The reactivity of gallium-(I), -(II) and -(III) heterocycles towards Group 15 substrates: attempts to prepare gallium–terminal pnictinidene complexes
Robert J.
Baker
a,
Cameron
Jones
*a,
David P.
Mills
a,
Damien M.
Murphy
*a,
Evamarie
Hey-Hawkins
b and
Robert
Wolf
ab
aCentre for Fundamental and Applied Main Group Chemistry, School of Chemistry, Main Building, Cardiff University, Cardiff, UK CF10 3AT. E-mail: jonesca6@cardiff.ac.uk
bInstitut für Anorganische Chemie der Universität Leipzig, Johannisallee 29, 04107, Leipzig, Germany
First published on 26th October 2005
Abstract
The reactivity of a series of Ga(I), Ga(II) and Ga(III) heterocyclic compounds towards a number of Group 15 substrates has been investigated with a view to prepare examples of gallium–terminal pnictinidene complexes. Although no examples of such complexes were isolated, a number of novel complexes have been prepared. The reactions of the gallium(I) N-heterocyclic carbene analogue, [K(tmeda)][:Ga{[N(Ar)C(H)]2}] (Ar = 2,6-diisopropylphenyl) with cyclo-(PPh)5 and PhN![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NPh led to the unusual anionic spirocyclic complexes, [{κ2P,P′-(PhP)4}Ga{[N(Ar)C(H)]2}]− and [{κ2N,C-PhNN(H)(C6H4)}Ga{[N(Ar)C(H)]2}]−, via formal reductions of the Group 15 substrate. The reaction of the digallane(4), [Ga{[N(Ar)C(H)]2}]2, with (Me3Si)N3 afforded the paramagnetic, dimeric imido–gallane complex, [{[N(Ar)C(H)˙]2}Ga{μ-N(SiMe3)}]2, via a Ga–Ga bond insertion process. In addition, the new gallium(III) phosphide, [GaI{P(H)Mes*}{[N(Ar)C(H)]2˙}], Mes* = C6H2But3-2,4,6; was prepared and treated with diazabicycloundecane (DBU) to give [Ga(DBU){P(H)Mes*}{[N(Ar)C(H)]2}], presumably via a gallium–terminal phosphinidene intermediate, [Ga{PMes*}{[N(Ar)C(H)]2˙}]. The possible mechanisms of all reactions are discussed, all new complexes have been crystallographically characterised and all paramagnetic complexes have been studied by ENDOR and/or EPR spectroscopy.
NPh led to the unusual anionic spirocyclic complexes, [{κ2P,P′-(PhP)4}Ga{[N(Ar)C(H)]2}]− and [{κ2N,C-PhNN(H)(C6H4)}Ga{[N(Ar)C(H)]2}]−, via formal reductions of the Group 15 substrate. The reaction of the digallane(4), [Ga{[N(Ar)C(H)]2}]2, with (Me3Si)N3 afforded the paramagnetic, dimeric imido–gallane complex, [{[N(Ar)C(H)˙]2}Ga{μ-N(SiMe3)}]2, via a Ga–Ga bond insertion process. In addition, the new gallium(III) phosphide, [GaI{P(H)Mes*}{[N(Ar)C(H)]2˙}], Mes* = C6H2But3-2,4,6; was prepared and treated with diazabicycloundecane (DBU) to give [Ga(DBU){P(H)Mes*}{[N(Ar)C(H)]2}], presumably via a gallium–terminal phosphinidene intermediate, [Ga{PMes*}{[N(Ar)C(H)]2˙}]. The possible mechanisms of all reactions are discussed, all new complexes have been crystallographically characterised and all paramagnetic complexes have been studied by ENDOR and/or EPR spectroscopy.
Introduction
Multiple bonds between the heavy p-block elements are of much current interest as such bonds often display very different chemistry to their “classical” first row analogues. Whilst there are now many examples of compounds containing “non-classical” homo- or heteronuclear multiple bonds between the heavier Group 14 and/or Group 15 elements,1 the chemistry of compounds possessing multiply bonded Group 13 metal centres is less well developed. In this area, several examples of compounds incorporating homonuclear Group 13 metal–metal interactions with a bond order of greater than one have been reported.1,2 The nature of the bonding in such compounds can be difficult to explain using standard models, and this has led to extensive debate in the literature. The extent of this debate is perhaps best exemplified by the unusual dianion, [Ar*GaGaAr*]2− Ar* = C6H3(C6H2Pri3-2,4,6)2-2,6; which has been described by a number of experimental and theoretical studies as having a Ga–Ga bond order of 1, 2 or 3.3 Compounds containing a heteronuclear multiple bond between a Group 13 metal and another heavy p-block element have also been studied. In this area, a handful of compounds incorporating Group 13 metal–Group 16 double bonds have been reported, e.g. the terminal gallium chalcogenides, [TptBuGaE] (TptBu = tris(3,5-di-tert-butylpyrazoloyl)hydroborate; E = S, Se, Te),4 though Group 13 metal–Group 15 element multiply bonded compounds have been more widely explored. These include the kinetically stabilised Group 13–terminal imide complexes, [Ar′MNAr″] M = Ga or In, Ar′ = C6H3(C6H3Pri2-2,6)2-2,6; Ar″ = C6H3(C6H2Me2But-2,6,4)2-2,6, which theoretical studies suggest have significant π-bonding inherent in their MN interactions.5 Varying levels of π-bonding have also been invoked for complexes containing interactions between gallium and the heavier pnictogens, as for example in [(Mes)P{Ga(C6H2Pri3-2,4,6)2}2] Mes = mesityl,6 [{But3SiP(H)Ga(μ-PSiBut3)}2]7 and [{Li(THF)3}2Ga2{As(SiPri3)}4].8
Most relevant to our endeavours in this area are the investigations by the groups of Roesky and Power into the reactivity of the neutral aluminium(I) and gallium(I) heterocycles, [:M{[N(Ar)C(Me)]2CH}], M = Al9 or Ga,10 Ar = C6H3Pri2-2,6, towards aryl and silyl azides. These have led to terminal imide complexes, [Ar*NM{[N(Ar)C(Me)]2CH}],11 but also to metal tetrazoles, [{κ2N,N′-N4(SiMe3)2}{M[N(Ar)C(Me)]2CH}],12 a gallium azide, [{(Me3Si)2N}(N3)Ga{[N(Ar)C(Me)]2CH}],12 and several other novel heterocyclic complexes13via different pathways which depend upon the metal and the azide precursor. We have developed a high yielding synthetic route to the anionic gallium(I) heterocycle, [K(tmeda)][:Ga{[N(Ar)C(H)]2}] 1,14 which is closely related to [:M{[N(Ar)C(Me)]2CH}] and behaves similarly to the valence isoelectronic N-heterocyclic carbene (NHC) class of ligand with respect to its main group and transition metal coordination chemistry.15,16 In the current study it was proposed to further examine the apparent analogy between 1 and both [:M{[N(Ar)C(Me)]2CH}] and NHCs by attempting to utilise it as a precursor to new gallium–terminal pnictinidene complexes which could potentially exhibit Ga–pnictogen multiple bond character. A closely related paramagnetic gallium(III) heterocycle, [GaI2{[N(Ar)C(H)˙]2}],17 and a diamagnetic digallane(4), [Ga{[N(Ar)C(H)]2}]2,18 containing gallium(II) centres, were also enlisted for this cause. Although no terminal gallium–pnictinidene complexes resulted, a variety of novel heterocyclic gallium–pnictide species were obtained, as discussed herein.
Results and discussion
N-Heterocyclic carbenes have been utilised as stabilising ligands in the preparation of a variety of novel main group complexes.19 One of the more interesting compound classes that has arisen from this work are the NHC–terminal pnictinidene complexes reported by Cowley et al., e.g. [(IMes)→ER], E = P or As; R = Ph, C6F5 or CF3, IMes = 1,3-dimesitylimidazol-2-ylidene.20 These were prepared in the reactions of IMes with cyclic pnictanes, e.g.cyclo-(PPh)5, and have been shown not to exist as pnictaalkenes with CE bonds and one lone pair on the pnictogen centre, but instead as NHC–pnictinidene adducts with two lone pairs on the pnictogen centre. In light of the previously described analogy between 1 and NHCs,16 it seemed that its reaction with cyclic pnictanes might lead to terminal gallium–pnictinidene complexes. In the reaction of 1 with (PhP)5 in THF under a range of stoichiometries, this did not prove to be the case and instead the novel ionic spirocyclic complex, 2, was formed in low to moderate yields after recrystallisation from toluene (Scheme 1). Monitoring of either the 1 : 1, 3 : 1 or 5 : 1 reactions by 31P NMR spectroscopy showed that 2 was the major reaction product, and in the latter two cases it was found after work-up that excess 1 remained unreacted. The mechanism of this reaction seemingly involves the oxidative insertion of the gallium(I) centre of 1 into a P–P bond of (PPh)5 with concomitant loss of one PPh fragment. The outcome of this fragment could not be determined but it most probably leads to cyclic oligomers, (PPh)n. The reaction that gave 2 is related to the treatment of the tetrameric gallium(I) diyl, [Ga{C(SiMe3)3}]4, with cyclo-(PBut)3, and the aluminium(I) heterocycle, [:Al{[N(Ar)C(Me)]2CH}], with P4, both of which lead to oxidative insertion of the Group 13 centre into a P–P bond of the phosphorus reactant and formation of cyclo-[{(SiMe3)3C}Ga(PBut)3]21 and [[Al{[N(Ar)C(Me)]2CH}]2(μ-P4)]22 respectively. It is interesting to note that 1 does not react with either (PBut)3 or (PhP)4CH2.
![i) cyclo-(PPh)5, THF; ii) PhNNPh, Et2O, [4 + 1]; iii) 1,3-H shift.](/image/article/2006/DT/b511451a/b511451a-s1.gif) | ||
| Scheme 1 i) cyclo-(PPh)5, THF; ii) PhNNPh, Et2O, [4 + 1]; iii) 1,3-H shift. | ||
The 31P{1H} NMR spectrum of 2 in THF displays a signal pattern indicative of an AA′BB′ spin system (δA = −80.2 ppm, δB = −6.1 ppm; where PA are attached to Ga), as has been seen for related heterocyclic systems, e.g. [As(PCy)4]−, Cy = cyclohexyl.23 The signals are, however, significantly broadened and as a result efforts to accurately simulate the spectrum were unsuccessful. It seems likely that the broadness of the signals results from a fluxional process in solution which could involve a cleavage and reformation of Ga–P and/or P–P bonds. Evidence for this proposal comes from the solid state structure of the compound (vide infra). Unfortunately, no light could be shed on the nature of the fluxional process occurring in solution as the 31P{1H} NMR spectrum of 2 did not resolve at temperatures as low as −90 °C.
Crystals of 2 suitable for an X-ray diffraction study were grown from a toluene solution and its molecular structure is depicted in Fig. 1. This shows it to contain an anionic spirocyclic system which coordinates the potassium counter-ion via a skewed η6-arene interaction with one of the GaN2C2 heterocycle's N-substituents. The potassium centre is additionally chelated by two phosphorus centres of the GaP4 ring and a molecule of tmeda. Interestingly, the phosphorus phenyl substituents are syn, anti, anti with respect to each other, a situation which renders all the phosphorus centres chemically inequivalent. This is at odds with the solution 31P{1H} NMR spectrum of 2 which displays only two broad signals, thus suggesting that the GaP4 ring of the compound undergoes a rapid, fluxional isomeric rearrangement via Ga–P and/or P–P bond cleavage and reformation. As in previously reported coordination complexes of the anion of 1,15,16 the Ga–N bond lengths of the GaN2C2 heterocycle in 2 [1.914 Å avg.] decrease, and its N–Ga–N angle [88.5(2)°] becomes more obtuse relative to those parameters in the free anionic ligand [Ga–N avg. = 1.970 Å; N–Ga–N = 83.02(11)°].14 It is believed this arises from the smaller covalent radius of the Ga(III) centre in 2 compared to that of the Ga(I) centre in 1. The average Ga–P and P–P bond lengths in 1 [2.402 Å and 2.209 Å respectively] are comparable to those in related compounds, e.g.cyclo-[{(SiMe3)3C}Ga(PBut)3] [2.360 Å avg. and 2.217 Å avg. respectively],21 whilst the P–K distances [3.452 Å avg.] are in the known range for such interactions (3.043–3.851 Å).24
 | ||
| Fig. 1 Molecular structure of 2 (isopropyl groups omitted for clarity). Selected bond lengths (Å) and angles (°): Ga(1)–N(1) 1.927(5), Ga(1)–N(2) 1.901(5), Ga(1)–P(1) 2.3818(18), Ga(1)–P(4) 2.4230(18), K(1)–P(2) 3.590(2), K(1)–P(4) 3.314(2), P(1)–P(2) 2.201(2), P(2)–P(3) 2.233(3), P(3)–P(4) 2.194(2), C(1)–C(2) 1.351(9); N(1)–Ga(1)–N(2) 88.5(2), N(1)–Ga(1)–P(1) 116.53(17), N(1)–Ga(1)–P(4) 104.77(17), N(2)–Ga(1)–P(1) 115.67(16), N(2)–Ga(1)–P(4) 124.80(16), P(1)–Ga(1)–P(4) 105.52(6), P(2)–K(1)–P(4) 54.76(5), Ga(1)–P(1)–P(2) 91.42(8), Ga(1)–P(1)–C(27) 104.8(2), P(1)–P(2)–P(3) 103.70(9), P(2)–P(3)–P(4) 91.99(9), P(3)–P(4)–Ga(1) 98.78(8). | ||
A number of other attempts were made to form a terminal gallium–pnictinidene complex using 1 as a precursor, but with limited success. For example, its reaction with Ar#PPEt3 (Ar# = C6H3Mes2-2,6), a known phosphinidene transfer reagent,25 led to an intractable mixture of many phosphorus containing products. The reactivity of 1 towards a variety of di-pnictenes was also investigated and, perhaps surprisingly, no reaction was observed with Mes*PPMes* (Mes* = C6H2But3-2,4,6)26 or Ar*EEAr* (E = As, Sb).27 Compound 1 did, however, react with azobenzene, PhNNPh, in 1 : 1 or 2 : 1 stoichiometries to give the unusual ionic spirocyclic system, 3, in good yields (Scheme 1). One possible mechanism for formation of 3 involves an initial orthometallation of azobenzene by the Ga(I) centre of 1 to give a short-lived Ga–H intermediate which reduces the NN moiety to give the observed product. It is noteworthy that the reduction of aromatic azo-compounds to hydrazoarenes by Group 13 hydride agents, e.g. Bui2AlH, is known.28 In addition, gallium centres have been shown to participate in intramolecular metallation reactions in similar systems. An example here is the reaction of Me2GaCl with the lithiated hydrazine, LiN(SiMe3)N(H)(Naph) (Naph = naphthyl),29 which leads to a diazagallole that is closely related to 3 and arises from a gallium mediated C–H activation (methane elimination) at the 2-position of the naphthyl substituent. Another possible, and perhaps more likely mechanism involves a [4 + 1] cycloaddition of the Ga(I) centre of 1 with the azobenzene, followed by a rapid 1,3-migration of the ortho-aryl proton to the nitrogen centre bearing the metallated phenyl group (Scheme 1). A precedent for this mechanism exits with the reaction of the iminophosphane, EtPNMes*, with azobenzene which affords the diazaphosphole, [(Mes*)N(Et)P{κ2N,P-N(Ph)N(H)(C6H4)}], via [4 + 1] cycloaddition and C–H activation processes.30 Whichever mechanism is in operation, it was found that 1 does not react with MesNNMes, which is devoid of ortho-aryl protons.
The spectroscopic data for 3 support its proposed formulation. Specifically, its infrared spectrum exhibits an N–H stretching absorption at 3486 cm−1, and a broad peak was observed in its 1H NMR spectrum at δ 7.47 ppm which clearly arises from the N–H proton. Additionally, the 13C NMR spectrum of the complex displays the expected number of resonances in the aromatic region. Crystals of 3 suitable for X-ray diffraction were grown from a saturated hexane solution and its molecular structure is shown in Fig. 2. As was the case for 2, this shows the complex to contain a spirocyclic anion. The hydrogen atom, H(4), bonded to N(4) was located from difference maps and isotropically refined, thus allowing the nitrogen centre, N(4), to be assigned as having a distorted tetrahedral geometry, which includes coordination to the potassium centre through its lone pair. This potassium also has a weaker interaction with the p-orbital lone pair of the trigonal planar centre, N(3), and an η2-attachment to the non-metallated phenyl ring of the diaza-ligand. The coordination sphere of the potassium centre is completed by one molecule each of diethyl ether and tmeda. Both heterocycles containing the distorted tetrahedral gallium centre are effectively planar and the bond lengths and angles within the heterocycle originating from 1 are similar to those in 2. The other heterocycle displays intracyclic N–N and N–C distances [1.442(5) Å and 1.427(6) Å respectively] that are normal for single bonded interactions and similar to those in the diazaphosphole, [(Mes*)N(Et)P{κ2N,P-N(Ph)N(H)(C6H4)}] [1.439(8) Å and 1.420(5) Å].30 Also within this heterocycle, the gallium–amide interaction [Ga(1)–N(3),1.964(4) Å] is significantly longer than those in the other GaN2C2 heterocycle, whilst the Ga–C bond length [1.969(5) Å] is in the normal range.24
 | ||
| Fig. 2 Molecular structure of 3. Selected bond lengths (Å) and angles (°): Ga(1)–N(1) 1.910(4), Ga(1)–N(2) 1.910(4), Ga(1)–N(3) 1.964(4), Ga(1)–C(38) 1.969(5), N(1)–C(1) 1.392(6), N(2)–C(2) 1.410(6), N(3)–N(4) 1.442(5), C(1)–C(2) 1.339(7); N(1)–Ga(1)–N(2) 87.78(17), N(1)–Ga(1)–N(3) 113.29(17), N(1)–Ga(1)–C(38) 128.06(18), N(2)–Ga(1)–N(3) 116.25(17), N(2)–Ga(1)–C(38) 128.06(18), N(3)–Ga(1)–C(38) 85.05(18), C(27)–N(3)–N(4) 114.8(4), C(33)–N(4)–N(3) 111.5(4). | ||
In consideration of the aforementioned reactivity of the neutral Al(I) and Ga(I) heterocycles, [:M{[N(Ar)C(Me)]2CH}], towards organic azides, the reactions of 1 with azides of varying steric bulk, RN3, R = SiMe3, 9-triptycenyl, Mes* or Ar*, were investigated. In all cases intractable mixtures were obtained. We considered that reaction of these azides with the digallane(4), [Ga{[N(Ar)C(H)]2}]24, obtainable in high yield via oxidative coupling of 1,15c could lead to the oxidative insertion of imide fragments into the Ga–Ga bond of 4. In this respect, we have recently shown that 4 will oxidatively add to low oxidation state transition metal fragments.15a The reaction of 4 with 2 equivalents of (Me3Si)N3 yielded the blue-green paramagnetic complex, 5, in good yield (79%) after stirring for 48 hours (Scheme 2). Repeating the reaction with a large excess of (Me3Si)N3 led to the same complex and did not appreciably increase the rate of reaction. Although, the formation of 5 is relatively slow (as judged by the colour change of the reaction mixture), no intermediates in its formation could be isolated. It is clear, however, that the mechanism involves single electron oxidations of the gallium heterocycles of 4. Compound 5 can be considered as a dimer of the gallium–terminal imide complex, [{[N(Ar)C(H)]2}GaN(SiMe3)], and can be compared to the related dimeric, diamagnetic imidogallane, [{(η1-Cp*)GaN(xylyl)}2], which arises from the reaction of (xylyl)N3 and (η5-Cp*)Ga:.31 There are also parallels between the formation of 5 and the singlet biradicaloid germanium imide complex, [{Ar′GeN(SiMe3)}2], which results from the reaction of the digermyne, Ar′GeGeAr′, with (Me3Si)N3.32 It is worthy of note that the reactions of 4 with either (9-triptycenyl)N3, Mes*N3 or Ar*N3 were carried out but all led to intractable mixtures of products.
 | ||
| Scheme 2 i) Excess Me3SiN3, hexane. | ||
As 5 is paramagnetic, no meaningful data could be obtained from its NMR spectra. However, X-band continuous wave EPR spectra for this compound were recorded at room temperature and 77 K. Only the spectra acquired at 298 K are shown in Fig. 3, as the anisotropic frozen solution spectrum did not yield any additional information. Owing to the poor resolution of the first derivative spectrum (Fig. 3a), the second derivative spectrum (Fig. 3b) was recorded. The resulting isotropic spectrum was simulated (Fig. 3c) based on the following spin Hamiltonian parameters; giso = 2.0035, aH = 5.8 G, aN = 6.0 G, a69Ga = 20.4 G and a71Ga = 26.0 G. Therefore, the strong EPR signal for 5 is centred close to that of free spin. In addition, it is typical for paramagnetic diazabutadiene–gallium complexes previously reported by us,33,34 in that it is dominated by isotropic hyperfine couplings (HFC) to two equivalent 1H nuclei, two equivalent 14N nuclei and a 69,71Ga nucleus. The relatively large HFCs to the gallium nucleus originate from the large theoretical isotropic hyperfine couplings for gallium (69Ga; I = 3/2, a0 = 4356 G, 60.1% natural abundance; 71Ga: I = 3/2, a0 = 5535 G, 39.9% natural abundance). This means that even the small electron spin density (0.47%) at the gallium nucleus of 5 produces easily observable HFCs to both gallium isotopes.
 | ||
| Fig. 3 X-Band (9.360 GHz) EPR spectrum of 5 recorded at 298 K in toluene. (a) First harmonic signal, (b) second harmonic signal, and (c) simulated spectrum. Inset figure shows the ΔMS = 2 transition due to the weakly interacting S = 1 triplet at half field. | ||
Evidence for the diradical nature of 5 can also be obtained from its EPR spectra. For two interacting unpaired electrons (S = ½), an S = 1 triplet ground state can be observed in the spectrum, provided the coupling between the two spin systems is sufficiently strong. In that case the zero field splitting term for the randomly oriented triplet should produce a characteristic pattern in the ΔMS = 1 region (centre field). Unfortunately, due to the intense nature of the signals arising from the individual S = ½ spins in 5, the zero field splitting parameter (D) could not be observed. However, the ΔMS = 2 transition at half field was seen at 1670 G (see inset in Fig. 3). While this half field transition is extremely weak, indicating that the two S = ½ spins are only weakly coupled, it nevertheless confirms the diradical nature of 5. The weak interaction between the two S = ½ spins is due to the fact that the unpaired electrons are primarily localised on the diazabutadiene backbone, and the tetrahedral bonding arrangement around the gallium centres prevents efficient spin–spin coupling.
The molecular structure of 5 is depicted in Fig. 4 and shows it to be dimeric with bridging imido ligands. Its gallium centres have heavily distorted tetrahedral geometries and are slightly displaced from the least squares planes defined by the chelating diazabutadiene ligands (0.375 Å avg.). In contrast, the Ga2N2 heterocycle is effectively planar and its Ga–N bonds (1.885 Å avg.) are significantly shorter than those to the diazabutadiene ligand (2.014 Å avg.) but longer than those seen in [{(η1-Cp*)GaN(xylyl)}2] (1.860 Å avg.),31 which possesses 3-coordinate Ga centres. The N-centres of the Ga2N2 heterocycle in 5 have distorted trigonal planar geometries (Σ angles = 358.3° avg.). An examination of the C–C and C–N bond lengths within the diazabutadiene ligands of 5 suggests a significant degree of delocalisation, as has previously been seen in related paramagnetic complexes employing this ligand, e.g. [GaI2{[N(Ar)C(H)]2˙}].17 Although the Ga⋯Ga separation [2.654(3) Å] is well within the sum of the van der Waals radii (3.8 Å),31 there is no evidence for a Ga–Ga bond in the compound, as has been previously discussed for similar compounds, e.g. [{(η1-Cp*)GaN(xylyl)}2] [Ga⋯Ga separation 2.6495(6) Å].31
 | ||
| Fig. 4 Molecular structure of 5 (isopropyl groups omitted for clarity). Selected bond lengths (Å) and angles (°): Ga(1)–N(1) 1.871(3), Ga(1)–N(2) 1.900(2), Ga(1)–N(3) 2.017(2), Ga(1)–N(4) 2.012(3), Ga(2)–N(1) 1.895(2), Ga(2)–N(2) 1.876(3), Ga(2)–N(5) 2.005(3), Ga(2)–N(6) 2.030(3), C(1)–C(2) 1.392(4), C(27)–C(28) 1.407(5), Si(1)–N(1) 1.706(3), Si(2)–N(2) 1.706(3); N(1)–Ga(1)–N(2) 90.50(11), N(1)–Ga(1)–N(3) 126.11(11), N(1)–Ga(1)–N(4) 123.45(11), N(2)–Ga(1)–N(3) 113.58(10), N(2)–Ga(1)–N(4) 123.45(11), N(3)–Ga(1)–N(4) 83.62(11), N(1)–Ga(2)–N(2) 90.50(11), N(1)–Ga(2)–N(5) 121.91(10), N(1)–Ga(2)–N(6) 112.90(11), N(2)–Ga(2)–N(5) 123.95(11), N(2)–Ga(2)–N(6) 127.50(11), N(5)–Ga(2)–N(6) 83.50(11), Ga(1)–N(1)–Ga(2) 89.61(11), Ga(1)–N(1)–Si(1) 135.09(15), Ga(2)–N(1)–Si(1) 133.12(15), Ga(1)–N(2)–Ga(2) 89.30(11), Ga(1)–N(2)–Si(2) 132.70(15), Ga(2)–N(2)–Si(2) 136.75(18). | ||
To the best of our knowledge, terminal phosphinidene complexes of gallium are unknown. If they were accessible, their potential use in, for example, cycloaddition and metathesis reactions is clear. Given that transition metal–terminal phosphinidene complexes can be readily made by the base assisted dehydrohalogenation of primary phosphido–metal halide complexes,35 we believed similar methodologies could be applied to appropriate Ga(III) heterocyclic complexes. To this end, a suitable gallium phosphide precursor to such a reaction, [GaI{P(H)Mes*}{[N(Ar)C(H)]2˙}] 6, was prepared in good yield by the 1 : 1 salt elimination reaction between [GaI2{[N(Ar)C(H)]2˙}] 7 and [LiP(H)Mes*] (Scheme 3). Considering the steric bulk of both the Ga and P substituents of 6, it was anticipated that its treatment with a base would lead to HI elimination and the formation of [Ga{PMes*}{[N(Ar)C(H)]2˙}]. Surprisingly, however, the reaction of 6 with an excess of DBU in THF gave rise to the diamagnetic complex, [Ga(DBU){P(H)Mes*}{[N(Ar)C(H)]2}] 8, in good isolated yield. The formation of this complex could conceivably occur via the expected phosphinidene complex, [Ga{P(Mes*)}{[N(Ar)C(H)]2˙}], which undergoes a rapid intramolecular single electron reduction of the diazabutadiene ligand to give [Ga{P˙Mes*}{[N(Ar)C(H)]2}], the radical P-centre of which abstracts an atom of hydrogen from the solvent. This would leave the unsaturated Ga-centre to be coordinated by a molecule of DBU. Such a proposal seems reasonable considering the well known red-ox activity of diazabutadienes.33,34
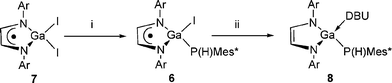 | ||
| Scheme 3 i) LiP(H)Mes*, THF; ii) excess DBU, THF. | ||
The infrared spectra of both 6 and 8 show characteristic P–H stretching absorptions at ν 2397 cm−1 and 2403 cm−1 respectively. No useful NMR information could be obtained from paramagnetic 6, but the data for 8 are consistent with its proposed formulation. Most diagnostically, its 31P NMR spectrum shows a doublet at high field, −148.5 ppm (1JP–H = 215 Hz), as would be expected for a metal–primary phosphide complex.
In order to obtain more information on 6, its room temperature isotropic EPR spectrum was acquired and is depicted in Fig. 5a. For higher resolution of the hyperfine couplings, the second harmonic signal is also shown (Fig. 5b). The profile of the spectrum was difficult to reproduce accurately in its simulation due to the broadened line-widths observed in the spectral wings (the mI dependency of the line-widths is influenced by the tumbling rates of the radical in solution). The hyperfine pattern is, however, dominated by a large isotropic coupling to the gallium isotopes, 69Ga and 71Ga, which in the case of 71Ga was estimated at 40 G (ca. 0.72% spin density on Ga) in order to account for the overall width of the spectrum. In addition, the magnitudes of the couplings to 1H and 14N in 6 were easily measured at 5.5 G and 5.8 G respectively, by inspection of the well resolved 1H and 14N hyperfine patterns associated with the −3/2 and +3/2 Ga mI states (Fig. 5b). The 127I coupling could not, however, be resolved. The values of the HFCs for 6 can be compared to those previously reported by us for 7 (1H ∼5.0 G, 14N ∼5.0 G, 69,71Ga ∼25 G, equating to ca. 0.45% spin density at Ga).34 The reason behind the increased coupling to the gallium centre in 6, relative to 7, lies with the electron withdrawing effect of the phosphido ligand which polarises the unpaired electron from the diazabutadiene backbone onto the gallium nucleus. We have previously observed similar effects in closely related complexes, e.g. [GaI{P(SiMe3)2}{[N(But)C(H)]2˙}].33
 | ||
| Fig. 5 X-Band cw-EPR spectrum of 6 recorded in hexane solution at 298 K. (a) First harmonic, and (b) second harmonic signals. | ||
The frozen solution EPR spectrum of 6 at X-band did not provide any additional resolution for the Ga hyperfine interaction (only a broad asymmetric spectrum was observed), suggesting that this interaction is dominated by the isotropic component. As a result, its X-band cw-ENDOR spectra revealed a powder type pattern regardless of the measuring field position. A representative 1H ENDOR spectrum of 6 is shown in Fig. 6. According to the room temperature EPR spectrum, the isotropic 1H hyperfine coupling was estimated to be 5.5 G (15.5 MHz). For a strongly coupled α-proton, principal values of the hyperfine tensor will occur at approximately a/2, a, and 3/2a, with slight deviations from those expected due to the delocalised nature of the spin density on the N–C–C–N unit. Depending on the magnitude of the coupling, the broadened outer lines are often not readily observed in the cw-ENDOR spectrum, as discussed in a recent publication.34 Therefore, the relatively small 1H ENDOR resonances in Fig. 6 arise primarily from the aryl substituents. The largest couplings of 4.90 MHz and 3.97 MHz can be assigned to the protons on the aryl ring, while the smaller inner couplings at 2.35 MHz and 1.20 MHz were assigned to the methyl protons of the aryl groups. These couplings are in good agreement with those previously reported by us for a series of related aryl substituted Ga–diazabutadiene complexes.34 While the P(H)Mes* substituent clearly affects the unpaired spin density on the gallium nucleus, it has little effect on the already small spin density within the aryl groups.

The X-ray crystal structure of 6 was obtained and its asymmetric unit was shown to contain two crystallographically independent molecules which have no significant geometric differences and, thus, only one is shown in Fig. 7. Unusually, the asymmetric unit also contains the reaction by-product, LiI, in the form of the dimeric molecule, [{Li2I2(THF)3.5(OEt2)0.5}], which exhibits coordinated solvent site disorder and has no appreciable contacts with either of the other two molecules of 6. An X-ray crystal structure analysis was also carried out on 8 and its molecular structure is depicted in Fig. 8. Both 6 and 8 are monomeric and exhibit distorted tetrahedral coordination environments for their gallium centres. Their Ga–P bond lengths are similar and in the normal range for gallium phosphide complexes, cf. 2.388 Å avg. in [(Mes*)Ga{P(H)Mes*}2]36 or 2.2991(11) Å in [GaI{P(SiMe3)2}{[N(But)C(H)]2˙}].33 The magnitudes of the C–C and C–N distances in their diazabutadiene ligands are, however, significantly different and suggest a degree of delocalisation over the NCCN fragment of 6, and localised CC and C–N bonds in 8. There are also differences between their Ga–N(diazabutadiene) bond lengths and their N–Ga–N angles, which are shorter and more obtuse respectively in 8. Not surprisingly, the Ga–N(DBU) bond length in this compound is more than 0.1 Å longer than its Ga–N(diazabutadiene) interactions.
 | ||
| Fig. 7 Molecular structure of 6. Selected bond lengths (Å) and angles (°): Ga(1)–I(1) 2.5512(11), Ga(1)–N(1) 1.976(5), Ga(1)–N(2) 1.972(6), Ga(1)–P(1) 2.337(2), N(1)–C(1) 1.347(9), N(2)–C(2) 1.324(5), C(1)–C(2) 1.402(10), P(1)–H(1A) 1.32(6); N(1)–Ga(1)–N(2) 84.0(2), N(1)–Ga(1)–I(1) 106.69(17), N(1)–Ga(1)–P(1) 123.97(17), N(2)–Ga(1)–I(1) 110.57(18), N(2)–Ga(1)–P(1) 114.43(19), P(1)–Ga(1)–I(1) 113.49(6), Ga(1)–P(1)–C(27) 102.0(2). | ||
 | ||
| Fig. 8 Molecular structure of 8 (isopropyl groups omitted for clarity). Selected bond lengths (Å) and angles (°): Ga(1)–N(1) 1.918(3), Ga(1)–N(2) 1.909(3), Ga(1)–N(3) 2.029(3), Ga(1)–P(1) 2.3675(12), C(1)–C(2) 1.339(6), N(1)–C(1) 1.413(5), N(2)–C(2) 1.405(5), P(1)–H(1A) 1.33(7); N(1)–Ga(1)–N(2) 88.72(13), N(1)–Ga(1)–N(3) 109.76(14), N(1)–Ga(1)–P(1) 115.81(10), N(2)–Ga(1)–N(3) 113.20(14), N(2)–Ga(1)–P(1) 111.67(10), P(1)–Ga(1)–N(3) 114.96(11), Ga(1)–P(1)–C(27) 114.32(13). | ||
Conclusion
The reactivity of a series of Ga(I), Ga(II) and Ga(III) heterocyclic compounds towards a number of Group 15 substrates has been investigated with a view to prepare examples of gallium–terminal pnictinidene complexes. Although no examples of such complexes were isolated, a gallium–terminal phosphinidene complex, [Ga{P(Mes*)}{[N(Ar)C(H)]2˙}], has been implicated as a short lived intermediate in one reaction. In addition, a number of unusual complexes containing gallium–pnictogen bonds have been prepared. These have highlighted the reducing ability of the anionic gallium(I) heterocycle, [:Ga{[N(Ar)C(H)]2}]−, the reactivity of which differs to that of valence isoelectronic NHCs in this study. Moreover, the reactivity of the Ga–Ga bond of the digallane(4), [Ga{[N(Ar)C(H)]2}]2, has been exploited in the synthesis of the dimeric imido–gallane complex, [{[N(Ar)C(H)˙]2}Ga{μ-N(SiMe3)}]2, which can be considered as a dimer of the gallium–terminal imide, [{[N(Ar)C(H)˙]2}GaN(SiMe3)]. ENDOR and/or EPR spectroscopy has shown that the unpaired electron of all paramagnetic complexes is primarily based on their diazabutadiene ligands. Studies of the reactivity of low oxidation state gallium heterocycles are ongoing in our laboratory.
Experimental
General considerations
All manipulations were carried out using standard Schlenk and glove box techniques under an atmosphere of high purity argon. Hexane, toluene and THF were distilled over potassium whilst Et2O was distilled over Na/K then freeze/thaw degassed prior to use. 1H, 13C and 31P NMR spectra were recorded on either a Bruker DXP400 or a Jeol Eclipse 300 spectrometer and were referenced to the residual 1H or 13C resonances of the solvent used or external 85% H3PO4, δ 0.0 ppm (31P NMR). A confident assignment of the 13C NMR spectrum of 2 could not be made due to the presence of many overlapping multiplet resonances. The continuous wave (cw) EPR/ENDOR spectra were recorded on an X-band Bruker ESP300E series spectrometer equipped with an ESP360 DICE ENDOR unit, operating at 12.5 kHz field modulation in a Bruker EN801 cavity. The ENDOR spectra were recorded at 10 K using an Oxford instruments ESR 900 continuous flow He cryostat. These spectra were obtained using 8 dB RF power from an ENI A-300 RF amplifier with 75 or 250 kHz RF modulation depth. Computer simulations were carried out using Bruker's Simfonia program.37 Mass spectra were recorded using a VG Fisons Platform II instrument under APCI conditions, or were obtained from the EPSRC National Mass Spectrometry Service at Swansea University. IR spectra were recorded using a Nicolet 510 FT-IR spectrometer as Nujol mulls between NaCl plates. Melting points were determined in sealed glass capillaries under argon, and are uncorrected. [K(tmeda)][:Ga{[N(Ar)C(H)]2}],14cyclo-(PPh)5,38 [Ga{[N(Ar)C(H)]2}]2,15c [I2Ga{[N(Ar)C(H)]2}]17 and Mes*PH239 were synthesised by literature procedures whilst all other chemicals were obtained from commercial sources and used as supplied.Syntheses
NPh (0.065 g, 0.35 mmol) in diethyl ether (20 cm3) at −80 °C was added a solution of [K(tmeda)][:Ga{[N(Ar)C(H)]2}] (0.40 g, 0.66 mmol) in diethyl ether (50 cm3) over 5 min. The resultant solution was allowed to warm to room temperature and stirred overnight. Volatiles were then removed in vacuo and the red residue extracted into hexane (20 cm3). Filtration, concentration and placement at −30 °C overnight gave orange/red crystals of 3 (0.17 g, 57% based on azobenzene). Mp 184–187 °C(decomp.); 1H NMR (400 MHz, C6D6): δ 1.22 (br. d, 2JHH = 6.6 Hz, 12H, CH3), 1.27 (t, 3JHH = 7.3 Hz, 6H, Et2O), 1.49 (br. d, 2JHH = 6.6 Hz, 12H, CH3), 2.08 (s, 12H, NCH3), 2.26 (s, 4H, NCH2), 3.11 (overlapping m., 4H, CH), 3.77 (q, 3JHH = 7.1 Hz, 4H, Et2O), 5.24 (s, 2H, NCH), 6.6–7.3 (m, 15H, Ar–H), 7.47 (br, 1H, NH); 13C NMR (75.57 MHz, C6D6): δ 15.3 (Et2O), 23.8 (CH3), 24.1 (CH3), 28.0 (CH), 28.2 (CH), 45.6 (NCH3), 57.9 (NCH2), 65.6 (Et2O), 119.6 (NC2H2N), 123.1, 123.3, 123.7, 128.2, 128.7, 129.3, 129.5, 136.6, 137.4, 140.6, 142.5, 144.2, 148.6, 149.1 (Ar–C); IR ν/cm−1 (Nujol): 3486 (br, NH), 1603 (m), 1493 (m), 1459 (s), 1382 (w), 1260 (s), 1024 (s), 801 (s), 693 (w), 668 (w); (MS/APCI) m/z: 377 [{N(Ar)C(H)}2H+, 100%].
X-Ray crystallography
Crystals of 2, 3, 5, 6 and 8 suitable for X-ray structural determination were mounted in silicone oil. Crystallographic measurements were made using a Nonius Kappa CCD diffractometer. The structures were solved by direct methods and refined on F2 by full matrix least squares (SHELX97)40 using all unique data. All non-hydrogen atoms are anisotropic with H-atoms included in calculated positions (riding model), except for the N–H hydrogen of 3 and the P–H hydrogen atoms of 6 and 8, which were refined isotropically. Crystal data, details of data collections and refinement are given in Table 1.CCDC reference numbers 280910–280914.
For crystallographic data in CIF or other electronic format see DOI: 10.1039/b511451a
| Compound | 2 | 3·C6H14 | 5 | 6·0.5{Li2I2(THF)3.5(OEt2)0.5} | 8 |
|---|---|---|---|---|---|
| Empirical formula | C56H72GaKN4P4 | C51H79GaKN6O | C58H90Ga2N6Si2 | C52H82.5GaI2LiNO2P | C53H82GaN4P |
| M | 1033.88 | 901.02 | 1066.98 | 1129.13 | 875.92 |
| T/K | 150(2) | 150(2) | 150(2) | 150(2) | 150(2) |
| Crystal system | Monoclinic | Monoclinic | Monoclinic | Monoclinic | Monoclinic |
| Space group | P2(1)/c | C2/c | P2(1)/c | P2(1)/n | P2(1)/n |
| a/Å | 21.808(4) | 20.663(4) | 15.592(3) | 10.088(2) | 11.127(2) |
| b/Å | 13.871(3) | 14.230(3) | 21.742(4) | 49.646(10) | 18.270(4) |
| c/Å | 20.504(4) | 35.603(7) | 17.818(4) | 22.378(5) | 24.459(5) |
| α/° | 90 | 90 | 90 | 90 | 90 |
| β/° | 114.97(3) | 101.76(3) | 102.34(3) | 97.94(3) | 94.49(3) |
| γ/° | 90 | 90 | 90 | 90 | 90 |
| V/Å3 | 5623(2) | 10249(4) | 5901(2) | 11100(4) | 4957.0(17) |
| Z | 4 | 8 | 4 | 8 | 4 |
| ρ calcd/Mg m−3) | 1.221 | 1.168 | 1.201 | 1.351 | 1.174 |
| μ(Mo-Kα)/mm−1 | 0.715 | 0.658 | 0.994 | 1.674 | 0.625 |
| F(000) | 2184 | 3880 | 2280 | 4636 | 1896 |
| No. of reflns collected | 18873 | 15119 | 47327 | 57446 | 30202 |
| No. of independent reflns | 9844 [R(int) = 0.0451] | 8869 [R(int) = 0.0700] | 10395 [R(int) = 0.0837] | 19185 [R(int) = 0.1030] | 8967 [R(int) = 0.0819] |
| Final R indices (I > 2σ(I)) | R1 = 0.0947 wR2 = 0.2364 | R1 = 0.0797 wR2 = 0.1526 | R1 = 0.0516 wR2 = 0.1022 | R1 = 0.0663 wR2 = 0.1283 | R1 = 0.0698 wR2 = 0.1461 |
Acknowledgements
We thank the EPSRC (postdoctoral fellowship for RJB, studentship for DPM) for financial support and for funding the National ENDOR Service (GR/R17980/01). Financial support from the Studienstiftung des Deutschen Volkes (PhD grant for R.W.) and the German Academic Exchange Service (313-ARC-XVI-02/10) is gratefully acknowledged. The EPSRC Mass Spectrometry Service at Swansea University is also thanked.References and notes
- (a) P. P. Power, Chem. Commun., 2003, 2091 RSC; (b) P. P. Power, Chem. Rev., 1999, 99, 3463 CrossRef CAS; (c) P. P. Power, J. Chem. Soc., Dalton Trans., 1998, 2939 RSC and references therein.
- (a) W. Uhl, Adv. Organomet. Chem., 2004, 51, 53 CrossRef CAS; (b) P. P. Power, Struct. Bonding, 2002, 103, 57 CAS; (c) W. Uhl, Coord. Chem. Rev., 1997, 163, 1 CrossRef CAS; (d) P. J. Brothers and P. P. Power, Adv. Organomet. Chem., 1996, 39, 1 CAS and references therein.
- (a) J. Su, X. W. Li, R. C. Crittendon, C. F. Campana and G. H. Robinson, Organometallics, 1997, 16, 4511 CrossRef CAS; (b) G. H. Robinson, Acc. Chem. Res., 1999, 32, 773 CrossRef CAS; (c) N. J. Hardman, R. J. Wright, A. D. Phillips and P. P. Power, J. Am. Chem. Soc., 2003, 125, 2667 CrossRef CAS; (d) R. Ponec, G. Yuzhakov, X. Gironés and G. Frenking, Organometallics, 2004, 123, 1790 CrossRef and references therein.
- (a) M. C. Kuchta and G. Parkin, Inorg. Chem., 1997, 36, 2492 CrossRef CAS; (b) M. C. Kuchta and G. Parkin, J. Chem. Soc., Dalton Trans., 1998, 2279 RSC.
- R. J. Wright, A. D. Philips, T. L. Allen, W. H. Fink and P. P. Power, J. Am. Chem. Soc., 2003, 125, 1694 CrossRef CAS.
- M. Petrie and P. P. Power, Inorg. Chem., 1993, 32, 1309 CrossRef CAS.
- S. Weinrich, H. Piotrowski, M. Vogt, A. Schulz and M. Westerhauser, Inorg. Chem., 2004, 43, 3756 CrossRef CAS.
- C. von Hänisch and O. Hampe, Angew. Chem., Int. Ed., 2002, 41, 2095 CrossRef CAS.
- C. Cui, H. W. Roesky, H.-G. Schmidt, M. Noltemeyer, H. Hao and F. Cimpoesu, Angew. Chem., Int. Ed., 2000, 39, 4274 CrossRef CAS.
- N. J. Hardman, B. E. Eichler and P. P. Power, Chem. Commun., 2000, 1991 RSC.
- N. J. Hardman, C. Cui, H. W. Roesky, W. H. Fink and P. P. Power, Angew. Chem., Int. Ed., 2001, 40, 2172 CrossRef CAS.
- (a) N. J. Hardman and P. P. Power, Chem. Commun., 2001, 1184 RSC; (b) C. Cui, H. W. Roesky, H.-G. Schmidt and M. Noltemeyer, Angew. Chem., Int. Ed., 2000, 39, 4531 CrossRef CAS.
- H. Zhu, J. Chai, V. Chandrashehkar, H. W. Roesky, J. Magull, D. Vidovic, H.-G. Schmidt, M. Noltemeyer, P. P. Power and W. A. Merrill, J. Am. Chem. Soc., 2004, 126, 9472 CrossRef CAS.
- R. J. Baker, R. D. Farley, C. Jones, M. Kloth and D. M. Murphy, J. Chem. Soc., Dalton Trans., 2002, 3844 RSC . NB: A synthesis of a related anion, [:Ga{[N(But)C(H)]2}]−, has been previously reported, see: E. S. Schmidt, A. Jockisch and H. Schmidbaur, J. Am. Chem. Soc., 1999, 121, 9358 Search PubMed.
- (a) R. J. Baker, C. Jones and D. M. Murphy, Chem. Commun., 2005, 1339 RSC; (b) R. J. Baker, C. Jones and M. Kloth, Dalton Trans., 2005, 2106 RSC; (c) R. J. Baker, C. Jones, M. Kloth and J. A. Platts, Organometallics, 2004, 23, 4811 CrossRef CAS; (d) R. J. Baker, C. Jones and J. A. Platts, Dalton Trans., 2003, 3673 RSC; (e) R. J. Baker, C. Jones and J. A. Platts, J. Am. Chem. Soc., 2003, 125, 10534 CrossRef CAS; (f) R. J. Baker, C. Jones, M. Kloth and J. A. Platts, Angew. Chem., Int. Ed., 2003, 43, 2660 CrossRef CAS.
- R. J. Baker and C. Jones, Coord. Chem. Rev., 2005, 149, 1857 CrossRef and references therein.
- T. Pott, P. Jutzi, W. Kaim, W. W. Schoeller, B. Neumann, A. Stammler, H. G. Stammler and M. Wanner, Organometallics, 2002, 21, 3169 CrossRef CAS.
- T. Pott, P. Jutzi, W. W. Schoeller, A. Stammler and H.-G. Stammler, Organometallics, 2001, 20, 5492 CrossRef CAS.
- (a) N. Kuhn and A. Al-Sheikh, Coord. Chem. Rev., 2005, 249, 829 CrossRef CAS; (b) W. Kirmse, Eur. J. Org. Chem., 2005, 237 CrossRef CAS; (c) C. J. Carmalt and A. H. Cowley, Adv. Inorg. Chem., 2000, 50, 1 CAS; (d) D. Bourissou, O. Guerret, F. P. Gabai and G. Bertrand, Chem. Rev., 2000, 100, 39 CrossRef and references therein.
- (a) A. J. Arduengo, J. C. Calabrese, A. H. Cowley, H. V. R. Rasika Dias, J. R. Goerlich, W. J. Marshall and B. Riegel, Inorg. Chem., 1997, 36, 2151 CrossRef; (b) A. J. Arduengo, C. J. Carmalt, J. A. C. Clyburne, A. H. Cowley and R. Pyati, Chem. Commun., 1997, 981 RSC.
- W. Uhl and M. Benter, J. Chem. Soc., Dalton Trans., 2000, 3133 RSC.
- Y. Peng, H. Fan, H. Zhu, H. W. Roesky, J. Magull and C. E. Hughes, Angew. Chem., Int. Ed., 2004, 43, 3443 CrossRef CAS.
- A. Bashall, F. García, A. A. Hopkins, J. A. Wood, M. McPartlin, A. D. Woods and D. S. Wright, Dalton Trans., 2003, 1143 RSC.
- As determined from a survey of the Cambridge Crystallographic Database, August, 2005.
- S. Shah and J. D. Protasiewicz, Chem. Commun., 1998, 1585 RSC.
- M. Yoshifuji, I. Shima, N. Inamoto, K. Hirotsu and T. Higuchi, J. Am. Chem. Soc., 1981, 103, 4587 CrossRef CAS.
- B. Twamley, C. D. Sofield, M. M. Olmstead and P. P. Power, J. Am. Chem. Soc., 1999, 121, 3357 CrossRef CAS.
- N. M. Yoon and Y. S. Gyoung, J. Org. Chem., 1985, 50, 2443 CrossRef CAS.
- H. Nöth and T. Seifert, Eur. J. Inorg. Chem., 2002, 602 CrossRef CAS.
- E. Niecke, M. Link and M. Nieger, Chem. Ber., 1992, 125, 2631 CrossRef.
- P. Jutzi, B. Neumann, G. Reumann and H.-G. Stammler, Organometallics, 1999, 18, 2037 CrossRef CAS.
- C. Cui, M. Brynda, M. M. Olmstead and P. P. Power, J. Am. Chem. Soc., 2004, 126, 6510 CrossRef CAS.
- K. L. Antcliff, R. J. Baker, C. Jones, D. M. Murphy and R. P. Rose, Inorg. Chem., 2005, 44, 2098 CrossRef CAS.
- R. J. Baker, R. D. Farley, C. Jones, D. P. Mills, M. Kloth and D. M. Murphy, Chem. Eur. J., 2005, 11, 2972 CrossRef CAS.
- K. Lammertsma and M. J. M. Vlaar, Eur. J. Inorg. Chem., 2002, 1127 CrossRef.
- W. P. Leung, C. M. Y. Chan, B. M. Wu and T. C. W. Mak, Organometallics, 1996, 15, 5179 CrossRef CAS.
- WINEPR SIMFONIA version 1.25, Brüker Analytische, Messtechnick, 1996 Search PubMed.
- J. J. Daly, J. Chem. Soc., 1964, 6147 RSC.
- A. H. Cowley, N. C. Norman and M. Pakulski, Inorg. Synth., 1990, 27, 235 CAS.
- G. M. Sheldrick, SHELX-97, University of Göttingen, Germany, 1997 Search PubMed.
| This journal is © The Royal Society of Chemistry 2006 |