Synthesis of mononuclear and dinuclear ruthenium(II) tris(heteroleptic) complexes via photosubstitution in bis(carbonyl) precursors†
Declan
Mulhern
a,
Sally
Brooker
b,
Helmar
Görls
c,
Sven
Rau
c and
Johannes G.
Vos
*a
aNational Centre for Sensor Research, School of Chemical Sciences, Dublin City University, Dublin, 9, Ireland. E-mail: han.vos@dcu.ie
bDepartment of Chemistry, University of Otago, P.O. Box 56, Dunedin, New Zealand
cInstitute für Anorganische und Analytische Chemie, Friedrich-Schiller-Universität, Jena, D-07743, Germany
First published on 26th October 2005
Abstract
A novel, and quite general, approach for the preparation of tris(heteroleptic) ruthenium(II) complexes is reported. Using this method, which is based on photosubstitution of carbonyl ligands in precursors such as [Ru(bpy)(CO)2Cl2] and [Ru(bpy)(Me2bpy)(CO)2](PF6)2, mononuclear and dinuclear Ru(II) tris(heteroleptic) polypyridyl complexes containing the bridging ligands 3,5-bis(pyridin-2-yl)-1,2,4-triazole (Hbpt) and 3,5-bis(pyrazin-2-yl)-1,2,4-triazole (Hbpzt) have been prepared. The complexes obtained were purified by column chromatography and characterized by HPLC, mass spectrometry, 1H NMR, absorption and emission spectroscopy and by electrochemical methods. The X-ray structures of the compounds [Ru(bpy)(Me2bpy)(bpt)](PF6)·0.5C4H10O [1·0.5C4H10O], [Ru(bpy)(Me2bpy)(bpzt)](PF6)·H2O (2·H2O) and [Ru(bpy)(Me2bpy)(CH3CN)2](PF6)2·C4H10O (6·C4H10O) are reported. The synthesis and characterisation of the dinuclear analogues of 1 and 2, [{Ru(bpy)(Me2bpy)}2bpt](PF6)3·2H2O (3) and [{Ru(bpy)(Me2bpy)}2bpzt](PF6)3 (4), are also described.
Introduction
The photophysical and photochemical properties of dinuclear Ru(II) metal complexes have received considerable attention for their potential to facilitate light-induced functions including energy and charge transfer.1–4 Ligand composition has been used widely to fine-tune the electronic and redox properties of such compounds5–9 for example to manipulate intercomponent interactions4,9 or to introduce specific functionalities to allow surface binding.10 Of particular interest would be compounds of the type shown in Fig. 1 where D is an electron donor and A an electron acceptor. With such compounds detailed studies of photoinduced electron transfer in dinuclear systems could be carried out as a function of the bridge. However, the synthesis of trisheteroleptic compounds of this type, especially with triazole type ligands, is by no means straightforward. Therefore detailed studies on the preparation of such complexes have been carried out in our laboratories. | ||
| Fig. 1 Prototype dinuclear acceptor–donor complex. | ||
It is just over 20 years since the first tris-heteroleptic Ru(II) polypyridyl complex was reported11 and the various ensuing synthetic routes produced only a relatively small number of complexes.12–15 However, the introduction of methods based on the chemical decarbonylation of a [Ru(L)(L′)(CO)2]2+ precursor or on the sequential addition of ligands to [Ru(DMSO)4Cl2] has seen a growing series of these complexes synthesised.16–21 A full and comprehensive review of the various synthetic routes to these complexes has recently been published.22 In this report we describe a new synthetic route based on the photochemical, rather than chemical, elimination of carbonyl ligands in ruthenium bis-carbonyl complexes, which we have successfully used to the prepare two dinuclear tris(heteroleptic) Ru(II) complexes containing the bridging ligands Hbpt and Hbpzt (for structures, see Fig. 2).
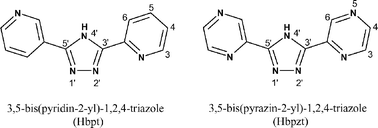 | ||
| Fig. 2 1,2,4-Triazole-based bridging ligands used in this study. | ||
This synthetic approach opens the way for the synthesis of compounds such as those outlined in Fig. 1 and hence the detailed investigation of photoinduced electron transfer processes in dinuclear acceptor–donor compounds of this type.
Experimental
Materials
All materials used were of reagent grade or better. Bpy (Aldrich) and Me2bpy (Fluka) were used as received. All solvents were HPLC grade or better. [Ru(bpy)(CO)2Cl2]23 and Hbpt and Hbpzt9,24 were synthesised according to previously reported procedures.Synthetic procedures
For further details see text.
Physical measurements
1H NMR spectra were performed on a Bruker Avance 400 NMR Spectrometer in CD3CN with TMS as reference. Free induction decay (FID) profiles were processed using XWIN-NMR software package. Mass spectra were recorded with a Bruker-EsquireLC_00050 electrospray ionisation mass spectrometer at positive polarity with cap-exit voltage of 167 V. Each spectrum was recorded by summation of 20 scans.HPLC measurements were performed on a JVA analytical HPLC system consisting of a Varian Prostar HPLC pump using a HiChrom Partisil P10SCX-3095 cation exchange column and Varian Prostar photodiode array and 280 nm detection. A 20 µl injection loop delivered the sample to the column using 0.08 M LiClO4 in MeCN–H2O (80/20) mobile phase at a flow rate of 1.8 cm3 min−1.
Infrared spectra were measured using a Perkin Elmer 2000 FTIR spectrometer. UV-vis absorption spectra were recorded in acetonitrile on a Shimadzu 3100 UV-Vis/NIR instrument with 1-cm quartz cells. Emission spectra were recorded at 298 K in MeCN using a Perkin-Elmer LS50B luminescence spectrometer equipped with a red sensitive Hamamatsu R928 detector. At 77 K, measurements were carried out in ethanol–methanol (4 : 1 v/v). Lifetime measurements were performed on an Edinburgh Analytical Instruments single photon counter with a T setting, using a lamp (nF900, in a nitrogen setting), monochromators (J-yA models), with a single photon photomultiplier detection system (model S 300), an MCA card (Norland N5000) and PC interface (Cd900 serial). Data correlation and manipulation was carried out using the program F900, Version 5.13. Samples were de-aerated for 15 min using Ar prior to analysis.
DPV electrochemical measurements were carried out using a CH Instruments CHI Version 2.07 software controlled potentiostat (CH Instruments Memphis 660). Solutions of the complex (typically 1 mM) to be tested were made up in a 0.1 M solution of TBABF4 (Aldrich) in dry MeCN. The solution was purged with Ar (10 min) and an Ar atmosphere was maintained throughout the experiment. A three compartment cell housed a platinum disc (working, 2 mm diameter), platinum wire (counter) and a Ag/Ag+ (acetonitrile + 10 mM AgNO3 + 0.1 M TBABF4) half-cell (reference). The instrument was calibrated before and after each session using the Fc/Fc+ couple.
Elemental analyses were carried out by the Microanalytical Department, University College Dublin.
Crystals suitable for X-ray studies of 1, 2 and 6 were grown by vapour diffusion of diethyl ether into an acetonitrile solution of the respective complex. For the three crystal structure determinations, the X-ray intensity data were collected on either a Bruker SMART CCD diffractometer (1·0.5C4H10O) or a Nonius KappaCCD diffractometer (2·H2O and 6·C4H10O). Data were corrected for Lorentz and polarization effects, but not for absorption effects in the case of 2·H2O and 6·C4H10O.25–28 The structures were solved by direct methods (SHELXS29) and refined by full-matrix least-squares techniques against Fo2 (SHELXL-9730). The hydrogen atoms were included at calculated positions with fixed thermal parameters except for the hydrogen atoms of the H2O molecule in 2 which could not be located. All non-hydrogen atoms were refined anisotropically. XP (SIEMENS Analytical X-ray Instruments, Inc.) was used for structure representations.
CCDC reference numbers 272204 (1·0.5C4H10O), 273899 (2·H2O) and 273900 (6·C4H10O).
For crystallographic data in CIF or other electronic format see DOI: 10.1039/b510751b
Results and discussion
Synthetic procedures
Synthesis of mononuclear and dinuclear Ru(II) tris(heteroleptic) complexes containing 1,2,4-triazole bridging ligands was successfully accomplished by using the product obtained by irradiating [Ru(bpy)(CO)2Cl2] in the presence of dry MeCN as precursor. The overall synthetic route is shown in Scheme 1. | ||
| Scheme 1 The synthetic route used to prepare the tris(heteroleptic) complexes 1–4. | ||
A second method based on the photolysis of precursors of the type [Ru(L1)(L2)(CO)2]2+ was considered but found less attractive (see below). Infrared spectroscopy was used to follow the course of the reaction by monitoring the loss of CO (see Fig. 3).
![Photolysis of [Ru(bpy)(CO)2Cl2] in MeCN.](/image/article/2006/DT/b510751b/b510751b-f3.gif) | ||
| Fig. 3 Photolysis of [Ru(bpy)(CO)2Cl2] in MeCN. | ||
Initially the two νCO stretching bands at 2064 and 2001 cm−1 indicative for the presence of [Ru(bpy)(CO)2Cl2] disappeared rapidly and were replaced by one band at 1969 cm−1, indicative of a monocarbonyl species as suggested by Eskelinen et al.31 This band gradually disappears. 1H NMR of the product obtained (see Fig. S1, ESI†) shows eight aromatic peaks each integrating to one and four aromatic peaks (shown by arrows) each integrating to a half, suggesting the formation of a mixture containing the symmetric [Ru(bpy)(MeCN)2Cl2] with the asymmetric [Ru(bpy)(MeCN)3Cl]Cl as the major species. This is confirmed by electrochemistry where the mixture shows two reversible oxidation potentials at E1/2 = 0.14 and 0.74 V (Ag/Ag+) in a 1 : 4 ratio which correspond to [Ru(bpy)(MeCN)2Cl2] and [Ru(bpy)(MeCN)3Cl]Cl respectively (see Fig. S1, ESI†). The assignment of the redox features is based on the reasonable assumption that replacement of a Cl− with a weaker σ-donor ligand such as MeCN shifts the oxidation potential of the metal centre to a more positive value.
Although the photolysis product was found to be a mixture of two species, pure [Ru(bpy)(Me2bpy)Cl2] was obtained after refluxing the mixture with Me2bpy in dry acetone for 15 h. Only the cis-dichloride species was observed and both the CHN and 1H NMR data indicate that no water of crystallisation was present. The 1H NMR spectrum of the dichloride is shown in the ESI, Fig. S2.† Reacting [Ru(bpy)(Me2bpy)Cl2] with excess Hbpt or Hbpzt in aqueous EtOH at reflux produced the tris(heteroleptic) mononuclear complexes [Ru(bpy)(Me2bpy)(bpt)](PF6)·H2O (1) and [Ru(bpy)(Me2bpy)(bpzt)](PF6)·2H2O (2), respectively.
The dinuclear complexes [{Ru(bpy)(Me2bpy)}2bpt](PF6)3·2H2O (3) and [{Ru(bpy)(Me2bpy)}2bpzt](PF6)3 (4) were obtained by reacting a greater than two-fold excess of the dichloride precursor, the 1 : 4 mixture of [Ru(bpy)(MeCN)2Cl2] and [Ru(bpy)(MeCN)3Cl]Cl, with the bridging ligand, Hbpt or Hbpzt. It was also possible to synthesise 3 and 4 by reacting 1 and 2 with excess [Ru(bpy)(Me2bpy)Cl2].
In all of these reactions HPLC analysis of the crude products showed that the product complex, 1–4, was contaminated by a small trace of the corresponding dinuclear or mononuclear species, 3, 4, 1 and 2, respectively. Pure samples of 1–4 were readily obtained by chromatography on alumina: afterwards only one peak was observed in the HPLC analysis.
The second method that was considered was based on the photolysis of compounds such as [Ru(bpy)(Me2bpy)(CO)2]2+ (5). This compound was obtained by reacting [Ru(Me2bpy)(CO)2Cl2] with excess bpy. Photolysis of 5 proved to be very efficient so this method is a viable alternative for chemical decarbonylation. The progress of the reaction was followed with infrared spectroscopy. The spectra show that the bands at 2099 and 2047 cm−1 observed for [Ru(Me2bpy)(bpy)(CO)2]2+ gradually disappear and are replaced by a single band at 2012 cm−1, indicative of an intermediate monocarbonyl species, which disappears upon further irradiation. After removal of the solvent, the 1H NMR and elemental analysis data on the product (see Experimental section) were consistent with the formation of the desired intermediate complex, [Ru(Me2bpy)(bpy)(CH3CN)2]2+6. This was confirmed by X-ray analysis (see below). Although some tris(heteroleptic) complexes were isolated by reacting this bis(acetonitrile) intermediate with an appropriate chelating ligand, several problems were identified with regard to the generality of this approach, such as the availability of bicarbonyl starting materials. For example, in our hands only the starting materials [Ru(bpy)2(CO)2]2+ and [Ru(Me2bpy)(bpy)(CO)2]2+ were obtained in reasonable yields. Against this background it was decided that the photo induced decarbonylation of [Ru(L)(CO)2Cl2] should be focussed on as the preferred synthetic route and the second route, via5, was not investigated any further.
X-Ray crystallography
The X-ray data for 1·0.5C4H10O, 2·H2O and 6·C4H10O are given in Table 1.| 1·0.5C4H10O | 2·H2O | 6·C4H10O | |
|---|---|---|---|
| Empirical formula | C36H33N9RuPF6O0.5 | C32H28N9RuPF6O | C30H36N6ORuP2F12 |
| M r | 845.75 | 828.69 | 887.66 |
| Colour | Red | Brown | Red–brown |
| Crystal source | MeCN–diethyl ether | MeCN–diethyl ether | MeCN–diethyl ether |
| T/K | 200(2) | 183(2) | 183(2) |
| Crystal size/mm | 0.42 × 0.24 × 0.20 | 0.09 × 0.07 × 0.06 | 0.03 × 0.03 × 0.02 |
| Space group | P21/c | P21/c | P21/c |
| a/Å | 13.95240(10) | 16.2156(4) | 11.1155(3) |
| b/Å | 12.24230(10) | 13.8572(4) | 18.1355(5) |
| c/Å | 23.4412(10) | 14.8161(4) | 18.3079(7) |
| β/° | 95.9090(10) | 92.117(2)° | 95.932(1) |
| V/Å3 | 3982.70(8) | 3326.95(16) | 3670.8(2) |
| D c/g cm−3 | 1.411 | 1.654 | 1.606 |
| Z | 4 | 4 | 4 |
| F(000) | 1716 | 1672 | 1792 |
| Radiation | Mo-Kα | Mo-Kα | Mo-Kα |
| Abs. coeff., µ/mm−1 | 0.501 | 0.60 | 0.61 |
| Abs. corr., T(min, max) | 0.40, 0.93 | None | None |
| 2θ limits/° | 1.47–26.38 | 2.35–27.44 | 2.25–27.48 |
| Number of reflections | 22545 | 13029 | 14401 |
| Number of unique reflections | 8079 | 7514 | 8418 |
| Number of parameters | 509 | 469 | 469 |
| R1 (observed data) | 0.059 | 0.049 | 0.056 |
| wR2 (observed data) | 0.1060 | 0.125 | 0.092 |
| Goodness of fit | 1.001 | 1.011 | 0.955 |
Selected bond lengths and angles are summarised in Table 2. The geometrical properties of the bis(acetonitrile) complex 6·C4H10O are in agreement with those of [Ru(bpy)2(CH3CN)2](PF6)2 reported by Heeg et al.32 In both cases an octahedral coordination mode is observed with the bipyridine ligands exhibiting acute bite-angles, in this case 78.70(14)° for the Me2bpy ligand and 78.74(14)° for the bpy ligand (See Fig. 4).
| 6·C4H10O | 1·0.5C4H10O | 2·H2O | |
|---|---|---|---|
| a N7 substituted for N6 in [Ru(bpy)(Me2bpy)(CH3CN)2](PF6)2 for comparision. | |||
| Ru–N1 | 2.067(3) | 2.059(4) | 2.061(3) |
| Ru–N2 | 2.046(3) | 2.054(4) | 2.050(3) |
| Ru–N3 | 2.043(3) | 2.064(4) | 2.070(3) |
| Ru–N4 | 2.067(3) | 2.065(4) | 2.055(3) |
| Ru–N5 | 2.032(4) | 2.105(4) | 2.069(3) |
| Ru–N7 | — | 2.048(4) | 2.046(3) |
| Ru–N6 | 2.037(4) | — | — |
| N7(6)–Ru–N3 | 89.66(13) | 174.82(14) | 173.99(12) |
| N2–Ru–N5 | 89.88(13) | 171.10(15) | 171.61(11) |
| N4–Ru–N1 | 171.14(13) | 173.35(15) | 175.05(11) |
| N7(6)–Ru–N5 | 90.35(13) | 77.82(15) | 78.03(11) |
| N7(6)–Ru–N4 | 88.80(13) | 98.24(16) | 97.68(11) |
| N7(6)–Ru–N1 | 96.65(14) | 87.73(15) | 86.40(11) |
| N7(6)–Ru–N2 | 175.34(14) | 93.81(15) | 93.65(11) |
| N4–Ru–N5 | 97.30(14) | 86.71(15) | 84.23(12) |
| N5–Ru–N3 | 176.04(14) | 97.85(15) | 96.75(11) |
| N1–Ru–N5 | 89.67(13) | 97.46(15) | 99.39(12) |
| N4–Ru–N3 | 78.74(14) | 78.56(16) | 78.69(12) |
| N2–Ru–N4 | 95.79(14) | 97.59(15) | 98.11(11) |
| N1–Ru–N3 | 94.26(13) | 95.69(16) | 97.46(12) |
| N2–Ru–N3 | 90.43(13) | 90.66(16) | 91.61(11) |
| N2–Ru–N1 | 78.70(14) | 79.01(15) | 78.78(11) |
![Molecular structure of [Ru(bpy)(Me2bpy)(CH3CN)2]2+, the cation of 6·C4H10O. Hydrogen atoms, anions and solvent have been omitted for clarity.](/image/article/2006/DT/b510751b/b510751b-f4.gif) | ||
| Fig. 4 Molecular structure of [Ru(bpy)(Me2bpy)(CH3CN)2]2+, the cation of 6·C4H10O. Hydrogen atoms, anions and solvent have been omitted for clarity. | ||
Both bidentate ligands exhibit similar the bond lengths between the metal and chelating nitrogen atoms. Ru–N bond lengths for bpy are (Ru–N3) 2.043(3) and (Ru–N4) 2.067(3) Å whereas those for Me2bpy are (Ru–N1) 2.067(3) and (Ru–N2) 2.046(3) Å. In each case the shorter bond length is that trans to a MeCN ligand as a consequence of the bipyridyl ligands being stronger π-acids than MeCN ligands. The presence of a methyl group on the 4- and 4′-position of one of the bipy ligands has little effect on the Ru–N bond lengths to that ligand. Ru–N bond lengths for the two MeCN ligands are 2.032(4) and 2.037(4) Å respectively.
The crystal structures of 1·0.5C4H10O and 2·H2O unambiguously confirm the presence of three different bis-chelating ligands around the metal centre (Fig. 5 and 6).
![Molecular structure of [Ru(bpy)(Me2bpy)(bpt)]+, the cation of 1·0.5C4H10O. The hydrogen atoms, counter ion and solvent have been omitted for clarity.](/image/article/2006/DT/b510751b/b510751b-f5.gif) | ||
| Fig. 5 Molecular structure of [Ru(bpy)(Me2bpy)(bpt)]+, the cation of 1·0.5C4H10O. The hydrogen atoms, counter ion and solvent have been omitted for clarity. | ||
![Molecular structure of [Ru(bpy)(Me2bpy)(bpzt)]+, the cation of 2·H2O. The hydrogen atoms have been omitted for clarity.](/image/article/2006/DT/b510751b/b510751b-f6.gif) | ||
| Fig. 6 Molecular structure of [Ru(bpy)(Me2bpy)(bpzt)]+, the cation of 2·H2O. The hydrogen atoms have been omitted for clarity. | ||
It is clear from these structures that the ruthenium centre binds to the three ligands in an octahedral fashion and via N(7) of the triazole ring. Importantly, for both compounds the triazole nitrogen is trans to the bpy ring. One PF6− cation is present in each case, confirming that the triazole ring deprotonates upon coordination.
The bond lengths and angles observed in the structures of 1 and 2 are typical of those found for other Ru(II) polypyridyl complexes containing triazoles.9,33 The Ru–bpy and Me2bpy bond lengths are in the range of 2.055–2.070 Å with their respective bite angles in the range of 78.56–79.01°. The biggest difference between the two complexes is the metal–triazole bond lengths. Although the Ru–N7(triazole) distances are approximately identical (2.048(4) Å for bpt− and 2.046(3) Å for bpzt−), a significant difference exists between the two Ru–N5 bond lengths. In the case of [Ru(bpy)(Me2bpy)(bpt)]+ this distance is 2.105(4) Å. For the pyrazine analogue this distance is more in line with the other polypyridyl ligands at 2.069(3) Å. This observation is in accordance with the properties of the two types of triazole ligands as has been described previously.34 Specifically, bpt− is a ligand with strong σ-donating and weak π-accepting properties. This weak π-accepting property leads to an increase in the Ru–N(5) bond length. On the other hand, bpzt− is a ligand that combines both the strong σ-donating capabilities of the triazole and the strong π-accepting properties of the pyrazine. Therefore, while in [Ru(bpy)(Me2bpy)(bpzt)]+ the Ru–N7(triazole) distance remains almost identical to that in [Ru(bpy)(Me2bpy)(bpt)]+, the Ru–N5 bond is shorter by 0.036 Å.
In [Ru(bpy)(Me2bpy)(bpzt)]+ a strong H-bond interaction is exhibited in the solid state as depicted in Fig. 6. The water (O1W) of crystallisation acts as H-bond donor to a pyrazine nitrogen (N6–O1W 2.815 Å) of the cationic complex and a fluorine (F3–O1W 2.856 Å) of the PF6− anion.
Spectroscopic and electrochemical characterisation
![1H NMR of aliphatic region of two samples of [Ru(bpy)(Me2bpy)(bpt)]+ in d3-MeCN. Sample (a) is that of the product after column chromatography on alumina. Sample (b) is the recrystallised sample used for the X-ray analysis.](/image/article/2006/DT/b510751b/b510751b-f7.gif) | ||
| Fig. 7 1H NMR of aliphatic region of two samples of [Ru(bpy)(Me2bpy)(bpt)]+ in d3-MeCN. Sample (a) is that of the product after column chromatography on alumina. Sample (b) is the recrystallised sample used for the X-ray analysis. | ||
For the mononuclear complexes, both N2′ and N4′ are available for binding but previous studies have shown that if a large substituent is present at C5′ on the triazole ring the N2′ binding mode is preferred for steric reasons and this is indeed observed (see above). However, the asymmetrical nature of the bridging ligands still allows for two positional isomers as shown in Fig. 8.
![Two positional isomers of [Ru(bpy)(Me2bpy)(bpt)]+. One isomer has the triazole ring trans to a Me2bpy ring (left) whereas the other has the triazole ring trans to a bpy ring (right).](/image/article/2006/DT/b510751b/b510751b-f8.gif) | ||
| Fig. 8 Two positional isomers of [Ru(bpy)(Me2bpy)(bpt)]+. One isomer has the triazole ring trans to a Me2bpy ring (left) whereas the other has the triazole ring trans to a bpy ring (right). | ||
In one instance the triazole ring is trans to a Me2bpy ring, whereas in the other it is trans to a bpy ring. Fig. 7(a) shows that initially two isomers are formed, while the recrystallised sample used for the X-ray analysis contains a single isomer (Fig. 7(b)). As the X-ray data show that in the crystal the triazole nitrogen is trans to the bpy ring. This indicates that the resonances observed at 2.42 and 2.44 ppm can be attributed to this particular isomer. The 1H NMR spectrum for this isomer in the aromatic range is shown in Fig. S3 (ESI†). Due to the complexity of the spectrum no efforts were made to assign the various peaks. It is proposed for the second isomer shown in Fig. 7(a) the triazole is trans to the Me2bpy ligand.
| Emission, λmax/nm (τ/µs) | E 1/2 c/V (vs. Fc/Fc+) | ||||
|---|---|---|---|---|---|
| Complex | Absorption,aλmax/nm (10−4ε/M−1 cm−1) | 298 Ka | 77 Kb | Ox | Red |
| a In MeCN, deaerated solutions. b In EtOH–MeOH (4 : 1, v/v) deaerated solutions. c In MeCN (0.1 M solution of TBABF4). Potentials vs. Fc/Fc+ represent one-electron transfer unless noted otherwise. d Measured in proprionitrile–butyronitrile (4 : 5). e Measured in aerated solutions. Data for compounds 7–10 obtained from references 9 and 34. | |||||
| 1 | 475 (1.10) | 686 (0.37) | 612 (3.0) | 0.42 | −1.90, −2.16 |
| 2 | 445 (1.45) | 647 (0.51) | 600 (8.1) | 0.64 | −1.80, −2.02 |
| 3 | 452 (2.18) | 645 (0.08) | 606 (3.8) | 0.62, 0.92 | −1.81 (2e), −2.11 (2e) |
| 4 | 451 (2.32) | 666 (0.09) | 602 (7.8) | 0.79, 1.05 | −1.66, −1.77, −1.91, −2.25 |
| 7 | 475 (1.13) | 678 (0.16) | 628 (2.8)d | 0.48 | −1.85, −2.10 |
| 8 | 453 (1.42) | 662 (0.10) | 610 (5.0)d | 0.60 | −1.78, −2.00 |
| 9 | 453 (2.26) | 648 (0.10)e | 608 (3.6)d | 0.66, 0.96 | −1.78, −2.00, −2.05 |
| 10 | 449 (2.65) | 690 (0.10)e | 610 (6.4)d | 0.78, 1.08 | −1.70, −1.76, −1.94, −2.07 |
Both mononuclear complexes 1 and 2 exhibit a one-electron reversible oxidation wave and two one-electron reversible reduction waves. Metal-based oxidation waves for 1 and 2 are found at 0.42 and 0.64 V, respectively, similar to those reported for 7 and 8. Two one-electron oxidation waves separated by about 300 mV are observed at 0.62 and 0.92 V for 3 and 0.79 and 1.05 V for 4. The complexes 1–4 exhibit intense metal to ligand charge transfer (MLCT) absorption bands in the visible part of the spectrum (see Table 3). The MLCT maximum for the compounds is red shifted compared to [Ru(bpy)3]2+ as the σ-donating ability of the triazole ligand raises the HOMO energy level on the metal thus lowering the HOMO–LUMO energy difference.9 When the triazole negative charge is shared between two metal centres, as in the case of 3, the relative HOMO level is lowered, thus raising the energy of λmax from 475 nm (mononuclear) to 452 nm (dinuclear). The same principles can be used to rationalise the shift in emission spectra from 686 nm for 1 to 606 nm for 3. Overall the electronic and electrochemical properties of the compounds are very similar to those reported for the bis(bpy) analogues 7–10. Small variations in the energy of the electronic transitions are obtained which can be attributed to the slight differences in the electronic properties of the polypyridyl ligands employed.
Conclusion
A new synthetic procedure for the preparation of tris(heteroleptic) complexes containing a triazole bridging ligand has been developed and successfully utilised to produce both mononuclear and dinuclear Ru(II) polypyridyl complexes. An alternative photochemical method based on [BzRuCl2]2 as starting material21 was recently reported. Our strategy is similar to that of Freedman et al. but a very different starting material is used for the photolysis step. The reaction scheme is general enough to bring a large number of tris(heteroleptic) complexes within synthetic reach. As outlined in the introduction the ultimate aim of this work is to investigate dinuclear complexes containing acceptor and donor moieties as shown in Fig. 1. In the present study the polypyridyl ligands were chosen to facilitate the development of the method and to be able to address the formation of isomers (i.e. the use of Me2bpy). Therefore, the complexes produced have properties which are very similar to the Ru bis(bpy) analogues previously reported. At present work is in progress on the use of different polypyridyl ligands such as biq and dpp to investigate whether the excited state in these dinuclear compounds can be located specifically on one of the polypyridyl ligands. In addition, the synthesis the type of compounds outlined in Fig. 1 is in progress. Overall, the synthetic procedure utilised here will allow for more in-depth studies of the interaction between different metal centres in these dinuclear compounds by systematically altering the nature of the polypyridyl ligands within the metal coordination sphere.Acknowledgements
We gratefully acknowledge Enterprise Ireland and the Electricity Supply Board of Ireland (ESB) for their financial support of this work. H. G. and S. R. acknowledge the DFG (RA1017/1 and SFB 436) and S. B. the Marsden Fund (Royal Society of New Zealand) for financial support. We thank T. Groutso and Professor G. R. Clark (University of Auckland) for the X-ray data collection on 1·0.5C4H10O.References
- V. Balzani and F. Scandola, Supramolecular Photochemistry, Ellis Horwood, Chichester, 1991 Search PubMed.
- A. Juris, V. Balzani, F. Barigelletti, S. Campagna, P. Belser and A. von Zelewsky, Coord. Chem. Rev., 1998, 84, 85–277 CrossRef.
- K. Kalyanasundaram, Photochemistry of Polypyridine and Porphyrin Complexes, Academic Press, New York , 1991 Search PubMed.
- V. Balzani, A. Juris and M. Venturi, Chem. Rev., 1996, 96, 759–833 CrossRef CAS.
- M. D. Ward and F. Barigelletti, Coord. Chem. Rev., 2001, 216–217, 127–154 CrossRef CAS.
- C. K. Ryu, R. Wang, R. H. Schmehl, S. Ferrere, M. Ludwikow, J. W. Merkert, C. E. L. Headford and C. M. Elliot, J. Am. Chem. Soc., 1992, 114, 430–438 CrossRef CAS.
- G. Denti, S. Serroni, S. Campagna, V. Ricevuto and V. Balzani, Coord. Chem. Rev., 1991, 111, 227–236 CrossRef CAS.
- D. P. Rillema, R. Sahai, P. Matthews, A. K. Edwards, R. J. Shaver and L. Morgan, Inorg. Chem., 1990, 29, 167–175 CrossRef CAS.
- R. Hage, J. G. Haasnoot, H. A. Nieuwenhuis, J. Reedijk, D. J. A. De Ridder and J. G. Vos, J. Am. Chem. Soc., 1990, 112, 9245–9251 CrossRef CAS.
- B. O'Regan and Grätzel, Nature, 1991, 335, 737–740 CrossRef.
- D. St. C. Black, G. B Deacon and N. C. Thomas, Inorg. Chim. Acta, 1982, 65, L75 CrossRef CAS.
- G. H. Allen, R. P. White, D. P. Rillema and T. J. Meyer, J. Am. Chem. Soc., 1984, 106, 2613–2620 CrossRef CAS.
- J. M. Miller, K. Balasanmugam, J. Nye and G. B. Deacon, Inorg. Chem., 1987, 26, 560–562 CrossRef CAS.
- R. P. Thummel, F. Lefoulon and S. Chirayil, Inorg. Chem., 1987, 26, 3072–3074 CrossRef CAS.
- H. B. Ross, M. Boldaji, D. P. Rillema, C. B Blanton and R. P. White, Inorg. Chem., 1989, 28, 1013–1021 CrossRef CAS.
- A. von Zelewsky and G. Gremaud, Helv. Chim. Acta, 1988, 71, 1108–1115 CrossRef.
- G. F. Strouse, P. A. Anderson, J. R. Schoonover, T. J. Meyer and F. R. Keene, Inorg. Chem., 1992, 31, 3004–3006 CrossRef CAS.
- S. M. Zakeeruddin, Md. K. Nazeeruddin, R. Humphry-Baker, M. Grätzel and V. Shklover, Inorg. Chem., 1998, 37, 5251–5259 CrossRef CAS.
- J. A. Treadway and T. J. Meyer, Inorg. Chem., 1999, 38, 2267–2278 CrossRef CAS.
- C. M. Kepert, A. M. Bond, G. B. Deacon, L. Spiccia, B. W. Skelton and A. H. White, Dalton Trans., 2004, 1766–1774 RSC.
- D. A. Freedman, J. K. Evju, M. K. Pomije and K. R. Mann, Inorg. Chem., 2001, 40, 5711 CrossRef CAS.
- L. Spiccia, G. B. Deacon and C. M. Kepert, Coord. Chem. Rev., 2004, 248, 1329–1341 CrossRef CAS.
- P. A. Anderson, G. B. Deacon, K. H. Haarmaan, F. R. Keene, T. J. Meyer, D. A. Reitsma, B. W. Skelton, G. F. Strouse, N. C. Thomas, J. A. Treadway and A. H. White, Inorg. Chem., 1995, 34, 6145–6157 CrossRef CAS.
- (a) E. J. Browne and J. B. Ploka, J. Chem. Soc., 1969, 824 Search PubMed; (b) W. Kuzmierkiewicz, H. Foks and H. Baranowski, Sci. Pharm., 1985, 53, 133 Search PubMed.
- COLLECT, Data Collection Software, Nonius B.V., Netherlands, 1998 Search PubMed.
- Z. Otwinowski and W. Minor, Methods Enzymol., 1997, 276, 307–326 CrossRef CAS.
- SMART, Software for the CCD Detector System, version 5.05: Bruker AXS, Madison, WI, 1998 Search PubMed.
- SAINT, Software for the CCD Detector System, version 5.05: Bruker AXS, Madison, WI, 1998 Search PubMed.
- (a) G. M. Sheldrick, Acta Crystallogr., Sect. A, 1990, 46, 467–473 CrossRef; (b) G. M. Sheldrick, Methods Enzymol., 1997, 276, 628–641 CAS.
- (a) G. M. Sheldrick, SHELXL-97 (Release 97-2), University of Göttingen, Germany, 1997; (b) G. M. Sheldrick and T. R. Schneider, Methods Enzymol., 1997, 277, 319–343 CrossRef CAS.
- E. Eskelinen, M. Haukka, T. Venäläinen, T. A. Pakkanen, M. Wasberg, S. Chardon-Noblat and A. Deronzier, Organometallics, 2000, 19, 163–169 CrossRef CAS.
- M. J. Heeg, R. Kroener and E. Deutsch, Acta Crystallogr., Sect. C, 1985, 41, 684–686 CrossRef.
- R. Hage, J. P. Turkenburg, R. A. G. De Graaff, J. G. Haasnoot, J. Reedijk and J. G. Vos, Acta Crystallogr., Sect. C, 1989, 45, 381–383 CrossRef.
- R. Hage, J. G. Haasnoot, J. Reedijk, R. Wang and J. G. Vos, Inorg. Chem., 1991, 30, 3263–3269 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1: 1H NMR (CD3CN) and DPV of the photolysis mixture obtained after irradiating [Ru(bpy)(CO)2Cl2] in MeCN for 1 h. Fig. S2: 1H NMR of [Ru(bpy)(Me2bpy)Cl2] in d6-DMSO. Fig. S3: 1H NMR of the aromatic region of the crystal sample of [Ru(bpy)(Me2bpy)(bpt)]+ in d3-MeCN. See DOI: 10.1039/b510751b |
| This journal is © The Royal Society of Chemistry 2006 |