Spectral signatures and structural motifs in isolated and hydrated monosaccharides: phenyl α- and β-L-fucopyranoside
Pierre
Çarçabal
a,
Theodosis
Patsias
a,
Isabel
Hünig
a,
Bo
Liu
a,
Cristina
Kaposta
a,
Lavina C.
Snoek
a,
David P.
Gamblin
b,
Benjamin G.
Davis
b and
John P.
Simons
*a
aDepartment of Chemistry, Physical and Theoretical Chemistry Laboratory, South Parks Road, Oxford, UK OX1 3QZ. E-mail: john.simons@chem.ox.ac.uk
bDepartment of Chemistry, Chemistry Research Laboratory, Mansfield Road, Oxford, UK OX1 3TA
First published on 14th November 2005
Abstract
The conformation and structure of phenyl-α-L-fucopyranoside (α-PhFuc), phenyl-β-L-fucopyranoside (β-PhFuc) and their singly hydrated complexes (α,β-PhFuc·H2O) isolated in a molecular beam, have been investigated by means of resonant two photon ionization (R2PI) spectroscopy and ultraviolet and infrared ion-dip spectroscopy. Conformational and structural assignments have been based on comparisons between their experimental and computed near IR spectra, calculated using density functional theory (DFT) and their relative energies, determined from ab initio (MP2) calculations. The near IR spectra of ‘free’ and hydrated α- and β-PhFuc, and many other mono- and di-saccharides, provide extremely sensitive probes of hydrogen-bonded interactions which can be finely tuned by small (or large) changes in the molecular conformation. They provide characteristic ‘signatures’ which reflect anomeric, or axial vs. equatorial differences, both revealed through comparisons between α/β-PhFuc and α/β-PhXyl; or similarities, revealed through comparisons between fucose (6-deoxy galactose) and galactose; or binding motifs, for example, ‘insertion’ vs. ‘addition’ structures in hydrated complexes. At the monosaccharide level (the first step in the carbohydrate hierarchy), these trends appear to be general. In contrast to the monohydrates of galactose (β-PhGal) and glucose (β-PhGlc), the conformations of α- and β-PhFuc are unaffected by the binding of a single water molecule though changes in the R2PI spectra of multiply hydrated α-PhFucW(n) however, may reflect a conformational transformation when n ≥ 3.
1. Introduction
To understand fully the nature of molecular recognition processes, a first requirement is knowledge about the structure of the molecular participants and their interaction, as well as the influence of other molecules that may also be involved, not least molecules of the ubiquitous solvent, water. L-Fucose, one of the components of glycans found in naturally occurring glycoconjugates, plays a crucial role in many specific recognition processes in biological systems. It is present for example, in the antigen determinants on red blood cell surfaces that are responsible for the differences in the ABO blood groups.1 Unlike most other glycan mono-saccharide components, which adopt a D-chirality it is found in the L-form. In addition the (more common) exocyclic hydroxymethyl group is replaced by a methyl group: it is the mirror image of 6-deoxy D-galactose. The pyranosyl form of L-fucose has been investigated experimentally by X-ray crystallography2–8 and in solution by NMR spectroscopy,9,10 and its conformational space (for α-L-fucose) has been explored computationally, in the ‘gas phase’, through ab initio11,12 and molecular mechanics calculations.13 To date, however, there have been no experimental studies in the gas phase, of its intrinsic conformation, structure or molecular interactions.A series of infrared spectroscopic investigations, combining double resonance spectroscopy experiments conducted at low temperature in a molecular beam, leading to the observation of the vibrational spectrum of each populated conformer, and a computational exploration of the conformational landscape has recently established the conformational structures of a variety of common monosaccharide units; these include the principal glycan components, phenyl-β-D-glucoside (β-PhGlc),14 phenyl-β-D-galactoside (β-PhGal),15,16 phenyl-α-D-mannoside (α-PhMan),15 the two anomers phenyl-α- and β-D-xylopyranoside17 (α- and β-PhXyl), and each of their singly hydrated complexes15,17,18—an essential first step along the road towards the structural investigation of extended oligo-saccharide and short glycan chains.15,19–21 The strategy has now been applied to the α- and β-anomers of phenyl L-fucopyranoside (α,β-PhFuc; see Fig. 1) and their singly hydrated complexes (α,β-PhFucW) to complete the ‘vibrational spectral library’ of the most common monosaccharide units, to identify their individual infrared spectral signatures and to enable the future investigation of the larger carbohydrates which incorporate them.
 | ||
| Fig. 1 (Top) Counter-clockwise conformation of (left) α-L-PhFuc, with the numbering of the OH groups followed in this paper, and (right) β-L-PhFuc. (Bottom) Chemical structure of each anomer in the most stable (1C4) chair conformation. | ||
2. Experimental and theoretical methods
Fully acetylated α/β-PhFuc was obtained from the reaction of peracetylated fucose and phenol in the presence of the Lewis acid BF3·OEt2 in 74% yield (α/β, 15∶1).22 The β-anomer was considerably less abundant, presumably because of the reversibility of the reaction under the Lewis acidic conditions23 which converts the kinetically favoured phenyl β-fucoside (due to the participatory O-2 acetyl)24 to the thermodynamically more stable α-fucoside.25,26 The two anomers were separated by recrystallisation and iterative flash column chromatography. Zemplen deacetylation of the acetylated phenyl fucosides gave the desired unprotected α-PhFuc and β-PhFuc products in 88% and 98% yields, respectively.27 The phenyl ‘tag’ provides the UV chromophore needed for spectroscopic interrogation of the molecular beam via resonant two photon ionization (R2PI), an essential part of the UV hole burn and IR ion dip spectroscopic techniques employed. It has been chosen for its good optical properties making it an ideal UV chromophore and because it is benign in the sense that it has a negligible effect on the conformation of the monosaccharide molecule to which it is attached.15,19A detailed description of the molecular beam experimental strategy has been published previously.28 α-PhFuc samples were vaporized at ∼115 °C, in an oven mounted in front of the nozzle (0.8 mm orifice) of a pulsed valve (General Valve) and seeded into a free jet argon expansion (∼3 bar Ar). Some samples were also vaporized into the expanding jet using a laser desorption source: the appearance of the phenol fragment ion in the R2PI mass spectrum of α-PhFuc at relatively low oven temperatures, reflected its unusual susceptibility to thermal decomposition, although both methods generated the same distribution of populated structures. Since β-PhFuc was more difficult to synthesize and therefore more scarce it was vaporized exclusively through laser desorption: a more ‘sample-conservative’ technique. Hydrated complexes were formed by seeding the argon with water vapor (∼0.25%). Collisional cooling during the molecular beam expansion process can freeze the high temperature free-energy driven conformational distribution if the barriers are high in comparison with the mean energy of the collisions experienced in the supersonic expansion, as in the case of the flexible α(1,6) linked mannose disaccharide, phenyl-α-D-mannopyranosyl-(1 → 6)-α-D-mannopyranoside.20 However, all the other carbohydrates and hydrated complexes studied so far19 appear to follow another cooling regime where the distributions relax into low potential energy structures that are unaffected by entropic effects. The high collision frequency and decreasing temperature also favour the formation of weakly bound carbohydrate inter-molecular complexes, allowing the investigation of their size-selected hydrate structures.15,17,18
The free jet expansion passed through a 1 mm skimmer to form a collimated molecular beam which was subsequently crossed by one, or two tunable laser beams in the extraction region of a linear time-of-flight mass spectrometer (Jordan). Resonant two photon ionization spectra were recorded using the frequency-doubled output of a pulsed Nd:YAG-pumped dye laser (Continuum Surelite III/Sirah PS-G, 1.5 mJ/pulse UV). Conformer-specific spectra were obtained through UV hole-burning (UV-HB) or IR ion dip experiments, employing UV-UV and IR-UV double resonance spectroscopy.28 The UV-HB experiments employed the frequency-doubled output of an excimer-pumped dye laser (Lambda-Physik EMG 201/Lambda-Physik FL3002, 0.5 mJ UV). IR experiments employed radiation in the range 3000–3800 cm−1, generated by difference frequency mixing of the fundamental of a Nd:YAG laser with the output of a dye laser in a LiNbO3 crystal (Continuum Powerlite 8010/ND6000/IRP module, 2 mJ pulse−1 IR); all laser pulses were ∼10 ns duration. In the IR ion dip and UV-HB experiments the 150 ns delay between the pump and the probe laser pulses guaranteed observation of the vibrational spectrum of the molecule of interest in its electronic ground state only, allowing direct comparisons with the calculated IR spectra.
Conformational and structural assignments have been made through comparisons between their experimental and computed near IR spectra calculated using density functional theory (DFT), and their relative energies determined from ab initio (MP2) calculations. Starting structures for ab initio and density functional theory (DFT) calculations were generated by performing molecular mechanics conformational searches using the Monte Carlo multiple minimization method (10 000 steps) as implemented in the MacroModel software (MacroModel v.8.5, Schrödinger, LLC21). The maximum intermolecular separation between the sugar and the water molecule was constrained by applying a force constant of 20 N m−1 when the distance between the ring oxygen (O5) and the water molecule exceeded 1.5 nm (and 0 N m−1 when the distance was within 1.5 nm). Nine conformers of α-PhFuc and 26 structures of its singly hydrated complexes were selected for DFT geometry optimization (B3LYP/6-31+G(d)) from the large set of conformers and structures generated using the MMFFs force field. In the case of the β-anomers, 11 conformers of the monomer and 32 water complex structures were selected for optimization. Relative energies, experience from previous studies of carbohydrates,14–18,21 and the desire to include all relevant conformations informed the selection. Harmonic vibrational frequencies of geometry optimized structures were also calculated at the B3LYP/6-31+G(d) level of theory. Their relative energies were obtained from single-point MP2/6-311++G(d,p) calculations, corrected for the zero-point energy (ZPE) derived from the B3LYP/6-31+G(d) frequency calculations. (Since DFT/B3LYP calculations do not take dispersion interactions into account the energies they provide are expected to be unreliable).29,30 All DFT and ab initio calculations were conducted using the Gaussian03 software package.31 The harmonic frequencies of the calculated spectra were scaled by 0.9734, a factor known to provide good agreement with experimental data for carbohydrates in the OH stretching region.14–18,20,21
3. Results and discussion
3.1. Isolated monosaccharides
The S0–S1 R2PI and UV-HB spectra of α-PhFuc, recorded in the molecular beam expansion between 36 650 and 37 050 cm−1 are displayed in Fig. 2a; they indicate the population of a single conformer only. Its near IR ion dip spectrum, shown in Fig. 3a, along with the calculated spectra of the lowest energy structure of α-PhFuc (Fig. 3b) displays three near equally spaced bands, centred at 3606, 3616 and 3626 cm−1, which reflect a modest degree of intramolecular hydrogen-bonded interaction. The dip in the central band is due to an absorption feature associated with atmospheric water which reduced the intensity of the IR laser. The central band must therefore be more intense than the experimental spectrum would otherwise suggest and both the intensity and frequency pattern of the experimental spectrum are in best accord with the calculated spectrum of the global minimum energy conformer. The large energy difference between it and its nearest calculated neighbour (∼6.5 kJ mol−1 higher in energy) is fully consistent with the observation of single conformer. The slightly increased spacing between the transitions in the experimental IR spectrum (∼10 cm−1 instead of the calculated 4 cm−1), may be due to insufficient account being taken, in the harmonic approximation, of the coupling between the σ2 and σ3 vibrations—the two calculated normal modes involve the motion of both H2 and H3. The three peripheral OH groups each adopt a counter-clockwise, ‘cc’ orientation, similar to that adopted in the minimum energy structures of phenyl β-D-gluco-14 and β-D-galactopyranoside16 and in the two anomers of phenyl α/β-D-xylopyranoside17 (though in the D-sugars, the pyranose ring adopts a 4C1, rather than the 1C4 chair conformation of L-fucose). | ||
| Fig. 2 (a) R2PI and UV-HB spectra of α-PhFuc. The UV-HB spectrum was recorded with the probe laser tuned to the origin band, 36 796 cm−1. Observations under different molecular beam conditions allow assignment of the weak broad bands observed at lower wavenumbers than the origin to hot bands. (b) R2PI spectrum of β-PhFuc. Its vibronic structure is virtually identical to that of α-PhFuc, indicating the population of a single conformer for this anomer also. | ||
 | ||
| Fig. 3 (a) and (c) Experimental IR ion dip spectra of the observed conformers of α- and β-PhFuc. The squares indicate the position of atmospheric water absorption bands that affect the intensities of the observed lines. (b) and (d) Predicted spectra of the calculated conformations to which the observed species are assigned. | ||
The S0–S1 R2PI spectrum of β-PhFuc (Fig. 2b) is closely similar to that of α-PhFuc, apart from a slight shift in its band origin, from 36 796 cm−1 to 36 837 cm−1. The absence of any supplementary vibronic band system in β-PhFuc suggests once again, the population in the molecular beam expansion, of a single conformer only. When intensity distortions due to atmospheric water absorption are taken into account its experimental IR ion dip spectrum is in very good agreement with the spectrum calculated for its lowest lying conformer (Fig. 3c and 3d), which is located ∼5 kJ mol−1 below its nearest neighbour, and it can be assigned unequivocally, to the calculated global minimum structure. Like the α-anomer, its three OH groups interact to create a counter-clockwise ‘cc’, intramolecular hydrogen bonded network. In spite of the conformational similarity however, their IR spectra are quite different: the band σ4 in each anomer is located at the same position but in α-PhFuc the other two bands, σ2 and σ3, are displaced towards lower wave numbers, see Fig. 4, and are significantly coupled. The difference is readily explained; the OH2⋯O1 distance in β-PhFuc is 2.52 Å but in the α-anomer it decreases to 2.28 Å, enabling a much stronger OH2 → O1 interaction which shifts the band σ2, towards lower wavenumbers by ∼50 cm−1. The stronger OH2 → O1 interaction in the α-anomer also enhances the induced dipole in OH2, strengthening the neighbouring interaction, OH3 → OH2; thus σ3 is also shifted towards lower wavenumbers with respect to the β-anomer, but to a much smaller extent, ∼10 cm−1.† The two bands, σ2 and σ3 in the α- and β-anomers of phenyl xyloside (α/β-PhXyl), also display the same behaviour, for the same reasons (compare Fig. 4b and 4d).17
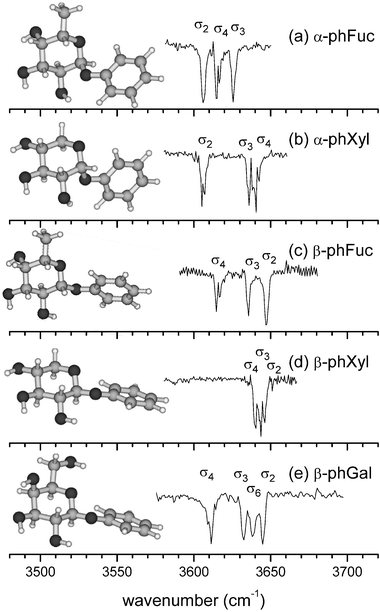 | ||
| Fig. 4 Comparison of the near IR ion dip spectra of the most stable conformers of (a) α-PhFuc, (b) α-PhXyl,17 (c) β-PhFuc, (d) β-PhXyl17 and (e) β-PhGal.16 In each one, the peripheral OH groups are oriented in a counter-clockwise hydrogen bonded network, OH4 → OH3 → OH2 → O1. | ||
Another structural effect is revealed by the relative position of the band σ4 in the xylosides and the fucosides: because of the structural constraints imposed by the pyranose ring, the minimum distance between two interacting OH groups is smaller if one of them is equatorial (eq) and the other is axial (ax) than if they are both equatorial. In α- and β-PhFuc, OH4 is involved in an ax → eq interaction with OH3 and σ4 is located 30 cm−1 below the corresponding band in the IR spectra of α- and β-PhXyl where the interaction is eq → eq. On the other hand, the location of σ4 is unaltered by the change in anomeric configuration: the inductive effect that shifts σ3 in the α-anomers is not strong enough to affect OH4.
Further insight into the correlation between hydrogen bonded structures in the monosaccharides and their IR signatures is revealed through comparison of β-L-PhFuc with β-D-PhGal, see Fig. 4c and 4e. Apart from the extra absorption band σ6, associated with the OH stretch of the hydroxymethyl group in β-PhGal, the IR spectrum of β-PhFuc duplicates that of β-PhGal (in its most stable, counter-clockwise ‘cc’ conformation).16 The hydrogen bonded networks linking their peripheral OH groups are identical despite the different chair configurations, 4C1 in D-galactose but 1C4 in L-fucose. This similarity is to be expected, since L-fucose is the mirror image of 6-deoxy-D-galactose but more importantly, this spectral pattern can be used as a benchmark for ‘galacto-like’ units embedded in larger systems. For example, the observation of any vibrational band deviating from this pattern in a galactose-fucose disaccharide would directly indicate, without needing any computational information, a transformation in one of the moieties of the system, e.g. an inter-ring H bond or conformational change.
3.2. Singly hydrated complexes
The S0–S1 R2PI and the UV-HB spectra of hydrated α-PhFuc, could be recorded in both the parent, α-PhFucW+ (α-PhFuc·H2O+) and the α-PhFuc+ fragment ion channels, reflecting efficient fragmentation of the hydrated ions. Spectra recorded over the range, 36 750–36 980 cm−1, shown in Fig. 5a, reveal two distinct components, labeled ‘A’ and ‘B’; they are assigned to isomers of the mono-hydrated complex, α-PhFucW. The relative intensities of their origin bands indicate an abundance ratio, A ∶ B ∼ 3 ∶ 1, (assuming similar resonant ionization efficiencies). The low signal to noise ratio limited UV-HB spectral recording to the more abundant complex, A, but all of the remaining set of peaks in the R2PI spectrum could readily be assigned by superimposing the vibronic pattern associated with structure A onto the origin band of the second structure, B (centred at 36 779 cm−1); their close correspondence identified A and B as the only two contributing mono-hydrate structures and their common vibronic spectral pattern suggests significant structural similarity between them, at least in the electronically excited state (S1). A third band origin, labeled ‘C’ is associated with a higher water complex that also fragments upon ionization: it will be discussed in section 3.3.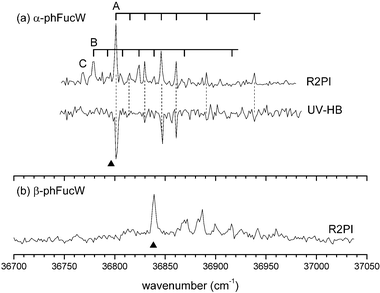 | ||
| Fig. 5 (a) R2PI and UV-HB spectra of hydrated, α-PhFuc. The spectrum of A was probed on its origin band at 36 801 cm−1; its vibronic structure is identical to that of the (down-shifted) second band system, B and both are assigned to mono-hydrated complexes. Band C is assigned to a larger water complex of α-PhFuc. (b) R2PI spectrum of hydrated β-PhFuc. The wedges indicate the position of the spectral band origins of the un-hydrated anomers. | ||
The IR ion dip spectra associated with the two mono-hydrate structures, A and B, are shown in Fig. 6a and 6c, respectively. The intensity and frequency pattern‡ of the experimental spectrum of A displays the characteristic signature of an insertion complex15 and it is in quite good agreement with the computed spectrum of the minimum energy structure, labeled ‘cc_ins3’, although the wavenumbers of the symmetric and asymmetric modes coupling σ3 and σwb, involving the ‘water binding’ OH groups, OH3 and OHw, are poorly predicted by the calculation. This is due a systematic error that overestimates the calculated vibrational shifts generated through intermolecular hydrogen bonding.15 The water molecule inserts into the most stable conformer of the isolated monosaccharide between OH3 and OH2, the site involving the weakest intramolecular H bond, to create the co-operatively linked hydrogen bonded chain, OH4 → OH3 → OHw → OH2 → O1. In the IR spectrum of the free molecule, α-PhFuc, the band σ3 is the least displaced. The preference for binding at the site of the weakest intramolecular hydrogen bond follows the general pattern displayed by all the singly hydrated complexes of monosaccharides characterised to date.15,17
 | ||
| Fig. 6 (a) Experimental IR ion dip spectrum of the most abundant conformer A, of α-PhFucW recorded in the α-PhFuc+ fragment ion channel. The black squares indicate the position of absorption bands associated with the unhydrated molecule. (b) Predicted spectrum and structure of the most stable calculated conformer of α-PhFucW. (c) Experimental IR ion dip spectrum of the minor conformer B, of α-PhFucW recorded in the α-PhFuc+ fragment ion channel. (d) and (e) Predicted spectra, structures and relative energies (in kJ mol−1) of the second and third most stable calculated conformers of α-PhFucW; relative energies calculated at the MP2/6-311++G**//B3LYP/6-31+G* level of theory. σwf and σwb refer to the free and bound OH stretching modes of the water molecule. For the structures ‘cc_ins3’ and ‘cc_ins4’, the bands labeled ‘s’ and ‘a’ correspond to the symmetric and asymmetric modes of the coupled stretches of the bound OH groups of the water molecule and of the monosaccharide. The calculated intensities have been normalized to the strongest band for each conformation. The structures are shown in a different orientation than those of the monomers in order to facilitate the display of the water molecule position. | ||
Structural assignment of the minor isomer, B, proved to be more difficult since its R2PI signal was much weaker and contamination of its IR ion dip spectrum by the spectrum of A (see Fig. 6c) did not make the task any easier. This contamination appeared as a strong positive signal in the spectrum recorded in the α-PhFuc+ fragment ion channel, and as a negative signal in the α-PhFuc+W parent ion channel (not shown): its appearance was attributed to vibrational predissociation of A following absorption of IR photons.
Despite these difficulties three IR absorption features associated with isomer B can be identified: a relatively strong and broad absorption band centred at ∼3490 cm−1, a weak and narrow band at ∼3730 cm−1 and a barely detectable unresolved group of weak absorption bands lying between 3580 and 3620 cm−1 (above the thick line shown in Fig. 6c). This vibrational pattern compares well with the predicted spectrum of the second most stable α-PhFucW structure (Erel ∼1.3 kJ mol−1) labeled ‘cc_add4’, see Fig. 6d. Instead of inserting between two OH groups, the water molecule is now bound by a single hydrogen bond to OH4, forming a so-called ‘addition’ complex, while the counter-clockwise, ‘cc’ conformation of the α-PhFuc moiety is retained.
The second lowest-lying conformer, ‘cc_ins4’, should also be considered since it is calculated to lie only 0.8 kJ mol−1 above ‘cc_add4’: Fig. 6d displays its computed spectrum. Although the two characteristic bands of a second insertion structure, ‘ins4’ might conceivably be overlapped and ‘hidden’ behind the two strong features at low wavenumbers, associated with the interference from A the preferential assignment of B to the addition structure, ‘cc_add4’ is favoured by considering the calculated IR spectral intensities. The band σwb, in the spectrum of ‘add4’ is predicted to be an order of magnitude less intense than the symmetric and antisymmetric doublet bands, σ3/σwb or σ4/σwb, associated with the insertion structures, ‘cc_ins3’ or ‘cc_ins4’. The intensities of the hole-burning signals for A and B confirm this trend: under identical experimental conditions the ion signal of A (‘cc_ins3’) was depleted by 60–70% when the IR pump laser was tuned to the antisymmetric band at 3520 cm−1 but the depletion of B (‘cc_add4’) when the laser was tuned to the band σwb at 3490 cm−1 was very much smaller, ≤10%.
The R2PI spectrum of β-PhFucW, recorded in the fragment ion channel, β-PhFuc+, is shown in Fig. 5b. Its vibronic structure suggests the population of one, very dominant conformer. Unfortunately, the R2PI signal recorded in the β-PhFucW+ ion channel was very weak and as with α-PhFucW, fragmentation of the complex following UV excitation was very efficient. Even more unfortunately, the recorded S0–S1 spectrum of β-PhFucW coincides, almost perfectly with that of the unhydrated molecule (the wedge in Fig. 5b indicates the position of its band origin) and it was not possible to record the UV-HB spectra of the hydrated complex in the β-PhFuc+ fragment ion channel since the ion signal associated with the free monosaccharide was two orders of magnitude more intense.
Despite these difficulties, by careful selection of the probe UV laser wavenumber within the strongest line of the R2PI spectrum, it was possible to record the associated IR ion dip spectrum directly in the mass channel of β-PhFucW+ (Fig. 7) although, because of the large difference in magnitude between the signals associated with the free monosaccharide and its mono-hydrate and the overlap of their R2PI spectra, observation of the latter could not be achieved without saturating the detection system with the former. Hence the recorded signal was superimposed on oscillations of the saturated MCP ion current. It displays features associated with both β-PhFucW and β-PhFuc but they have opposite signs. This has a subtle origin. When β-PhFuc absorbs resonant IR photons, the consequent ion dip reduces the level of saturation and the observed contamination of the β-PhFucW IR spectrum appears as a positive-going signal, opposite in sign to the IR ion dip signal associated with β-PhFucW. This allows an easy distinction to be made between the IR absorption bands of the hydrated cluster and the free mono-saccharide.
 | ||
| Fig. 7 (a) Experimental IR spectrum of β-PhFucW. The horizontal dashed line indicates the baseline level. The bands below this line are associated with the hydrated molecule while those above the line are associated with the bare monosaccharide (see text). (b) and (c) Predicted spectra, structures and relative energies (in kJ mol−1) of the two most stable calculated structures. | ||
There is a good match between the experimental IR spectrum and that calculated for insertion at the most favourable site, to create the global minimum hydrate structure, ‘cc_ins2’, which retains the counter-clockwise conformation of the mono-saccharide host (Fig. 7). The spectrum predicted for the third most stable structure ‘cc_ins3’ also matches the observed spectrum well but its much higher relative energy (6 kJ mol−1) makes it unlikely to be the most abundant hydrate structure in the expansion. The large difference of energy between ‘cc_ins2’ and the second most stable conformer ‘c_ins4’ (4.9 kJ mol−1) is also consistent with the observation of a unique conformer. The observed hydrate can be assigned with some confidence, to the structure, ‘cc_ins2’.
The conformational landscape of β-PhFucW, although very different to that of β-PhGalW where at least five different structures lie within 1.5 kJ mol−1 of the global minimum,15 continues in one important respect, to follow the same trend as the other monosaccharides: in every case, the water molecule preferentially binds to the OH group involved in the weakest intramolecular interaction in the bare molecule (OH2 in this case). Another common trend is revealed through comparisons between the α- and β-anomers. The c_ins4 structure of β-PhFucW is calculated to be the second most stable structure, but the lowest-lying clockwise ‘c’, rather than ‘cc’ oriented conformer of α-PhFucW is found only at the sixth position; in β-PhGlc and β-PhGal the switch from a ‘cc’ to a ‘c’ orientation is observed in the most stable hydrate structures. This probably reflects differences in the OH2 → O1 interaction, against which the intermolecular H bonds have to compete if the ‘c’ conformation is to be favoured. Their anomeric vibrational signatures reveal an OH2 → O1 interaction that is weaker in the β configuration than in the α.
3.3. Multiply hydrated complexes
The observation of an R2PI band (band C in Fig. 5a) associated with a multiply hydrated complex of α-PhFuc was noted in section 3.2. The detection of larger complexes is favoured by increasing the delay between the molecular beam expansion and the UV laser pulse and a series of R2PI spectra recorded at different delays is shown in Fig. 8. The evolution of the relative intensities of the bands associated with systems A, B and C with increasing delay (spectra 1, 2 and 3) supports the association of C with a doubly hydrated complex, α-PhFucW2. More interestingly, the band system(s) labeled D which appear at the longest delays (spectra 3 and 4), and the increase of the baseline signal identified by the dashed lines below the resonant transitions, point to the formation and observation of much larger clusters. The band labeled ‘d1’ is tentatively assigned to a triply hydrated complex, α-PhFucW3 (though it might be associated with a larger hydrated cluster experiencing multiple photofragmentation).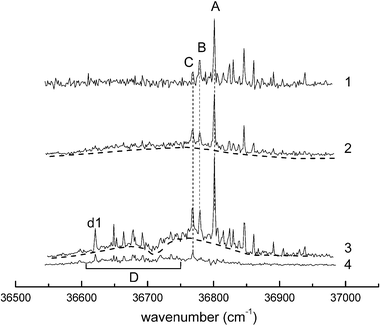 | ||
| Fig. 8 Mass selected R2PI spectra of the hydrated complexes of α-PhFuc observed at different delays (t) between the pulsed molecular beam expansion and the ionizing UV laser pulse. (1) t = 520 μs, (2) t = 550 μs, (3) and (4) t = 600 μs. (1), (2) and (3) recorded in the α-PhFucW+ ion channel and (4) recorded in the α-PhFucW2+ ion channel. | ||
3.4. Some general observations
The mono-hydrated complex of β-PhGal presents a structural landscape very different from those of the corresponding complexes of α- and β-PhFuc. In hydrated β-PhGal (where the exocyclic group is hydroxymethyl rather than methyl) several alternative structures are populated but in the most favoured, the bound water molecule creates a cooperative structure in which the peripheral OH groups switch their orientation from counter-clockwise to clockwise.15 The exocyclic hydroxymethyl group plays an important role in increasing the conformational flexibility of the monosaccharides and in adapting its conformation to optimize their interactions with the environment.The band system D, shown in Fig. 8 and attributed to large hydrated complexes, α-PhFucWn≥3, is strongly shifted towards low wavenumbers (by ∼200 cm−1) with respect to the corresponding bands of α-PhFuc and α-PhFucW1,2. Similar shifts have been observed in the R2PI spectra of singly hydrated complexes of β-PhGal and β-PhGlc;15 in these two cases the shifts correlated with a structural transformation, from the ‘cc’ conformation in the free mono-saccharides to the ‘c’ conformation in their hydrated complexes, stabilized by an extended, cooperative chain of hydrogen bonds. The present observation suggests that a similar conformational change only occurs in fucose when it is multiply hydrated (n ≥ 3). Lacking the flexible hydroxymethyl group, fucose would need to interact with more water molecules before the cooperative chain of hydrogen bonds, OH2 → OH3 → OH4 → OHw1…OHwn → Oring was long enough to stabilize the clockwise conformation. One of the water molecules would then ‘mimic’ the exocyclic OH group of galactose. As shown previously,15 the water molecule in hydrated complexes can act as a ‘decoy’ of a ‘missing group’, to achieve the stabilization of a particular conformation: in α-PhGalW it plays the same role as OH6 in the glucose component of phenyl lactoside, to stabilize in each case, the clockwise conformation of the galactose unit. Also, to some extent, the water molecule in the c_ins4 structure of β-PhFucW can be considered as a substitute for the OH6 group of the previously observed conformer cG-g+ of β-PhGal.16
The work reported here completes a systematic exploration (pioneered a few years ago in our laboratory with the study of β-PhGlc14) of the conformational landscapes of the principal monosaccharides found in glycans (mannose, glucose, galactose, xylose and fucose) and of the influence of bound water molecules on their landscapes. This systematic “building block” approach has revealed some general trends that apply to all of these systems, and also some significant differences. The counter-clockwise, ‘cc’ structure is preferentially adopted by all monosaccharides except the mannoside, where the axial OH2 group, and an α-anomeric (O1 axial) configuration, facilitates the clockwise sequence, OH2 → OH3 → OH4.15 Bound water molecules create extended, cooperatively hydrogen bonded chains that favour the conformational switch from a ‘cc’ to a ‘c’ orientation,15 though more water molecules may be required in the less flexible deoxy-monosaccharides for these structures to be dominant. α-PhMan is the only monosaccharide preferentially adopting a ‘c’ conformation without any surrounding water.
4. Conclusions
Comparisons between the near IR spectra of the two anomeric forms of phenyl L-fucopyranoside and their hydrated complexes recorded in a molecular beam environment, and their conformational landscapes and vibrational spectra computed ab initio, have led to the assignment of the intrinsically favoured conformation of these species. The α- and β-anomers of phenyl L-fucopyranoside (α,β-PhFuc) each adopt a single preferred conformation, with their three peripheral OH groups oriented into a counter-clockwise, ‘cc’ configuration. This simplicity contrasts with the conformational flexibility displayed by the majority of mono-saccharides19 that, unlike fucose, are endowed with an exo-cyclic hydroxymethyl group and have all been observed in several distinct conformations.In their early computational study on the conformation of α-L-fucose,11,12 Csonka et al. predicted the preference of clockwise or counter-clockwise OH conformations, stabilised by concerted H-bonds; they also suggested that such H-bonded networks could enable carbohydrates to be efficient ‘information encoders’. Our conclusions confirm their early insight and show that IR spectroscopy provides an extremely powerful probe of their H-bonded conformational structures and a means of ‘reading’ the information they contain.
The ‘cc’ configuration is retained in their singly hydrated complexes, again in contrast to ‘hydroxymethyl monosaccharides’ where a conformational switch is promoted, in order to maximize the length of the encircling chain of cooperatively hydrogen-bonded OH groups.15 As in the case of all the singly hydrated monosaccharides observed so far, the water molecule ‘selects’ the weakest link in the chain as its preferred binding site.
The comparison of the conformational assignments of β-PhGlc,14 β-PhGal16 and α-PhMan,15 with those of α/β-PhFuc and α/β-PhXyl17 has allowed two important conclusions to be drawn. The hydroxymethyl group is key to the conformational flexibility, complexity and adaptability of the monosaccharides that comprise this group and less predictably, the interaction between OH2 and the anomeric oxygen O1 is key to the conformational choice of all the monosaccharides studied. When the OH2 → O1 interaction is possible all the monosaccharides preferentially adopt a counter-clockwise conformation. Although the interaction is more favoured in the α anomers, in mannose the axial orientation of its OH2 group prevents an OH2 → O1 interaction; α-PhMan is the only isolated monosaccharide favouring a clockwise conformation.
The near IR spectra of ‘free’ and hydrated α- and β-PhFuc, and many other mono- and di-saccharides provide extremely sensitive probes of hydrogen-bonded interactions,19 which can be finely tuned by small (or large) changes in the molecular conformation. They provide characteristic ‘signatures’ which can reflect anomeric, or axial vs. equatorial differences, both revealed through comparisons between α/β-PhFuc and α/β-PhXyl; or similarities, revealed through comparisons between fucose (6-deoxy galactose) and galactose; or binding motifs, for example, ‘insertion’ vs. ‘addition’ structures in hydrated complexes. At the monosaccharide level (the first step in the carbohydrate hierarchy), these trends appear to be general. The evolution of these correlations and the recognition of characteristic signatures will greatly aid the interpretation and understanding of larger oligosaccharide and glycan structures in the future.20
Acknowledgements
The authors are grateful to Rebecca A. Jockusch for her many insights and helpful suggestions. Financial support was provided by the EPSRC, the Leverhulme Trust (Grant F/08788D) and the Royal Society (LCS). We also acknowledge support from the Physical and Theoretical Chemistry Laboratory in Oxford and the CLRC Laser Support Facility at the Rutherford Appleton Laboratory.References
- P. Greenwell, Glycoconjugate J., 1997, 14(2), 159 CrossRef CAS.
- G. Audette, D. Olson, A. Ross, J. Quail and L. Delbaere, Can. J. Chem., 2002, 80, 1010 CrossRef CAS.
- W. Cook and C. Bugg, Biochim. Biophys. Acta, 1975, 389, 428 CAS.
- L. Eriksson, R. Stenutz and G. Widmalm, Acta Crystallogr., Sect. C, 2000, C56, 702 CAS.
- P. Longchamnon and H. Gillier-Pandraud, Acta Crystallogr., Sect. B, 1977, B33, 2094 CrossRef.
- P. Longchamnon, J. Ohanessian, D. Avenel and N. A, Acta Crystallogr., Sect. B, 1975, B31, 2623 CrossRef.
- D. Lamba, A. Segre, G. Fabrizi and B. Matsuhiro, Carbohydr. Res., 1993, 243, 217 CrossRef CAS.
- D. Watt, D. Brasch, D. Larsen, L. Melton and J. Simpson, Carbohydr. Res., 1996, 285, 1 CAS.
- K. Bock and H. Thorgersen, Annu. Rep. NMR Spectrosc., 1982, 13, 1 CAS.
- S. Angyal and V. Pickles, Aust. J. Chem., 1972, 25, 1711 CrossRef CAS.
- G. Csonka, K. Elias, I. Kolossvary, C. Sosa and I. Csizmadia, J. Phys. Chem. A, 1998, 102, 1219 CrossRef CAS.
- G. Csonka and K. Elias, J. Comput. Chem., 1996, 18, 330.
- W. Rockey, M. Dowd, P. Reilly and A. D. French, Carbohydr. Res., 2001, 335, 261 CrossRef CAS.
- F. O. Talbot and J. P. Simons, Phys. Chem. Chem. Phys., 2002, 4, 3562 RSC.
- P. Çarçabal, R. A. Jockusch, I. Hunig, L. C. Snoek, R. T. Kroemer, B. G. Davis, D. P. Gamblin, I. Compagnon, J. Oomens and J. P. Simons, J. Am. Chem. Soc., 2005, 127, 11414 CrossRef CAS.
- R. A. Jockusch, F. O. Talbot and J. P. Simons, Phys. Chem. Chem. Phys., 2003, 5, 1502 RSC.
- I. Hunig, A. Painter, R. A. Jockusch, P. Çarçabal, E. M. Marzluff, L. C. Snoek, D. P. Gamblin, B. G. Davis and J. P. Simons, Phys. Chem. Chem. Phys., 2005, 7, 2474 RSC.
- R. A. Jockusch, R. T. Kroemer, F. O. Talbot and J. P. Simons, J. Phys. Chem. A, 2003, 107, 10725 CrossRef CAS.
- J. P. Simons, R. A. Jockusch, P. Carcabal, I. Hunig, R. T. Kroemer, N. A. Macleod and L. C. Snoek, Int. Rev. Phys. Chem., 2005 Search PubMed , in press.
- P. Carcabal, I. Hunig, B. Liu, D. P. Gamblin, R. A. Jockusch, R. T. Kroemer, L. C. Snoek, B. G. Davis, A. J. Fairbanks and J. P. Simons, J. Am. Chem. Soc., 2006 Search PubMed , submitted.
- R. A. Jockusch, R. T. Kroemer, F. O. Talbot, L. C. Snoek, P. Çarçabal, J. P. Simons, M. Havenith, J. M. Bakker, I. Compagnon, G. Meijer and G. von Helden, J. Am. Chem. Soc., 2004, 126, 5709 CrossRef CAS.
- V. M. Sokolov, V. I. Zakharov and E. P. Studentsov, Russ. J. Gen. Chem., 2002, 72, 806 CrossRef CAS.
- E. M. Montgomery, N. K. Richtmyer and C. S. Hudson, J. Am. Chem. Soc., 1942, 64, 690 CrossRef CAS.
- G. Wulff and G. Rohle, Angew. Chem., Int. Ed. Engl., 1974, 13, 157 CrossRef CAS.
- J. T. Edward, Chem. Ind., 1955, 1102 Search PubMed.
- R. U. Lemieux and N. J. Chu, Abstr. Pap. Am. Chem. Soc., 1958, 133, 31N.
- G. Zemplen, Ber. Dtsch. Chem. Ges., 1920, 60, 1555.
- E. G. Robertson and J. P. Simons, Phys. Chem. Chem. Phys., 2001, 3, 1 RSC.
- T. van Mourik and R. Gdanitz, J. Chem. Phys., 2002, 116, 9620 CrossRef CAS.
- S. Cybulski and C. Sevensern, J. Chem. Phys., 2005, 122, 014117 CrossRef.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheesemam, J. J. A. Montgomery, T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Lyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, C. Gonzalez and J. A. Pople, in GAUSSIAN 03 (Revision B03), Gaussian, Inc., Pittsburgh, PA, 2003 Search PubMed.
Footnotes |
| † The better prediction of the band positions by the DFT calculation for the β anomer may also be a consequence of the weaker OH2 → O1 H-bond, which reduces the inductive contribution to the interaction, OH2 → OH3 and also the coupling between σ2 and σ3. |
| ‡ The experimental spectrum, recorded in the mass channel of the ion α-PhFuc+, is contaminated by the absorption bands of unhydrated α-PhFuc (squares in Fig. 6a) which appear as positive-going features. They are generated through non-resonant two-photon ionisation, which is enhanced by the ‘heating’ of the molecule through the resonant IR absorption. This leads to an increase in the non-resonant R2PI signal: IR-UV ion dip measurements generate a decrease in a resonant R2PI signal. |
| This journal is © the Owner Societies 2006 |