum was recorded. This process was repeated until a final temperature of 90 °C was reached. The NMR cell was cooled to room temperature.
Similar techniques were used for: [Cp*Rh(PEt3)(CO)] (0.06 g, 0.20 mmol), [Rh(Cp(CH2)2PEt2)(CO)] (0.04 g, 0.13 mmol), and [Co(Cp′(CH2)2PEt2)(CO)] (0.09 g, 0.28 mmol). Similar studies were also carried out on [Co(Cp′(CH2)2PEt2)(CO)] (0.04 g, 0.13 mmol) in 2 cm3 of a stock solution of deuteriated methanol (5 cm3) : methyl iodide (0.5 cm3) : water (0.35 cm3).
Catalytic experiments
These experiments were carried out in a Hastelloy autoclave (30 cm3) attached via a mass flow controller to a ballast vessel (37.8 cm3) so that constant pressure and temperature could be maintained within the autoclave. The autoclave was fitted with a stirrer, heater, catalyst injector, internal thermocouple and pressure transducer for the autoclave and ballast vessel.Throughout the course of these experiments the catalyst was either introduced to the autoclave via the catalyst injector at temperature and pressure or added to the autoclave initially. For the latter procedure methyl iodide was added via the catalyst injector at temperature and pressure to initiate the reaction. A solution was added to the thoroughly carbon monoxide flushed autoclave (through the autoclave side-arm, via a syringe and needle while under continuous CO flush). The autoclave was flushed with three cycles of approximately 30 bar of carbon monoxide and finally pressurised to 10 bar. The autoclave was heated to approximately 150 °C (internal temperature) and once semi-stabilised at approximately 150 °C, the pressure in the autoclave was raised to 23 bar and the temperature again allowed to stabilise. A second solution was added through the catalyst injector and the pressure brought up to 27 bar. The pressure in the ballast vessel was monitored electronically every five seconds. After 2 h the autoclave was cooled and vented.
X-Ray crystallography
Data were collected using a Bruker SMART diffractometer, graphite monochramated MoKα radiation (λ = 0.71073 Å). Lorentzian polarization and absorption corrections were performed. The structures were solved by direct methods. The non-hydrogen atoms were refined anisotropically, the hydrogen atoms were idealized and refined isotropically using a riding model. Structural refinements were performed with the full-matrix least-squares method on F2 for all data.25 Full details in Table 1. The structure determination of [Ir(Cp′(CH2)2PEt2)(CO)(Me)]I proved difficult due to poor crystal quality. We examined several different crystals and the reported data are the most satisfactory, there are some high residual peaks in the final Fourier map which reflects the difficulties associated with this data collection.| Compound | [Ir(Cp′(CH2)2PEt2)(CO)] | [Co(Cp′(CH2)2PEt2)(CO)] | [Ir(Cp′(CH2)2PEt2)(CO)(Me)]Ia | [Co(Cp′(CH2)2PEt2)(COMe)I] | [Co2(Cp′H(CH2)2Ph2)2(CO)6] | [Co2(C5Et4HSi(Me)2CH2PMe2)2(CO)6] |
|---|---|---|---|---|---|---|
| a Co-crystallises with [Ph3PMe]I (1 : 1). | ||||||
| Empirical formula | C16H26IrOP | C16H26CoOP | C36H47I2IrOP2 | C17H29CoIP | C52H52Co2O6P2 | C42H68Co2O6P2Si2 |
| Formula weight | 457.54 | 324.27 | 1003.68 | 466.20 | 952.74 | 904.94 |
| Temperature/K | 293(2) | 125(2) | 125(2) | 125(2) | 293(2) | 293(2) |
| System | Monoclinic | Orthorhombic | Triclinic | Monoclinic | Monoclinic | Triclinic |
| Space group | P21/c | P212121 |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P21/c | P21/c |
P |
| a/Å | 7.1629(4) | 7.1336(13) | 9.030(2) | 16.750(7) | 9.273(2) | 8.700(3) |
| b/Å | 16.6122(9) | 13.999(3) | 10.604(3) | 8.612(4) | 18.559(4) | 10.216(3) |
| c/Å | 14.3026(8) | 16.244(3) | 20.661(6) | 14.429(6) | 15.020(3) | 14.368(5) |
| α/° | 90 | 90 | 93.879(4) | 90 | 90 | 92.900(7) |
| β/° | 102.5260(10) | 90 | 101.951(4) | 114.170(7) | 106.736(5) | 96.994(6) |
| γ/° | 90 | 90 | 105.618(4) | 90 | 90 | 100.669(6) |
| Volume/Å3 | 1661.38(16) | 1622.2(5) | 1847.9(8) | 1898.8(14) | 2475.6(8) | 1242.2(7) |
| Z | 4 | 4 | 2 | 4 | 4 | 2 |
| μ/mm−1 | 8.124 | 1.147 | 5.396 | 2.611 | 0.781 | 0.820 |
| Reflections collected | 8179 | 6927 | 9137 | 7800 | 10612 | 6358 |
| Independent reflections | 2373 [R(int) = 0.0252] | 2276 [R(int) = 0.0697] | 5167 [R(int) = 0.1184] | 2655 [R(int) = 0.1279] | 3556 [R(int) = 0.2206] | 3569 [R(int) = 0.1209] |
| Max./min. transmission | 1.00000 and 0.559729 | 1.00000 and 0.762953 | 1.00000 and 0.525955 | 1.00000 and 0.460717 | 1.000000 and 0.765508 | 0.4229 and 0.2258 |
| Final R | R1 = 0.0212, wR2 = 0.0585 | R1 = 0.0467, wR2 = 0.1124 | R1 = 0.1076, wR2 = 0.2442 | R1 = 0.0695, wR2 = 0.1514 | R1 = 0.0716, wR2 = 0.0893 | R1 = 0.0890, wR2 = 0.2104 |
| Largest diff. peak and hole/e Å−3 | 0.724 and −0.705 | 0.875 and −0.742 | 3.748 and −2.237 | 1.665 and −1.335 | 0.282 and −0.229 | 0.593 and −0.925 |
The experimental details for the structure determinations of [Rh(Cp′(CH2)2PEt2)(CO)], [Rh(Cp′(CH2)2PPh2)(CO)], [Rh(Cp(CH2)2PEt2)(CO)], [Rh(Cp′(CH2)2PEt2)(COMe)I], [Rh(Cp′(CH2)2PEt2)Cl2], [Rh(Cp′(CH2)2PEt2)I2] (compounds 1, 2, 3, 9, 10 and 11 in ref. 19, CCDC reference numbers 197739–197744) have already been reported.26
CCDC reference numbers 282064–282069.
For crystallographic data in CIF or other electronic format see DOI: 10.1039/b512054c
Results and discussion
Ligand synthesis
Tertiary (phosphanylalkyl)cyclopentadienyl ligands have been prepared by a variety of routes including reactions of fulvenes with LiPR2 (R = aryl),27–31 alkylation of cyclopentadienyl anions with 3-chloropropyldiphenyl phosphine,32 reaction of a substituted cyclopentadienone with allylMgBr, followed by hydroboration and oxidative hydrolysis to give the hydroxypropylcyclopentadienyl complex which was reacted with TsCl (Ts = 4-MeC6H4SO2) followed by LiPPh2,33 and reactions of spirohydrocarbons with LiPR2 (R = Ph, Pri or But).22,34–37 Related ligands with an aryl bridge have been synthesised by HF elimination from coordinated cyclopentadienyl and fluorinated aryl phosphine ligands.38–42 We attempted many of these routes but there were problems with all of them except that starting from the spirohydrocarbon.Deprotonation of tetramethylcyclopentadiene followed by reaction with X(CH2)2Cl (X = Br or OTs) can give three isomeric products (Scheme 1).43 There is some dispute in the literature36 as to the relative amounts of these products, but it seems clear that largely the geminal isomers, which are unsuitable for coordination since they cannot form cyclopentadienyl ligands by deprotonation, are formed when using TsO(CH2)2Cl, although subsequent reaction with LiPPh2 does give some of the desired 1-(diphenylphosphanylethyl)-2,3,4,5-tetramethylcyclopentadiene.34,35 Better results have been obtained using 1-bromo-3-chloroethane, with the desired non-geminal isomer being formed in 84% yield and the geminal isomers making up the remaining 16%.22 In our hands, the ratio of geminal to non-geminal isomers was closer to 1 : 1 at full conversion (30 days). Reaction of this mixture with BuLi produced a mixture of the desired spirohydrocarbon and other products, which were separated by column chromatography. The spirohydrocarbon was obtained in 45% yield. The desired (diethylphosphanylethyl)-2,3,4,5-tetramethylcyclopentadienyl lithium was synthesised by the reaction of the spirohydrocarbon with lithium diethylphosphide in THF at −60 °C. Similar methods were used to prepare R2P(CH2)2C5R′4Li (R = Et or Ph, R′ = H or Me). Me2PCH2SiMe2C5Et4HLi was synthesised from the reaction of [C5Et4H]− with Me2SiCl2 followed by LiCH2PMe2.
Complexation
| Compound | δP (JRhP/Hz) | ν CO (hexane)/cm−1 |
|---|---|---|
a Pentane.
b Light petroleum.
c
ν
C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O.
d CH2Cl2. O.
d CH2Cl2.
|
||
| Li[C5H4(CH2)2PEt2] | −25.1 | |
| Li[Cp′(CH2)2PEt2] | −21.9 | |
| Li[Cp′H(CH2)2PPh2] | −15.1 | |
| C5Et4HSiMe2CH2PMe2 | −53.9 | |
| [Rh(C5H4(CH2)2PEt2)(CO)] | 80.3 (191) | 1938 |
| [CpRh(PEt3)(CO)] | 1944 | |
| [Rh(C5H4(CH2)2PPh2)(CO)] | 1947a | |
| [CpRh(PPh3)(CO)] | 1953a | |
| [Rh(Cp′(CH2)2PEt2)(CO)] | 76.3 (189) | 1924 |
| [Cp*Rh(PEt3)(CO)] | 1920 | |
| [Rh(Cp′(CH2)2PPh2)(CO)] | 52.1 (175) | 1933 |
| [Cp*Rh(PPh3)(CO)] | 1947a | |
| [Rh(C5Et4SiMe2CH2PMe2)(CO)] | 15.2 (185) | 1928b |
| [Co(Cp′(CH2)2PEt2)(CO)] | 87.6 | 1910b |
| [Ir(Cp′(CH2)2PEt2)(CO)] | 37.7 | 1910b |
| [Rh(Cp′(CH2)2PEt2)(C(O)Me)I] | 68.6 (168) | 1629b,c |
| [Co(Cp′(CH2)2PEt2)(C(O)Me)I] | 69.1 | |
| [Ir(Cp′(CH2)2PEt2)Me(CO)]I | 32.1 | 2038d |
| [Rh(Cp(CH2)2PEt2)Me(CO)]BF4 | 72.2 (130) | 2059d |
| [Rh(Cp′(CH2)2PEt2)Cl2] | 72.8 (147) | |
| [Rh(Cp′(CH2)2PPh2)Cl2] | 51.5 (157) | |
| [Rh(Cp′(CH2)2PEt2)I2] | 63.7 (148) | |
| [Co2(CO)6(Cp′H(CH2)2PPh2)2] | 58.2, 57.6, 55.8 | 2078, 2009, 1993, 1951 |
| [Co2(CO)6(C5Et4HSiMe2CH2PMe2)2] | 32.3 | 1969, 1951b |
An aliquot of the solution taken before the start of the reflux period showed, in addition to resonances from the final product, extra doublets around δ 40. These may arise from dimeric complexes with bridging Cp′(CH2)2PEt2 ligands. Such dimers have previously been reported to be intermediates in the synthesis of [Rh(C5H4(CH2)2PPh2)(CO)] and exist in cisoid and transoid forms.21
The structures of each of the previously unreported complexes, [Rh(C5R′4(CH2)2PR2)(CO)] (R = Et, R′ = H or Me, R = Ph, R′ = Me) were confirmed crystallographically. For R = Et, R′ = Me, the structure is shown in Fig. 1, whilst for the other complexes they are in the ESI.†
![Single crystal X-ray molecular structure and numbering scheme for [Rh(Cp′(CH)2PEt2)(CO)].](/image/article/2006/DT/b512054c/b512054c-f1.gif) | ||
| Fig. 1 Single crystal X-ray molecular structure and numbering scheme for [Rh(Cp′(CH)2PEt2)(CO)]. | ||
The complex [Rh(C5Et4SiMe2CH2PMe2)(CO)] was also synthesised and spectroscopically characterised as having a similar composition to those of the other complexes. The P atom resonates at δ 15 compared with −53.9 for the free ligand and 32.6 for the ligand bound only through P to cobalt. In this case, it is less clear that the complex is monomeric and a dimer (Fig. 2) similar to those formed initially during the synthesis of [Rh(C5H4(CH2)2PPh2)(CO)]21 may predominate. The IR spectrum of [Rh(C5Et4SiMe2CH2PMe2)(CO)] contains a strong carbonyl band at 1929 cm−1, assigned to the complex, but two weaker bands at 2029 and 1967 cm−1. The similarity of these absorptions to those of [Cp*Rh(CO)2] (2027, 1964 cm−1) suggest that this may arise from [Rh(C5Et4SiMe2CH2X)(CO)2] (X = Cl or PMe2). There is some evidence that LiCH2PMe2 deprotonates C5Et4HSiMe2Cl in competition with substitution of the SiCl bond since the 31P NMR spectrum of a crude sample of C5Et4SiMe2CH2PMe2 also contained a resonance from PMe3.
![Possible dimeric structure for [Rh(C5Et4SiMe2CH2PMe2)(CO)].](/image/article/2006/DT/b512054c/b512054c-f2.gif) | ||
| Fig. 2 Possible dimeric structure for [Rh(C5Et4SiMe2CH2PMe2)(CO)]. | ||
The values of νCO for the rhodium complexes are shown in Table 2 along with those of the analogous complexes which do not contain the bridge between the cyclopentadienyl ring and the phosphine. In most cases, except [Rh(Cp′(CH)2PEt2)(CO)], the complexes containing the bidentate ligands have lower νCO values. As expected on the basis of their better electron donating properties, ethyl groups on P cause lower νCO than phenyl groups and methyl substituents on the cyclopentadienyl moiety also lower νCO, the latter effect being slightly more marked (compare [Rh(Cp′(CH2)2PPh2)(CO)] (1933 cm−1) with [Rh(C5H4(CH)2PEt2)(CO)] (1937 cm−1)). The drop in νCO on introducing the bridge arises when an extra electron donating group is introduced into the complexes, the (CH2)2 bridge replacing either a phenyl group on P or an H on the cyclopentadienyl ring. In contrast, for [Rh(Cp′(CH)2PEt2)(CO)], the bridge replaces an ethyl group on P and a methyl group on the cyclopentadienyl ring, reducing the overall number of electron donating methyl or methylene groups and leading to a slightly decreased electron density and higher value for νCO.
Reactions of phosphanylalkylcyclopentadienes with [Co2(CO)8] led to the formation of dimeric products of the form [Co2(CO)6L2], which were crystallographically characterised for L = Cp′H(CH)2PPh2 (Fig. 3) and C5Et4HSiMe2CH2PMe2 (ESI†), even if they were carried out in the presence of cyclohexene, which assists the formation of cyclopentadienyl complexes in reactions of this kind by acting as a hydrogen acceptor.46 The X-ray structures of both complexes show that the ligand is coordinated only through P and that the P–Co–Co–P is almost linear (P–Co–Co = 177°), as is usual for complexes of this type,47–49 with the carbonyl ligands occupying the equatorial positions of a trigonal bipyramid. The 31P NMR spectrum of [Co2(CO)6(Cp′H(CH)2PPh2)2] shows 6 different resonances. These arise because the protonated form of the ligand exists as a mixture of 3 different isomers, depending on the site of ring protonation (see Scheme 1). In the dimeric complex, the two ligands can be the same (3 resonances) or different (6 resonances). Presumably some of the expected 9 resonances overlap. 31P and 1H NMR studies suggest that the related complex, [Co2(CO)7(Et2P(CH2)2Cp′H)] (31P 55.8, 57.6, 58.2, from the three isomers of the protonated ligand, νCO 2078, 2009, 1993, 1951) forms from [Co2(CO)8] and Cp′H(CH2)2PEt2 at 100 °C.
![X-Ray molecular structure and numbering scheme for [Co2(CO6)(Cp′H(CH)2PPh2)2].](/image/article/2006/DT/b512054c/b512054c-f3.gif) | ||
| Fig. 3 X-Ray molecular structure and numbering scheme for [Co2(CO6)(Cp′H(CH)2PPh2)2]. | ||
[Co(Cp′(CH2)2PEt2)(CO)] was successfully prepared, however, by the reaction of [Co2(CO)8] with Li[Cp′(CH2)2PEt2] (1 : 1) in THF at 170 °C in an autoclave. The complex was characterised crystallographically (see ESI). The 31P NMR resonance was at δ 87.6 and νCO at 1910 cm−1. IR and NMR studies of this reaction suggest that the first complex formed at room temperature may be [Co2(CO)7(Et2P(CH2)2Cp′)]− (31P δ 56.9), but that this converts into the desired product by attack of the [Me4C5]− moiety onto the cobalt atom which is already bound to P with [Co(CO)4]− (inferred from a strong absorption in the IR at 1890 cm−1, lit.,50 1888 cm−1) acting as the leaving group (Scheme 2). In this mechanism, the phosphine and cyclopentadienyl ligands are coordinated to the cobalt in the opposite order to that in the reaction of [CoI(CO)4] with [C5H4(CH2)2PBut2]−, because the lack of a good anionic leaving group on Co makes CO displacement by the phosphine more facile than nucleophilic attack of the cyclopentadienyl moiety.
![Proposed mechanism of formation of [Co(Cp′(CH2)2PEt2)(CO)].](/image/article/2006/DT/b512054c/b512054c-s2.gif) | ||
| Scheme 2 Proposed mechanism of formation of [Co(Cp′(CH2)2PEt2)(CO)]. | ||
Catalytic carbonylation of methanol
Various of the rhodium complexes and some standards were tested for methanol carbonylation at 150 °C under CO (27 bar) in the presence of water and methyl iodide. The results are collected in Table 3. Linear gas uptake plots were observed, suggesting zero order dependence on [methanol] and good catalyst stability. At higher concentrations of [Rh(Cp′(CH2)2PEt2)(CO)] (0.105 mol dm−3, initial rate 11.4 mol dm−3 h−1) [Rh(Cp′(CH2)2PEt2)I2] (X-ray structure in Fig. 4) was isolated from the final solution, suggesting that the ligand remains intact and coordinated in a bidentate fashion to Rh. This complex is presumably formed by reaction of [Rh(Cp′(CH2)2PEt2)(CO)] with 2 mol of HI and is analogous to [RhI4(CO)2]−, which can be formed from methanol carbonylation solutions containing [RhI2(CO)2]− at low water concentrations2 or on cooling and depressurising. [Rh(Cp′(CH2)2PEt2)I2] was inactive for methanol carbonylation. In the case of [Cp*Rh(PEt3)(CO)], the active species is believed to be [Cp*Rh(CO)2] because HPNMR studies (see later) show that all the phosphine is oxidised. Interestingly, the most active of all the catalysts is [Rh(Cp′(CH2)2PPh2)(CO)], giving a rate (0.81 mol dm−3 h−1, [Rh] = 1.25 × 10−3 mol dm−3) slightly higher than from [RhI2(CO)2]− (0.64 mol dm−3 h−1). The most electron rich of the new complexes, [Rh(Cp′(CH2)2PEt2)(CO)] gives a lower rate of 0.38 mol dm−3 h−1, which is similar to that for [Cp*Rh(PEt3)(CO)], which decomposes to [Cp*Rh(CO)2].| Catalyst | Rate/mol dm−3 h−1 |
|---|---|
| a For conditions, see Experimental section. | |
| [Rh(CO)I2]− | 0.64 |
| [Cp*Rh(PEt3)(CO)] | 0.30 |
| [Rh(Cp′(CH2)2PEt2)(CO)] | 0.38 |
| [Rh(Cp′(CH2)2PPh2)(CO)], | 0.81 |
| [Rh(Cp′(CH2)2PEt2)I2] | 0 |
![X-Ray molecular structure and numbering scheme for [Rh(Cp′(CH2)2PEt2)I2].](/image/article/2006/DT/b512054c/b512054c-f4.gif) | ||
| Fig. 4 X-Ray molecular structure and numbering scheme for [Rh(Cp′(CH2)2PEt2)I2]. | ||
At 150 °C, [Ir(Cp′(CH2)2PEt2)(CO)] showed no activity for methanol carbonylation, but slow gas uptake was observed at 190 °C. At this temperature, the rate was similar to that obtained with [IrCl(CO)3]n, suggesting that [Ir(Cp′(CH2)2PEt2)(CO)] is not stable at this high temperature. [Co(Cp′(CH2)2PEt2)(CO)] also showed no activity for methanol carbonylation under any of the following conditions (120 °C, 100 bar; 150 °C, 30 bar or 150 °C, 100 bar), which is consistent with the observations from the high pressure 31P NMR studies, which show decomposition of the complex under catalytic conditions at 60–90 °C. These results suggest that the very active but unstable catalyst obtained14 from [Cp*Co(CO)2] and PEt3 may not be [Cp*Co(CO)(PEt3)] as previously proposed. A small amount of activity was obtained under low water conditions, but it ceased after a short time. At the end of the reaction, the solution was pale pink, suggesting complete catalyst degradation.
Stoichiometric reactions
In order to gain more insight into the catalytic reactions, we have carried out a variety of stoichiometric reactions of the catalyst precursors, backing them up with high pressure spectroscopy (see later). Our intention was to prepare very highly nucleophilic metal complexes in order to speed up the oxidative addition of methyl iodide during methanol carbonylation. An initial indication of the very high nucleophilicity of the rhodium centre came from the observation that [Rh(Cp′(CH2)2PEt2)(CO)] converted into [Rh(Cp′(CH2)2PEt2)Cl2] on standing in CD2Cl2 for 24 h. This may proceed by initial nucleophilic attack on CD2Cl2, a reaction which only occurs for highly nucleophilic metal centres51 or possibly by a free radical reaction.52 [Rh(Cp′(CH2)2PEt2)Cl2] was fully characterised crystallographically (ESI†), the structure being very similar to that for [Rh(Cp′(CH2)2PEt2)I2]. That the chlorides come from CD2Cl2 rather than HCl impurities was confirmed by repeating the reaction using CD2Cl2, which had been pretreated with NaHCO3. [Rh(Cp′(CH2)2PEt2)Cl2] was still formed.Reaction of [M(Cp′(CH2)2PEt2)(CO)] (M = Co or Rh) with MeI led smoothly to the formation of [M(Cp′(CH2)2PEt2)(C(O)Me)I], which were characterised crystallographically (Fig. 5, M = Co, ESI† for M = Rh). These ethanoyl complexes are formed by nucleophilic attack of the metal centre on MeI followed by methyl migration to CO and coordination of I− (see Scheme 3). An interesting feature of the 1H NMR spectra of the ethanoyl complexes is that all four methyl groups on the cyclopentadienyl ring are now inequivalent because there is no plane of symmetry in the molecule. For the Rh complex, 1H{31P} studies showed that only the two low frequency signals (δ 1.34 and 1.43) are coupled to rhodium, whilst both sets of signals are coupled to P, the resonances at δ 2.09 and 1.43 have the higher coupling constants to P (5.4 and 2.7 Hz respectively) compared with 1.2 (δ 1.34) and 0.9 (δ 1.74). The reaction of [Ir(Cp′(CH2)2PEt2)(CO)] with MeI led to the methyl complex, [Ir(Cp′(CH2)2PEt2)Me(CO)]I¶ (Fig. 6), confirming the lack of migration in this iridium complex, which can be attributed to the high strength of the Ir–C bond. In the [IrI2(CO)2]− catalysed carbonylation of methanol, the rate of methyl migration on Ir(III) is the rate determining step. In the commercial process an iodide scavenging promoter, such as [RuI2(CO)3], aids dissociation of I− from [IrI3(CO)2Me)]− to allow coordination of CO giving neutral [IrI2(CO)3Me], in which the methyl migration occurs.9,53 Despite the positive charge on [Ir(Cp′(CH2)2PEt2)Me(CO)]+ migration does not readily occur, again confirming the very high electron density placed on the metal by the Cp′(CH2)2PEt2 ligand.
![X-Ray molecular structure and numbering scheme for [Co(Cp′(CH2)2PEt2)(C(O)Me)I].](/image/article/2006/DT/b512054c/b512054c-f5.gif) | ||
| Fig. 5 X-Ray molecular structure and numbering scheme for [Co(Cp′(CH2)2PEt2)(C(O)Me)I]. | ||
![X-Ray structure and numbering scheme for [Ir(Cp′(CH2)2PEt2)Me(CO)]+.](/image/article/2006/DT/b512054c/b512054c-f6.gif) | ||
| Fig. 6 X-Ray structure and numbering scheme for [Ir(Cp′(CH2)2PEt2)Me(CO)]+. | ||
![General mechanism for the reactions of MeI with [M(C5R4(CH2)2PR′2)(CO)]. For M = Co and Rh the acetyl complex is isolated whereas for M = Ir the methyl complex is obtained.](/image/article/2006/DT/b512054c/b512054c-s3.gif) | ||
| Scheme 3 General mechanism for the reactions of MeI with [M(C5R4(CH2)2PR′2)(CO)]. For M = Co and Rh the acetyl complex is isolated whereas for M = Ir the methyl complex is obtained. | ||
The proposed rhodium methyl complex, [Rh(C5H4(CH2)2PEt2)Me(CO)]+ was observed as an intermediate during IR studies of the oxidative addition reaction (see below) and could also be generated as its tetrafluoroborate salt, [Rh(C5H4(CH2)2PEt2)Me(CO)]BF4 (νCO 2059 cm−1) from the reaction of [Rh(C5H4(CH2)2PEt2)(C(O)Me)I] with AgBF4 in CH2Cl2. The 1H NMR spectrum of [Rh(C5H4(CH2)2PEt2)Me(CO)]BF4 displayed a doublet of doublets for the methyl ligand, due to coupling with both P and Rh. Separate signals were present due to each of the four inequivalent hydrogens on the cyclopentadienyl ring. Addition of Bu4NI to a CH2Cl2 solution of [Rh(C5H4(CH2)2PEt2)Me(CO)]BF4 caused conversion back into [Rh(C5H4(CH2)2PEt2)(C(O)Me)I], as judged by IR spectroscopy.
Kinetic measurements of the reactions of MeI with several of the Rh complexes were conducted by IR spectroscopy and a representative example of a series of spectra, for [Rh(C5H4(CH2)2PEt2)(CO)], is given in Fig. 7. As the parent rhodium(I) complex (νCO 1918 cm−1) decays, a νCO band due to the methyl intermediate, [Rh(C5H4(CH2)2PEt2)Me(CO)]+ (2056 cm−1), grows and decays with the formation of the final product, [Rh(C5H4(CH2)2PEt2)(C(O)Me)I]. The νCO band of this compound is split into two component peaks (1640, 1663 cm−1, see Fig. 7), probably due to the ethanoyl ligand adopting two different rotameric conformations with respect to the Rh centre. The time dependences of the relative intensities of the IR signals of the various species are shown in Fig. 8.
![Series of IR spectra during the reaction of [Rh(C5H4(CH2)2PEt2)(CO)] with MeI (0.16 mol dm−3 in CH2Cl2, 16 °C).](/image/article/2006/DT/b512054c/b512054c-f7.gif) | ||
| Fig. 7 Series of IR spectra during the reaction of [Rh(C5H4(CH2)2PEt2)(CO)] with MeI (0.16 mol dm−3 in CH2Cl2, 16 °C). | ||
![Plot of absorbance vs. time for ν(CO) bands of Rh complexes during the reaction of [Rh(C5H4(CH2)2PEt2)(CO)] with MeI (0.08 M in CH2Cl2, 11 °C).](/image/article/2006/DT/b512054c/b512054c-f8.gif) | ||
| Fig. 8 Plot of absorbance vs. time for ν(CO) bands of Rh complexes during the reaction of [Rh(C5H4(CH2)2PEt2)(CO)] with MeI (0.08 M in CH2Cl2, 11 °C). | ||
The exponential decay of the reactant νCO band indicates a first order dependence of rate on the parent Rh(I) complex. Values of the pseudo first order rate constant, kobs, were measured over a range of MeI concentrations and temperatures and are listed in the ESI.† A linear dependence of kobs on [MeI] (Fig. 9) demonstrates that the reaction is also first order in MeI and the second order rate constant, k1, was obtained from the slope of this plot. Rate constants for reactions of MeI with [Rh(C5H4(CH2)2PEt2)(CO)] and [Rh(Cp′(CH2)2PPh2)(CO)] are listed in Table 4, together with comparative values for [Cp*Rh(CO)2] and [CpRh(CO)(PPh3)].55 These data display a clear correlation between the electron density on the metal (as revealed by νCO in the starting material) and the rate constant (k1) for reaction with methyl iodide. The electron donating power of the methyl groups on the ring and of the phosphine contribute to making the relative rate constants (krel) increase in the order [Cp*Rh(CO)]2 (1) < [CpRh(CO)(PPh3)] (3.3) < [Rh(C5H4(CH2)2PEt2)(CO)] (74) < [Rh(Cp′(CH2)2PPh2)(CO)] (2100). To our knowledge, the rate constant for oxidative addition of MeI to [Rh(Cp′(CH2)2PPh2)((CO)] is the highest yet reported for a Rh(I) carbonyl complex, surpassing that recently found for a square planar complex containing a tridentate bis(imino)carbazolide ligand (k = 1.66 dm3 mol−1 s−1).56 Qualitative observations indicated that the rate for [Rh(Cp′(CH2)2PEt2)(CO)] is even faster, but quantitative measurements have not been made using this complex. Activation parameters for the oxidative addition reactions, as determined from Eyring plots (ESI†) are also shown in Table 4. The large negative entropies of activation are consistent with an SN2 mechanism for attack of the rhodium centre at the C atom of MeI. The faster oxidative addition for [Rh(Cp′(CH2)2PPh2)(CO)] relative to [Rh(C5H4(CH2)2PEt2)(CO)] can be attributed to a smaller activation enthalpy for the more electron rich complex, perhaps indicating a stronger Rh–Me partial bond in the SN2 transition state.
![Plot of kobsvs. [MeI] for the reaction of [Rh(C5H4(CH2)2PEt2)(CO)] with MeI in CH2Cl2 at 16 °C.](/image/article/2006/DT/b512054c/b512054c-f9.gif) | ||
| Fig. 9 Plot of kobsvs. [MeI] for the reaction of [Rh(C5H4(CH2)2PEt2)(CO)] with MeI in CH2Cl2 at 16 °C. | ||
The experimental activation parameters can be used to extrapolate rate constants for oxidative addition at higher temperatures, corresponding to the conditions of the catalytic experiments. At 150 °C, calculated rate constants are 15 and 150 dm3 mol−1 s−1 for [Rh(C5H4(CH2)2PEt2)(CO)] and [Rh(Cp′(CH2)2PPh2)(CO)] respectively, which are 3–4 orders of magnitude higher than that calculated for [RhI2(CO)2]− (0.014 dm3 mol−1 s−1) at the same temperature. These very fast rates make it unlikely that oxidative addition remains the rate determining step in the catalytic carbonylation cycle, as discussed later.
The rate constants for migration of the methyl group onto CO in [Rh(C5H4(CH2)2PEt2)Me(CO)]+, [Rh(Cp′(CH2)2PPh2)Me(CO)]+ and [Rh(Cp′(CH2)2PEt2)Me(CO)]+ were also measured. The methyl complexes were generated in situ by dissolving their Rh(I) precursors in CH2Cl2 containing a high concentration (2 mol dm−3) of MeI. Under these conditions the oxidative addition step was effectively complete in the time of mixing and the methyl migration step could be monitored in isolation. The νCO bands of the products [Rh(Cp′(CH2)2PPh2)(C(O)Me)I] and [Rh(Cp′(CH2)2PEt2)(C(O)Me)I] formed at 1628 and 1623 cm−1 respectively. The former exhibits a high frequency shoulder, possibly due to a rotameric form. The lack of well resolved splitting of the νCO bands for these two complexes may indicate a stronger preference (than in [Rh(C5H4(CH2)2PEt2)(C(O)Me)I)]) for one rotameric conformation of the ethanoyl ligand, due to the steric bulk of the C5Me4 moiety. This is supported by inspection of a space-filling model of the X-ray structure of [Rh(Cp′(CH2)2PEt2)(C(O)Me)I)], in which the ethanoyl ligand is oriented so as to avoid clashes with methyl and ethyl substituents of the Cp′(CH2)2PEt2 ligand. The kinetic parameters are collected in Table 5, along with those for [Cp*Rh(CO)2Me.57
| Complex | ν CO/cm−1 | 103k2/s−1 | ΔH‡/kJ mol−1 | ΔS‡/J K−1 mol−1 |
|---|---|---|---|---|
| [Rh(C5H4(CH2)2PEt2)(CO)Me]+ | 2056 | 14.7 | 69 ± 2 | −47 ± 7 |
| [Rh(Cp′(CH2)2PPh2)(CO)Me]+ | 2048 | 17.5 | 74 ± 3 | −29 ± 9 |
| [Rh(Cp′(CH2)2PEt2)(CO)Me]+ | 2044 | 6.7 | 80 ± 2 | −20 ± 5 |
[Cp*Rh(CO)2Me]+![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 57 57 |
2118, 2087 | 220 | 72 ± 2 | −16 ± 6 |
The more electron rich metal complexes have slower rates of methyl migration, than [Cp*RhMe(CO)2]+, but the rate constant for [Rh(C5H4(CH2)2PEt2)Me(CO)]+ is slightly lower than for [Rh(Cp′(CH2)2PPh2)Me(CO)]+, despite the lower electron density of the former. This may be because the higher steric bulk of the methylated cyclopentadienyl ring and of the phenyl groups on P promote migration to relieve steric strain. The rate constant for methyl migration in [Rh(Cp′(CH2)2PEt2)Me(CO)]+ is the slowest of the ones measured in this study. Activation parameters for methyl migration obtained from Eyring plots (ESI†) are shown in Table 5. The value of ΔH‡ tends to increase with the donor strength of the (C5R4(CH2)2PR′2) ligand, indicating that increased Rh → CO π back donation (accompanied perhaps by Rh–Me bond strengthening) raises the enthalpic barrier to methyl migration.
High pressure spectroscopic studies
Because the rate constants for oxidative addition of MeI for the phosphanylethylcyclopentadienyl ligands are so high, it is possible that the rate determining step for methanol carbonylation may no longer be oxidative addition of methyl iodide, especially as the rate of reductive elimination of MeC(O)I from the highly electron rich ethanoyl complexes is likely to be substantially reduced. We, therefore, carried out spectroscopic studies under methanol carbonylation conditions (CO (30 bar) in ethanoic acid containing methyl ethanoate, methyl iodide and deuteriated water, with the same composition as used for the catalytic studies), but at slightly lower temperature, in order to try to observe the species present during catalysis, but also to investigate the stability of the complexes.Although [Rh(C5H4(CH2)2PEt2)(CO)] and [Cp*Rh(CO)(PEt3)] gave 31P NMR signals from unidentified rhodium complexes at room temperature under CO, heating the solutions to 90 °C led to the formation of broad singlets at δ 82 (75 at 25 °C) and 82 (75.5 at 25 °C), which we assign to the phosphine oxides, which will give these high shifts in a protic environment.11 In both cases small amounts of rhodium complexes were present, but these could not be assigned to any of the complexes in the catalytic cycle. In contrast, [Rh(Cp′(CH2)2PEt2)(CO)] at room temperature showed four doublets from rhodium containing complexes. Two of these (δ 73.8, d, JRhP = 116 Hz, 71.0, d JRhP = 134 Hz) were from unidentified complexes, whilst the other two were from [Rh(Cp′(CH2)2PEt2)(C(O)Me)I] (δ 65.2, d, JRhP = 165 Hz) and [Rh(Cp′(CH2)2PEt2)I2] (δ 61.5, d, JRhP = 146 Hz). On heating to 90 °C, all of these resonances were still present, with those at δ 71.0 and 61.5 dominating, but they all converted into [Rh(Cp′(CH2)2PEt2)I2] over 90 min at 90 °C. These results not only confirm that the [Rh(Cp′(CH2)2PEt2)] unit is more stable than its analogue without the bridge between the cyclopentadienyl and phosphine moieties, [Cp*Rh(PEt3)(CO)], but also that the methyl groups on Cp are essential for this extra stability. The observation that [Rh(Cp′(CH2)2PEt2)(C(O)Me)I] is present but not [Rh(Cp′(CH2)2PEt2)(CO)], suggests that the rate of oxidative addition has been accelerated so much in this system that reductive elimination of MeC(O)I has become the rate determining step of the catalytic reaction. The formation of [Rh(Cp′(CH2)2PEt2)I2], which was also isolated from catalytic reactions, is a problem, however, because, unlike [RhI4(CO)2]−, it does not appear to be reintroduced into the catalytic cycle by reaction with water. This is because regeneration of the Rh(I) species in these systems occurs by nucleophilic attack of water onto coordinated CO, which is not possible for [Rh(Cp′(CH2)2PEt2)I2], because it does not contain CO.58
The rate of methanol carbonylation using [Rh(Cp′(CH2)2PPh2)(CO)] was found to be higher than for [Rh(Cp′(CH2)2PEt2)(CO)], despite the fact that the latter carries higher electron density. This may arise if reductive elimination of MeC(O)I is rate determining in these systems, as suggested by the HPNMR studies using [Rh(Cp′(CH2)2PEt2)(CO)], since increasing the electron density on the metal would be expected to inhibit reductive elimination.
High pressure NMR studies on [Co(Cp′(CH2)2PEt2)(CO)] in the catalytic solution at room temperature showed a single resonance (δ 75.2, s), which did not correspond to either [Co(Cp′(CH2)2PEt2)(CO)] or [Co(Cp′(CH2)2PEt2)(C(O)Me)I] and was not from oxidised phosphine because it changed on heating to several peaks at δ 31–59, which probably correspond to complexes which do not contain the ligand bound through both the cyclopentadienyl and the phosphine moieties. Similar results were obtained under low water conditions. The signals in the final solution are not from phosphine oxide (expected signal at δ > 70), but could be from phosphine quaternised by MeI or HI.
X-Ray crystallography
Several of the complexes have been studied by X-ray crystallography (Table 6, Figs. 1 and 2–5). For complexes containing the same ligand, the M–P bonds are shorter in the cobalt complexes than in the Rh or Ir complexes, as expected on the basis of the sizes of the elements. The M–P bond lengths in [Rh(Cp′(CH2)2PEt2)(CO)] (2.2157(19)), [Rh(Cp′(CH2)2PPh2)(CO)] (2.2306(10)) and [Rh(Cp(CH2)2PEt2)(CO)] (2.1838(11)) can be rationalised by considering the σ-donation. The less donating PPh2 group results in the longest M–P bond, whilst the higher electron density on rhodium in [Rh(Cp′(CH2)2PEt2)(CO)] compared with [Rh(Cp(CH2)2PEt2)(CO)] also leads to a longer Rh–P bond. Steric effects may also influence these bond lengths. In all of the complexes containing η5 cyclopentadienyl ligands, the substituents at C3, the C atom which binds the phosphanylethyl group to the Cp ring, is planar, suggesting that there is no strain at this position. In the CH2CH2 bridge, it is apparent that C11C10C1 is relatively strain free in all of the complexes, though the other angles in the bridge (MPC11 and PC11C10) are generally contracted in the complexes where the cyclopentadienyl ring is coordinated when compared with the cobalt dimers where the CpH ring is not coordinated. The one exception is [Rh(Cp′(CH2)2PEt2)(CO)], for which only RhPC11 deviates significantly from that in [Co2(Cp′H(CH2)2Ph2)2(CO)6]. The optimum dihedral angles of the bridge are presumably 180° (anti) for PC11C10C1 and 60° (gauche) for MPC11C10 close to the values (177.6° and 66.2°) observed for [Co2(Cp′H(CH2)2Ph2)2(CO)6], in which the cyclopentadiene moiety is not coordinated. For the bidentate ligands a dihedral angle close to 180° for PC11C10C1 is not possible and both this angle and MPC11C10 are usually closer to 30°, indicating significant distortion towards an eclipsed conformation.| Compound | Bond lengths/Å | Bond angles/° | Dihedral angles/° | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| M–P | M–C(O) | C–O | P–M–C(O) | M–P–C11 | P–C11–C10 | C11–C10–C1 | Σ angles at C1 | P–C11–C10–C1 | M–P–C11–C10 | |
| a P–C11–Si. b C11–Si–C1. c P–C11–Si–C1. d Co–P–C11–Si. | ||||||||||
| [Rh(Cp′(CH2)2PEt2)(CO)] | 2.2157(19) | 1.814(8) | 1.163(8) | 94.9(2) | 103.2(2) | 112.6(5) | 112.7(6) | 360.0 | 36.6(8) | −28.8(6) |
| [Rh(Cp′(CH2)2PPh2)(CO)] | 2.2306(10) | 1.825(5) | 1.157(5) | 98.94(14) | 102.41(13) | 109.2(3) | 111.5(3) | 359.9 | 45.6(4) | −37.9(3) |
| [Ir(Cp′(CH2)2PEt2)(CO)] | 2.2145(12) | 1.820(5) | 1.158(6) | 93.46(15) | 103.81(18) | 103.81(18) | 113.7(4) | 359.9 | 36.6(6) | −28.4(5) |
| [Co(Cp′(CH2)2PEt2)(CO)] | 2.1177(15) | 1.710(6) | 1.166(7) | 93.64(18) | 103.9(2) | 108.6(5) | 110.8(5) | 359.9 | 35.3(6) | −36.8(7) |
| [Rh(Cp(CH2)2PEt2)(CO)] | 2.1838(11) | 1.800(4) | 1.171(6) | 92.81(14) | 99.4(2) | 100.5(6) | 110.6(6) | 53.4(12) | −53.6(6) | |
| [Rh(Cp′(CH2)2PEt2)Cl2] | 2.259(4) | 101.9(5) | 106.8(10) | 113.4(13) | 359.6 | −42.7(16) | 40.7(12) | |||
| [Rh(Cp′(CH2)2PEt2)I2] | 2.272(2) | 101.5(4) | 109.3(7) | 112.8(8) | 359.9 | 39.7(12) | −38.9(8) | |||
| [Rh(Cp′(CH2)2PEt2)(COMe)I] | 2.254(2) | 2.4036(7) | 1.0533 | 93.23(7) | 100.7(3) | 109.5(7) | 112.7(8) | 359.7 | 44.7(12) | −42.9(8) |
| [Ir(Cp′(CH2)2PEt2)(CO)(Me)]I | 2.273(2) | 1.846(8) | 1.163(9) | 97.0(3) | 102.4(3) | 108.8(4) | 112.8(7) | 359.6 | −38.0(8) | 40.0(6) |
| [Co(Cp′(CH2)2PEt2)(COMe)I] | 2.1872(11) | 2.064(5) | 1.011(5) | 94.88(11) | 102.94(12) | 108.2(3) | 110.2(4) | 360.0 | 38.8(4) | −39.6(3) |
| [Co2(Cp′H(CH2)2Ph2)2(CO)6] | 2.179(3) | 1.757 (av) | 1.157 (av) | 113.9(3) | 114.9(6) | 114.6(9) | 350.9 | −177.6(9) | 66.2(7) | |
| [Co2(C5Et4HSi(Me)2CH2PMe2)(CO)6] | 2.188(4) | 118.8(4) | 125.4(6)a | 106.2(5)b | 366.2 | −159.9(7)c | −58.3(8)d | |||
Conclusions
Highly electron rich complexes of Rh, Co and Ir have been synthesized by the combined use of permethylcyclopentadienyl and alkyl phosphine groups. These complexes have been stabilized by joining the phosphine to the cyclopentadienyl ligand via a C2 bridge. These new ligands all bind in a bidentate manner and the chelate effect ensures that, at least the rhodium complexes are more stable towards phosphine loss than e.g. [Cp*Rh(PEt3)(CO)]. This extra stability allows some of the complexes, such as [Rh(Cp′(CH2)2PEt2)(CO)] and [Rh(Cp′(CH2)2PPh2)(CO)] to give active catalysts which survive the harsh conditions of methanol carbonylation. The most active methanol carbonylation catalyst is [Rh(Cp′(CH2)2PPh2)(CO)]. Kinetic studies show that the rates of oxidative addition of methyl iodide to the rhodium(I) complexes are amongst the highest yet recorded, but the migration of the methyl group onto the carbonyl is retarded. HPNMR studies on methanol carbonylation catalysed by [Rh(Cp′(CH2)2PEt2)(CO)] suggest that reductive elimination of MeC(O)I, rather than oxidative addition of MeI is rate determining, thus explaining why [Rh(Cp′(CH2)2PPh2)(CO)] gives a higher rate of methanol carbonylation than [Rh(Cp′(CH2)2PEt2)(CO)]. These results therefore show that there is an optimum electron density on rhodium for obtaining high rates of methanol carbonylation. Below this optimum the rate determining step is oxidative addition of MeI whilst above it becomes reductive elimination of MeC(O)I.Acknowledgements
We thank BP Chemicals and the EPSRC for a studentships (A. E. C. M., P. I. P. E) and the EPSRC and Sasol for support of an upgrade of the NMR machine.References
- C. M. Thomas and G. Suss-Fink, Coord. Chem. Rev., 2003, 243, 125 CrossRef CAS.
- M. J. Howard, M. D. Jones, M. S. Roberts and S. A. Taylor, Catal. Today, 1993, 18, 325 CrossRef.
- M. J. Howard, G. J. Sunley, A. D. Poole, R. J. Watt and B. K. Sharma, Stud. Surf. Sci. Catal., 1999, 121, 61 CAS.
- R. T. Eby and T. C. Singleton, in Applied Industrial Catalysis, ed. B. E. Leach, Academic Press, New York, 1983 Search PubMed.
- T. W. Dekleva and D. Forster, J. Am. Chem. Soc., 1985, 107, 3568 CrossRef CAS.
- T. W. Dekleva and D. Forster, J. Mol. Catal., 1985, 33, 269 CrossRef CAS.
- D. Forster, J. Am. Chem. Soc., 1976, 98, 846 CrossRef CAS.
- T. W. Dekleva and D. Forster, Adv. Catal., 1986, 34, 81 CAS.
- G. J. Sunley and D. J. Watson, Catal. Today, 2000, 58, 293 CrossRef CAS.
- D. Forster, J. Am. Chem. Soc., 1975, 87, 951 CrossRef.
- J. Rankin, A. C. Benyei, A. D. Poole and D. J. Cole-Hamilton, J. Chem. Soc., Dalton Trans., 1999, 3771 RSC.
- J. Rankin, A. D. Poole, A. C. Benyei and D. J. Cole-Hamilton, Chem. Commun., 1997, 1835 RSC.
- A. C. Marr, P. Lightfoot, E. J. Ditzel, A. D. Poole, G. P. Schwarz, D. F. Foster and D. J. Cole-Hamilton, Inorg. Chem. Commun., 2000, 3, 617 CrossRef CAS.
- A. C. Marr, E. J. Ditzel, A. C. Benyei, P. Lightfoot and D. J. Cole-Hamilton, Chem. Commun., 1999, 1379 RSC.
- C. M. Thomas, R. Mafua, B. Therrien, E. Rusanov, H. Stoeckli-Evans and G. Suss-Fink, Chem.-Eur. J., 2002, 8, 3343 CrossRef CAS.
- C. A. Carraz, E. J. Ditzel, A. G. Orpen, D. D. Ellis, P. G. Pringle and G. J. Sunley, Chem. Commun., 2000, 1277 RSC.
- M. J. Baker, M. F. Giles, A. G. Orpen, M. J. Taylor and R. J. Watt, J. Chem. Soc., Chem. Commun., 1995, 197 RSC.
- L. Gonsalvi, H. Adams, G. J. Sunley, E. Ditzel and A. Haynes, J. Am. Chem. Soc., 2002, 124, 13597 CrossRef CAS.
- C. M. Thomas, A. Neels, H. Staeckli-Evans and G. Suss-Fink, Eur. J. Inorg. Chem., 2001, 3005 CrossRef CAS.
- A. E. C. McConnell, D. F. Foster, P. Pogorzelec, A. M. Z. Slawin, D. J. Law and D. J. Cole-Hamilton, Dalton Trans., 2003, 510 RSC.
- I. Lee, F. Dahan, A. Maisonnat and R. Poilblanc, Organometallics, 1994, 13, 2743 CrossRef CAS.
- J. A. M. van Beek and G. J. M. Gruter, Eur. Pat. Appl., 1997, 0805142.
- S. Doring and G. Erker, Synthesis, 2001, 43 CrossRef CAS.
- G. J. M. Gruter, J. A. M. van Beek, R. Green and E. G. Ijpeij, US Pat., 2000, 6117811.
- G. M. Sheldrick, SHELXTL Version 6.10, Bruker AXS, Madison, WI, 2001 Search PubMed.
- A. E. C. McConnell, D. F. Foster, P. Pogorzelec, A. M. Z. Slawin, D. Law and D. J. Cole-Hamilton, J. Chem. Soc., Dalton Trans., 2002, 510 Search PubMed.
- T. A. Mobley and R. G. Bergman, J. Am. Chem. Soc., 1998, 120, 3253 CrossRef CAS.
- B. Bosch, G. Erker and R. Frohlich, Inorg. Chim. Acta, 1998, 270, 446 CrossRef CAS.
- T. Heidemann and P. Jutzi, Synthesis, 1994, 777 CrossRef CAS.
- D. M. Bensley and E. A. Mintz, J. Organomet. Chem., 1988, 353, 93 CrossRef.
- N. E. Schore and B. E. Labelle, J. Org. Chem., 1981, 46, 2306 CrossRef CAS.
- R. T. Kettenbach, W. Bonrath and H. Butenschon, Chem. Ber./Recl., 1993, 126, 1657 CrossRef CAS.
- D. M. Bensley, E. A. Mintz and S. J. Sussangkarn, J. Org. Chem., 1988, 53, 4417 CrossRef.
- D. P. Krut'ko, M. V. Borzov, E. N. Veksler, R. S. Kirsanov and A. V. Churakov, Eur. J. Inorg. Chem., 1999, 1973 CrossRef CAS.
- D. P. Krut'ko, M. V. Borzov, E. N. Veksler, E. M. Myshakin and D. A. Lemenovskii, Russ. Chem. Bull., 1998, 47, 956 CrossRef CAS.
- P. Jutzi and J. Dahlhaus, Synthesis, 1993, 684 CrossRef CAS.
- T. Kauffmann, J. Ennen, H. Lhotak, A. Rensing, F. Steinseifer and A. Woltermann, Angew. Chem., Int. Ed. Engl., 1980, 19, 328 CrossRef.
- R. M. Bellabarba and G. C. Saunders, Polyhedron, 2004, 23, 2659 CrossRef CAS.
- R. M. Bellabarba, M. Nieuwenhuyzen and G. C. Saunders, Organometallics, 2003, 22, 1802 CrossRef CAS.
- R. M. Bellabarba, M. Nieuwenhuyzen and G. C. Saunders, Organometallics, 2002, 21, 5726 CrossRef CAS.
- R. M. Bellabarba and G. C. Saunders, J. Fluorine Chem., 2001, 112, 139 CrossRef CAS.
- R. M. Bellabarba, M. Nieuwenhuyzen and G. C. Saunders, Inorg. Chim. Acta, 2001, 323, 78 CrossRef CAS.
- J. Szymoniak, J. Besancon, A. Dormond and C. Moise, J. Org. Chem., 1990, 55, 1429 CrossRef CAS.
- P. E. Garrou, Chem. Rev., 1981, 81, 229 CrossRef CAS.
- M. Tasi, T. Ranga and G. Palyi, in Organometallic Synthesis, ed. J. J. Eisch, Academic Press, New York, 1988 Search PubMed.
- P. Jutzi and T. Redeker, Eur. J. Inorg. Chem., 1998, 663 CrossRef CAS.
- A. Polas, J. Wilton-Ely, A. M. Z. Slawin, D. F. Foster, P. J. Steynberg, M. J. Green and D. J. Cole-Hamilton, Dalton Trans., 2003, 4669 RSC.
- R. F. Bryan and A. R. Manning, Chem. Commun., 1968, 1316 RSC.
- J. A. Ibers, J. Organomet. Chem, 1968, 14, 423 CrossRef CAS.
- Y. Misumi, Y. Ishii and M. Hidai, Organometallics, 1995, 14, 1770 CrossRef CAS.
- W. S. Weston, P. Lightfoot and D. J. Cole-Hamilton, J. Organomet. Chem., 1998, 553, 473 CrossRef CAS.
- D. M. Heinekey, M. H. Voges and D. M. Barnhart, J. Am. Chem. Soc., 1996, 118, 10792 CrossRef CAS.
- A. Haynes, P. M. Maitlis, G. E. Morris, G. J. Sunley, H. Adams, P. W. Badger, C. M. Bowers, D. B. Cook, P. I. P. Elliott, T. Ghaffar, H. Green, T. R. Griffin, M. Payne, J. M. Pearson, M. J. Taylor, P. W. Vickers and R. J. Watt, J. Am. Chem. Soc., 2004, 126, 2847 CrossRef CAS.
- A. Haynes, unpublished observations.
- A. J. Hart-Davis and W. A. G. Graham, Inorg. Chem., 1970, 9, 2658 CrossRef.
- J. A. Gaunt, V. C. Gibson, A. Haynes, S. K. Spitzmesser, A. J. P. White and D. J. Williams, Organometallics, 2004, 23, 1015 CrossRef CAS.
- A. Haynes, C. E. Haslam, K. J. Bonnington, L. Parish, H. Adams, S. E. Spey, T. B. Marder and D. N. Coventry, Organometallics, 2004, 23, 5907 CrossRef CAS.
- P. M. Maitlis, A. Haynes, G. J. Sunley and M. J. Howard, J. Chem. Soc., Dalton Trans., 1996, 2187 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Kinetic data and crystal structures. See DOI: 10.1039/b512054c |
| ‡ Six isomers of this product exist depending on the position of the double bond in the cyclopentadienyl ring. |
| § Unable to assign the exact methylene groups due to complicated pattern. |
| ¶ The measured crystal was of a double salt with [Ph3PMe]I. Presumably some PPh3 from the starting [IrCl(CO)(PPh3)2] must have co-crystallised with the batch of [Ir(Cp'(CH2)2PEt2)(CO)] used for the reaction with MeI. |
| This journal is © The Royal Society of Chemistry 2006 |