Statistical vs. non-statistical deactivation pathways in the UV photo-fragmentation of protonated tryptophan–leucine dipeptide
G.
Grégoire
*b,
H.
Kang
a,
C.
Dedonder-Lardeux
a,
C.
Jouvet
a,
C.
Desfrançois
b,
D.
Onidas
c,
V.
Lepere
c and
J. A.
Fayeton
c
aLaboratoire de Photophysique Moléculaire du CNRS-Université Paris-Sud 11, Bât. 210, 91405, Orsay, France
bLaboratoire de Physique des Lasers du CNRS-Université Paris 13, Institut Galilée, 93430, Villetaneuse, France. E-mail: gregoire@lpl.univ-paris13.fr
cLaboratoire des Collisions Atomiques et Moléculaires CNRS-Université Paris-Sud 11, 91405, Orsay, France
First published on 24th October 2005
Abstract
The excited state dynamics of protonated tryptophan–leucine ions WLH+, generated in an electrospray source, is investigated by photo-induced fragmentation in the gas phase, using femtosecond laser pulses. Two main features arise from the experiment. Firstly, the initially excited ππ* state decays very quickly with 2 time constants of 1 and 10 ps. Secondly, the transient signals recorded on different fragments are not the same which indicates two competing primary fragmentation processes. One involves a direct dissociation from the excited state that gives evidence for a non-statistical deactivation path. The other is attributed to a statistical decay following internal conversion to the ground electronic surface.
1. Introduction
The intrinsic properties of the macromolecules can be studied through their fragmentation patterns. These fragmentations can be induced by different methods. Fifteen years ago, photo-induced dissociation mass spectrometry of protonated peptides emerged from the combination of electrospray ionization source with UV lasers. McLafferty et al. first proved the feasibility of such an experiment1 and several other groups2–5 then pushed forward in the comparison of photo-induced dissociation with a more common mass spectrometry technique, i.e. collision induced dissociation.6 The role of the non-ergodic dissociation in the peptide fragmentation has been quite controversial7,8 and it appears now that by using electron capture9 or photon absorption10 some fragmentations can follow a non-statistical pathway.The UV spectroscopy and fragmentation pattern of protonated tryptophan (TrpH+) have been recently investigated in an ion trap. The reported absorption is very similar to that of the neutral Trp molecule.11 One particular fragmentation channel, the H atom loss channel, seems to be specific to photo-induced fragmentation.5,12 This leads to the formation of a radical cation that further fragments into the m/z 130 ion after Cα–Cβ bond cleavage.5 The pump–probe experiments on a femtosecond timescale have pointed to a short excited state lifetime due to the coupling of the initially excited ππ* state with a πσ* state.13 This state is characterized by an electron localized on the protonated amino group and can be viewed as an electron transfer from the indole ring to the C–NH3+ group. The localization of the electron on this group leads to the formation of a hypervalent C–NH3 radical similar to the ammonium radical NH4. Like NH4, this radical is unstable and undergoes a fast dissociation along the NH coordinate.14–16 On the way out, the πσ* excited state will cross the ground state potential energy surface (PES) through a conical intersection. This crossing leads to a branching ratio between an H atom loss and internal conversion due to the H atom recombination with the ground electronic state. In the case of protonated tryptamine17 or tryptophan,12,13 the H atom loss channel is clearly observed and the branching ratio between H atom loss and internal conversion is found to be about 50%. In order to understand the fragmentation dynamics of larger peptides we have initiated a study on tryptophan-containing dipeptides and we here present results on the UV photofragmentation of protonated tryptophan–leucine (WLH+) that clearly show evidence for both statistical and non-statistical deactivation pathways.
The role of the long time fragmentation or the fact that in mass spectrometry the fragment ions are detected at a finite time after the deposition of the energy in the system is also an important parameter to take into account in the understanding of the mass spectra and on the energetics of the fragmentation processes.18,19 With the large excess energy deposited by the photons (UV and UV + IR), both primary and secondary fragments are observed in the photofragmentation pattern of WLH+. Since the fragmentation kinetics span a large range of rate constants, the fragment ion yields depend on the detection time window of the experiment. We will show that selecting a detection time window allows one to discriminate fast fragmentation kinetics from slower ones.
2. Experimental
The experimental setup, depicted in Fig. 1, includes an electrospray ionization source which is interfaced with an hexapole ion guide, an electrostatic lens and a time-of-flight mass spectrometer. | ||
| Fig. 1 Scheme of the experimental setup. The experimental setup consists of an electrospray ion source coupled to a hexapole ion trap, an electrostatic einzel lens and a home made time-of-flight mass spectrometer. The laser beams are focused at the exit of the hexapole. Typical voltages used for the optics are +10 V for the hexapole exit electrode and (−100/−500/0 V) on the electrodes of the einzel focusing lens. There is a field-free region between the einzel lens and the extraction grids of the time-of-flight mass spectrometer. The distance between the hexapole exit electrode and the extraction grids is 10 cm and the delay between the laser pulse and the extraction voltage can be varied. | ||
A 50/50 water–methanol solution of dipeptide (500 μmol l−1) is sprayed in front of a 20 cm long capillary. A buffer gas (air, 2 atm) heated at 200 °C is injected through the capillary to assist the desolvation process and finally, the protonated ions are trapped for 1 ms in the hexapole (5.2 MHz RF, ±500 V peak-to-peak) by the exit electrode, set a few volts above the voltage of the hexapole. An ion bunch of 400 ns duration is ejected from the guide 1 μs before the laser pulse.
The pump and probe laser beams are respectively issued from the 3rd harmonic (266 nm, 50–100 μJ) and the fundamental (800 nm, 150–250 μJ) of a Ti:Sa femtosecond laser (150 fs) operating at 1 kHz. The zero-delay time and cross-correlation of the lasers have been monitored through resonant ionization of neutral toluene that provides a temporal laser width of 250 ± 50 fs. The lasers are delayed in time by scanning an optical delay line by 50 fs steps and the spectra are recorded by averaging 10 scans with 100 laser shots per scan step.
Both parent and fragment ions are focused by means of an electrostatic lens, between the extraction grids of the time-of-flight mass spectrometer located 10 cm downstream from the hexapole. Typical voltages used for the optics are +10 V for the hexapole exit electrode and (−100/−500/0 V) on the electrodes of the einzel focusing lens. The ions spend about 5–7 μs in the lens and about 12 μs in the field-free region between the third grounded electrode of the lens and the extraction grids of the mass spectrometer. When the ions arrive between the extraction grids, the extraction voltage is switched on to send the ions perpendicular to the detector (micro channel plates) through the acceleration region and the field-free region. The delay between the laser pulses and the extraction voltage can be varied.
The laser beams are focused just at the exit of the hexapole located 1 cm before the first electrode of the einzel lens. In such a configuration, the laser interacts with the ion bunch in a non-zero field region. Therefore, fragment ions that are created within this area will be accelerated toward the einzel lens and will arrive in the extraction region of the mass spectrometer at a specific delay depending on their mass. Typical arrival times are 20 μs for the protonated dipeptide parent ions WLH+ (m/z 318) and 16 μs for lighter ions such as protonated tryptamine at m/z 161.
In Fig. 2 trajectory simulations along the electrostatic lens axis for a fragmenting ion are presented. A parent ion of m/z 300 is allowed to fragment into m/z 160 ions with different time constants varying from 100 to 500 ns. In the conditions used here, the ions are accelerated up to the second electrode of the einzel lens and then decelerated between the second and third electrodes. Since the kinetic energy of the fragment ion is given by Ekfrag = Ekparent(Mfrag/Mparent), if the ion breaks in the acceleration region, the fragment may not have enough energy to pass through the deceleration region.
 | ||
| Fig. 2 Ion trajectory simulations from the hexapole exit electrode to the entrance of the mass spectrometer for a parent ion at mass m/z 300 which fragments into mass m/z 160 at different times. The two rectangles define two delays between the laser pulses and the extraction voltage used in the experiment. When the fragmentation time is 500 ns (thin solid line), the fragment has not enough kinetic energy to exit the electrostatic lens. | ||
For a very fast dissociation (within 100 ns), both parent and fragment ions experience the same acceleration zone and the lighter fragments reach the entrance of the mass spectrometer at shorter times than the heavy parent ions. As the fragmentation time increases, the ion begins to be accelerated as a parent ion, fragmentation occurs, the fragment continues its acceleration as a fragment and the arrival time shifts to a longer delay. For a fragmentation time longer than 500 ns, the fragment kinetic energy is not large enough, so the fragment ion cannot get out of the focusing lens and these ions will never be observed. Fragments produced around 400 ns can get out of the lens but with a very small kinetic energy and they will need a very long time to reach the extraction region of the time-of-flight mass spectrometer. Therefore, selecting an extraction delay (delay between the laser pulses and the extraction voltage) is equivalent to selecting a fragmentation time window.
3. Results
3.1 Mass spectra
The UV photodissociation mass spectra (laser on–laser off) of protonated tryptophan–leucine (WLH+), recorded for two different delays between the laser pulse and the extraction voltage of the time-of-flight mass spectrometer, are plotted in Fig. 3. In the upper spectrum, the extraction delay is 15 μs, corresponding to the arrival time of medium size fragments produced in fast fragmentation events. In the lower spectrum, the extraction delay is set at 19 μs to detect the parent mass peak and the heavy fragment at m/z 301 corresponding to the loss of ammonia. Interestingly, even with such a long extraction delay, some light fragments are still detected (mainly m/z 144, 130 and 86) together with the higher masses. These light ions are probably issued from late fragmentation processes, either in or after the einzel lens (in this field-free region, a daughter ion has the same speed as the parent ion and will arrive at the same time in the extraction region). On the contrary, fragments at mass m/z 159 and 132 are missing for the long extraction delay time. These two ions should be produced in a fast fragmentation reaction.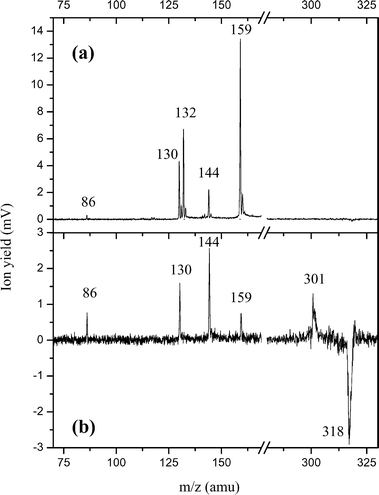 | ||
| Fig. 3 UV photodissociation mass spectra of WLH+ for two delays between the laser pulse and the pulsed extraction voltage of the mass spectrometer. Without the UV laser, the ion pulse emerging from the hexapole contains mainly the parent ion and is recorded as the original ESI mass spectrum. When we shine the UV laser on the emerging ion pulse, several fragments are observed in the photo-induced mass spectrum. By subtracting the original ESI mass spectrum from this photo-induced mass spectrum, we obtain the photodissociation spectrum here displayed that shows the main dissociation channels. The parent ion peak (m/z 318) is depleted and appears as a negative signal while the corresponding photofragments come out as positive mass peaks. (a) The extraction delay is set at 15 μs to detect light fragments produced with fast dissociation rate constants. (b) The extraction delay is set at 19 μs to detect the depletion of the parent ion and the formation of a high mass fragment at m/z 301 (ammonia loss). Note that light fragments at mass m/z 144, 130 and 86 are also detected. | ||
3.2 Femtosecond transients
In previous reports13,17,20 we have shown that the excited state lifetime of protonated molecules can be probed by monitoring the effect of a probe photon on the fragmentation efficiency. Fig. 4 presents the time evolution of the ion signals, recorded on different photofragments of WLH+, that show clear dynamics (m/z 301, 144, 130 and 86). The fragments at m/z 159 and 132, although they are the two most intense UV photo-induced ions, have a very poor signal-to-noise ratio in the pump–probe experiment and are not reported here. For each fragment, the extraction delay has been adjusted to maximize the signal. All the transients can be fitted by a bi-exponential decay function with the same time constants of 1.0 ± 0.2 ps and 10 ± 2 ps. The fitting function is obtained after convolution by a Gaussian like function to take into account the width of the laser (250 fs) and can be written as:| S(t) = A0(1 + A1f(t,t1) + A2f(t,t2) + A3c(t)) |
 | ||
| Fig. 4 Time evolution of the fragment ion signals (open circles) as a function of the pump (266 nm)–probe (800 nm) time delay, recorded during 15 ps. The extraction delay is adjusted for each fragment in order to select the fast dissociation events (around 19 μs for the m/z 301 ion and 15 μs for the lighter ions). The fitted curves (heavy solid line) are the sum of two exponential decrease functions with 1 ps (dotted line) and 10 ps (dashed line) time constants. For the tryptophan side chain cation at m/z 130, a step function (thin solid line) is added which is assigned to the absorption of the probe photon by the WL radical cation formed after H atom loss. | ||
While the fragment coming from the ammonia loss channel is depleted by the probe photon absorption, all the other fragmentation channels increase. As was already the case in the pump–probe experiments on protonated tryptophan, the tryptophan side chain fragment at m/z 130 exhibits a dominant short time component and a plateau at long pump–probe delays. The other fragments do not exhibit such a plateau at long delays and have identical pre-exponential factor ratios within the experimental uncertainty (see Table 1).
| m/z | A 1/A2 | t 1/ps | t 2/ps |
|---|---|---|---|
| 130 | 5 | 1 ± 0.2 | 10 ± 2 |
| 301 | 0.75 | 1 ± 0.2 | 10 ± 2 |
| 144 | 0.75 | 1 ± 0.2 | 10 ± 2 |
| 86 | 0.75 | 1 ± 0.2 | 10 ± 2 |
Fig. 5 shows the time evolution of the fragment ion intensity versus the delay between the pump and probe pulses for the three light fragments at m/z 144, 130 and 86 that are detected with the long extraction delay (19 μs as in the mass spectrum of Fig. 3b). We are thus looking at fragments that are produced in a late fragmentation event. The pump–probe signals are now totally different from those observed in Fig. 4. The absorption of a probe photon does not lead to an increase of the ion signal any longer, but to a decrease. The signal-to-noise ratio in this case is not as good as in the previous experiment but the time constants measured are indeed the same as in Table 1, within the experimental error. Furthermore, all these fragments have equal pre-exponential factor ratios: at this long extraction delay, the tryptophan side chain cation (m/z 130) does not show a dominant short time component and a plateau as previously.
 | ||
| Fig. 5 Time evolution of the fragment ion signals (open circles) as a function of the pump (266 nm)–probe (800 nm) time delay, when the mass spectrometer extraction delay is set to select the slow dissociation events (19 μs). All the fragment ion signals are now depleted and can be fitted with the same bi-exponential function with equal pre-exponential factor ratios. | ||
4. Discussion
4.1 General mechanism
It is important to stress that, in this experiment, there are two timescales related to two different processes: (a) a short timescale (from 100 fs to ns) which corresponds to the excited state lifetimes measured with a femtosecond pump–probe scheme; (b) a long timescale (100 ns to μs) which corresponds to the fragmentation kinetics which can affect the ion trajectory and therefore the detection efficiency in the mass spectrometer device.It has been shown in a previous study that the excited state dynamics of protonated tryptophan exhibits an ultrafast decay with two time constants of 400 fs and 15 ps.13 These short lifetimes are accounted for by assuming that the excited ππ* state is predissociated by a nearby Rydberg-like πσ* state. A possible explanation for the bi-exponential decrease here observed would be to invoke the well known electronic excited states of indole, La and Lb having a ππ* nature, which would have different lifetimes. This hypothesis can be ruled out because protonated tryptamine, derived from the decarboxylation of tryptophan, whose electronic properties are very similar to those of indole and tryptophan,21,22 does not show such a bi-exponential decay under the same experimental conditions.17 A second explanation, generally adopted in fluorescence studies in liquid phase, would be to attribute the multiple fluorescence lifetimes to ground state conformers. The rotamer model proposes that the multiple lifetimes represent several ground-state rotamers of the tryptophan side chain.23 Electrospray sources produce ions at room temperature and a distribution of conformations is thus expected. As assumed for protonated tryptophan, the bi-exponential decrease recorded for WLH+ may be assigned to two classes of isomers that differ from each other in the localization of the πσ* predissociating state versus the ππ* excited states.
The different fragments can be sorted into two groups regarding the fragmentation mechanism as proposed in Scheme 1. H atom loss reaction is evidenced by the detection of the Trp side chain cation at m/z 130 after Cα–Cβ bond cleavage, which is a specific dissociation channel of tryptophan cation5 and tryptophan-containing radical cation peptides.24,25 This reaction occurs directly from the WLH+ excited state and statistical energy redistribution cannot account for such an excited state reaction. The formation of a radical cation following H atom loss evidences a non-ergodic fragmentation channel which is further emphasized by the specific transient signal recorded on m/z 130 ion (see discussion below).
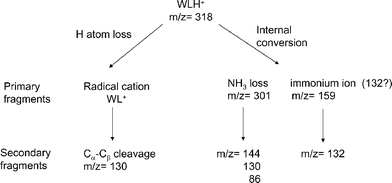 | ||
| Scheme 1 | ||
On the other hand, internal conversion following H atom recombination leads to protonated dipeptides in the ground electronic state with a large excess energy brought by the absorbed photons (UV or UV + IR). In such a scheme, the fragmentation patterns, i.e. A,B/Y ions and ammonia or water loss, are those observed and explained in the collision induced dissociation (CID) experiment .26,27 The large excess energy not only induces primary fragmentation in the ground electronic surface (ammonia loss at m/z 301 and immonium ion formation at m/z 159) but secondary dissociation possibly occurs with kinetics that span a large range of rate constants. The UV photo-induced mass spectrum reported in Fig. 3a corresponds to prompt fragmentations (primary and secondary) with dissociation time constants faster than 100 ns. Secondary fragmentations with slower rate constants are evidenced in our experiment by the detection of light fragments at m/z 144, 130 and 86 with the long extraction delay corresponding to the ammonia loss channel at m/z 301 (Fig. 3b). The ion at m/z 132 is thought to be a secondary fragment of the tryptophan immonium ion5,28 but, since it is observed at short extraction delay, it might also be a primary product.
4.2 Interpretation of the femtosecond dynamics
The analysis of the transient recorded on each fragment can help to differentiate a direct dissociation in the excited state from a fragmentation following internal conversion. Internal conversion should result in statistical energy redistribution via intramolecular vibrational relaxation in the ground electronic surface while a direct dissociation in the electronic excited state should be more specific. The basis of statistical (ergodic) dissociation is that the system does not keep the memory of the initial conditions. After internal conversion, the hot protonated molecules will fragment statistically and independently of their initial conformations. The multiple exponential decays observed for WLH+ may be assigned to different conformers initially excited by the pump photon which have different excited state lifetimes. All the fragments will show the same multiexponential transient with relative amplitudes proportional to the initial conformer populations. However, the m/z 130 ion displays a different time dependent signal than those recorded on the other fragments: the short time component is more pronounced, and there is a plateau at long pump–probe delays. This is a clear indication that another fragmentation mechanism, namely H atom loss reaction, is involved for this deactivation channel. As for tryptophan, the plateau can be understood in terms of absorption/dissociation of the radical cations produced after H atom loss: radical cation of aromatic molecules have an open shell electronic structure and thus low lying electronic states in the near-infrared can be probed by absorption of the 800 nm probe photon.294.3 Effect of the probe photon on the fragmentation yield
Depending on the fragmentation time windows (extraction delays) used in the experiment, the probe laser has a different effect on the fragmentation yield of a given photofragment. A schematic plot of the ion fragment intensity as a function of time for two internal energies (pump only and pump + probe) is drawn in Fig. 6 with three detection windows that will lead to different pump–probe transients:(a) the pump–probe signal will be reflected as an increase of the signal of the ion fragment.
(b) the pump–probe signal will show a very bad contrast.
(c) the pump–probe signal will be reflected as a decrease of the signal of the ion fragment.
(d) another case that will lead to the absence of pump–probe transient is when the fragmentation is very fast and ion yield does not change with the probe photon absorption.
 | ||
| Fig. 6 Schematic representation of the fragment ion yield as a function of the dissociation time for two WLH+ internal energies corresponding to 4.6 eV (full line, pump UV only) and 6.2 eV (dotted line, pump UV + probe IR photons). The rectangles a, b and c define three detection windows for which the absorption of the probe photon will lead to an increase, a poor two-colour signal-to-noise ratio and a decrease of the fragment intensity, respectively. | ||
The tryptophan side chain cation (m/z 130) detected at long extraction delay does not show either a dominant short time component or a plateau, which have been assigned, when the extraction delay is short, to the formation of the radical cation after the H atom loss reaction. This means that the H atom loss reaction occurs rather rapidly after UV excitation. Since the transients detected at long extraction delay are all similar, the m/z 130 ion can be a secondary fragment of an ion produced via internal conversion, which corroborates that several channels can lead to this fragment.5,28
5. Conclusions
The UV excitation of WLH+ leads to a competition between fragmentation following internal conversion and direct H atom loss reaction. Primary fragmentation channels have a fast rate constant while secondary fragmentation can span a wide range of dissociation times. The femtosecond pump–probe study of WLH+ reveals that the excited state lifetime is short. All the fragments produced after internal conversion exhibit the same pump–probe signal that supports the hypothesis of statistical energy redistribution in the ground electronic state surface. However, the tryptophan side chain cation, formed via Cα–Cβ bond cleavage in the WL radical cation, exhibits a dominant short time component. This emphasizes a direct dissociation reaction experienced in the excited state with a non-statistical decay enhanced by the probe pulse excitation. The detailed analysis of the WLH+ excited states properties according to its conformers is presently under study with the help of high-level ab initio calculations and will be published in a forthcoming paper.Acknowledgements
The authors thank Dr P. Dugourd and his collaborators for communicating their results before publication and for many helpful discussions. Dr H. Kang thanks the Université Paris-Sud for financial support. The authors are grateful to F. Gobert for the maintenance of the femtosecond laser. This work has been performed at the Centre de Cinétique rapide ELYSE (Bât. 349, Université Paris-Sud, 91405 Orsay, France).References
- E. R. Williams, J. J. P. Furlong and F. W. McLafferty, J. Am. Soc. Mass Spectrom., 1990, 1, 288 CrossRef CAS.
- J. U. Andersen, H. Cederquist, J. S. Forster, B. A. Huber, P. Hvelplund, J. Jensen, B. Liu, B. Manil, L. Maunoury, S. B. Nielsen, U. V. Pedersen, J. Rangama, H. T. Schmidt, S. Tomita and H. Zettergren, Phys. Chem. Chem. Phys., 2004, 6, 2676 RSC.
- W. Gabryelski and L. Li, Rev. Sci. Instrum., 1999, 70, 4192 CrossRef CAS.
- Z. Q. Guan, N. L. Kelleher, P. B. O'Connor, D. J. Aaserud, D. P. Little and F. W. McLafferty, Int. J. Mass Spectrom. Ion Processes, 1996, 158, 357 CrossRef.
- F. Talbot, T. Tabarin, R. Antoine, M. Broyer and P. Dugourd, J. Chem. Phys., 2004, 122, 74310.
- R. Aebersold and D. R. Goodlett, Chem. Rev., 2001, 101, 269 CrossRef CAS.
- P. Hvelplund, B. Liu, S. B. Nielsen and S. Tomita, Int. J. Mass Spectrom., 2003, 225, 83 CrossRef CAS.
- F. Turecek, J. Am. Chem. Soc., 2003, 125, 5954 CrossRef CAS.
- K. F. Haselmann, T. J. D. Jorgensen, B. A. Budnik, F. Jensen and R. A. Zubarev, Rapid Commun. Mass Spectrom., 2002, 16, 2260 CrossRef CAS.
- Y. J. Hu, B. Hadas, M. Davidovitz, B. Balta and C. Lifshitz, J. Phys. Chem. A, 2003, 107, 6507 CrossRef CAS.
- D. Nolting, C. Marian and R. Weinkauf, Phys. Chem. Chem. Phys., 2004, 6, 2633 RSC.
- H. Kang, C. Dedonder-Lardeux, C. Jouvet, S. Martrenchard, G. Grégoire, C. Desfrançois, J.-P. Scherman, M. Barat and J. A. Fayeton, Phys. Chem. Chem. Phys., 2004, 6, 2628 RSC.
- H. Kang, C. Dedonder-Lardeux, C. Jouvet, G. Grégoire, C. Desfrançois, J.-P. Schermann, M. Barat and J. A. Fayeton, J. Phys. Chem. A, 2005, 109, 2417 CrossRef CAS.
- K. Daigoku, N. Miura and K. Hashimoto, Chem. Phys. Lett., 2001, 346, 81 CrossRef.
- K. Fuke and R. Takasu, Bull. Chem. Soc. Jpn., 1995, 69, 3303.
- E. Kassab and E. M. Evleth, J. Am. Chem. Soc., 1987, 109, 1653 CrossRef CAS.
- H. Kang, C. Dedonder-Lardeux, C. Jouvet, S. Martrenchard, G. Grégoire, C. Desfrançois, J. P. Schermann, M. Barat and J. A. Fayeton, J. Chem. Phys., 2005, 122, 084307 CrossRef CAS.
- J. Laskin, J. M. Behm, K. R. Lykke and C. Lifshitz, Chem. Phys. Lett., 1996, 252, 277 CrossRef CAS.
- J. Laskin and J. H. Futrell, Mass Spectrom. Rev., 2005, 24, 135 CrossRef CAS.
- H. Kang, C. Jouvet, C. Dedonder-Lardeux, S. Martrenchard, G. Grégoire, C. Desfrançois, J.-P. Schermann, M. Barat and J. A. Fayeton, Phys. Chem. Chem. Phys., 2005, 7, 394 RSC.
- B. C. Dian, A. Longarte and T. S. Zwier, J. Chem. Phys., 2003, 118, 2696 CrossRef CAS.
- E. G. Robertson and J. P. Simons, Phys. Chem. Chem. Phys., 2001, 3(1), 1 RSC.
- J. R. Lakowicz, Principles of fluorescence spectroscopy, Kluwer Academic/Plenum, New York, 2nd edn, 1999 Search PubMed.
- R. Weinkauf, P. Schanen, D. Yang, S. Soukara and E. W. Schlag, J. Phys. Chem., 1995, 99, 11255 CrossRef CAS.
- I. K. Chu, C. F. Rodriquez, A. C. Hopkinson, K. W. M. Siu and T. C. Lau, J. Am. Soc. Mass Spectrom., 2001, 12, 1114 CrossRef CAS.
- A. G. Harrison, I. G. Csizmadia, T.-H. Tang and Y.-P. Tu, J. Mass Spectrom., 2000, 35, 683 CrossRef CAS.
- F. Pingitore, M. J. Polce, P. Wang and C. Wesdemiotis, J. Am. Soc. Mass Spectrom., 2004, 15, 1025 CrossRef CAS.
- H. El Aribi, G. Orlova, A. C. Hopkinson and K. W. M. Siu, J. Phys. Chem. A, 2004, 108, 3844 CrossRef CAS.
- R. Weinkauf, P. Schanen, A. Metsala, E. W. Schlag, M. Bürgle and H. Kessler, J. Phys. Chem., 1996, 100, 18567 CrossRef CAS.
| This journal is © the Owner Societies 2006 |