Palladium-coated nickel nanoclusters: new Hiyama cross-coupling catalysts
Laura Durán Pachón, Mehul B. Thathagar, František Hartl and Gadi Rothenberg*
Van ‘t Hoff Institute of Molecular Sciences, University of Amsterdam, Nieuwe Achtergracht 166, 1018 WV, Amsterdam, The Netherlands. E-mail: gadi@science.uva.nl
First published on 17th November 2005
Abstract
The advantages of bimetallic nanoparticles as C–C coupling catalysts are discussed, and a simple, bottom-up synthesis method of core–shell Ni–Pd clusters is presented. This method combines electrochemical and ‘wet chemical’ techniques, and enables the preparation of highly monodispersed structured bimetallic nanoclusters. The double-anode electrochemical cell is described in detail. The core–shell Ni–Pd clusters were then applied as catalysts in the Hiyama cross-coupling reaction between phenyltrimethoxysilane and various haloaryls. Good product yields were obtained with a variety of iodo- and bromoaryls. We found that, for a fixed amount of Pd atoms, the core–shell clusters outperform both the monometallic Pd clusters and the alloy bimetallic Ni–Pd ones. THF is an excellent solvent for this process, with less than 2% homocoupling by-product. The roles of the stabiliser and the solvent are discussed.
Introduction
Assembling functional nano-sized objects is one of the key objectives of nanotechnology, with applications ranging from controlled drug release to improved television screens.1–3 Due to their size, there is no clear-cut argument for building nano-structures using either a top-down or a bottom-up approach. On one hand, top-down methods (such as chemical etching or machining), usually give more precise and controllable results. On the other hand, bottom-up protocols, especially those based on self-assembly, are attractive because they can enable, in the long run, the inexpensive manufacturing that is needed for technological applications.4–7One exciting area of application for metal nanoparticles is catalysis.8 Owing to their ‘intermediate’ size, too big to be ‘homogeneous catalysts’ but too small to be ‘bulk metals’, nanoparticles (referred to also as nanoclusters9,10) can sometimes catalyse reactions that are not accessible to their homogeneous or heterogeneous counterparts, as we and others recently showed in the case of Suzuki11–14 and Sonogashira15 cross-coupling. Moreover, the large surface area can give a catalytic advantage over conventional systems.13,16,17 Bimetallic catalysts18,19 are especially interesting for several reasons: combining two metals may provide control over the catalytic activity, selectivity and stability, and some combinations may exhibit synergistic effects.20–22 Moreover, by controlling the type of cluster synthesised, one can improve the “catalyst atom economy”.23,24
Using bottom-up synthesis, one can envisage three types of cluster mixtures (Fig. 1): alloy particles, core–shell particles, and segregated particles. The core–shell species are particularly interesting, as catalysis occurs on the shell surface, so one can envisage a cluster where the core is an inexpensive, inactive metal, and the shell made from an active (noble) metal.25 In this paper, we report the first combined chemical/electrochemical cluster synthesis approach to make core–shell Ni–Pd nanoparticles. We then show that not only these clusters are active in Hiyama cross-coupling, but they are also superior to the monometallic and bimetallic alloy structures. The pros and cons of the synthesis method and the possible applications of such clusters in other catalytic systems are discussed.
 | ||
| Fig. 1 Different compositions and structures of metal nanoclusters. | ||
Results and discussion
Preparation and characterization of metal clusters
Homometallic cluster suspensions of Ni and Pd were electrochemically prepared in the presence of TOAB (tetraoctyl ammonium bromide) as surfactant to avoid particle agglomeration. Dark brown suspensions were obtained for both Ni clusters and Pd clusters, as observed previously in the literature.20 The Pd clusters were stable for long periods (several months) when kept under nitrogen, without observing any agglomeration. In contrast, the Ni clusters remained stable only for ca. two weeks, and only under a dry and non-oxidising atmosphere.The bimetallic alloy Ni–Pd clusters were prepared in a similar way by simultaneous electrolyses of the corresponding metal wire electrodes. Good yields and high stabilities were obtained. This observation already points to one advantage of the alloy preparation: it enables the study and application of Ni-containing clusters in a stable configuration.
To synthesise the structured core–shell Ni–Pd particles, we combined electrochemical and ‘wet chemical’ methods.26 First, homometallic Ni particles were prepared electrochemically as above. Then, the Pd shells were grown on these Ni cores from solution, using chemical reduction (see below). This method leads to spherical bimetallic nanoparticles with a narrow size distribution and no aggregation, with a mean diameter of 4.9 nm (see Fig. 2).
 | ||
| Fig. 2 Transmission electron micrograph (top, magnification ×200 000) and corresponding size distribution (bottom, based on 100 particles counted) of the core–shell Ni–Pd nanoparticles. | ||
UV–visible spectroscopy was also used to determine the presence of Pd2+ in the core–shell Ni–Pd clusters. Fig. 3 shows that the UV absorption of PdCl2 (λmax ∼ 370 nm) is not observed in the cluster solution. Thus, we assume that all of the Pd(II) is reduced to Pd0 atoms. Control experiments confirmed that Ni0 was not UV–visible active. Separate measurements yielded the expected spectra for the alloy Ni–Pd clusters and for Pd0 clusters (all in DMF).27
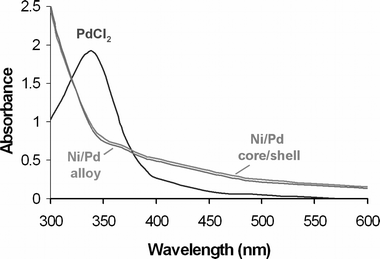 | ||
| Fig. 3 UV-Vis spectra showing the absorbance of PdCl2, core–shell Ni–Pd clusters and alloy Ni–Pd clusters (all solutions are in DMF). | ||
Regarding the formation of the Ni cores, the mechanism of electrochemical cluster synthesis has been described previously by Reetz and co-workers.20 It involves formation of adatoms in the vicinity of the cathode, followed by cluster formation and stabilization. The self-organisation of the ammonium salt around the metal core is essential for preventing undesired metal powder formation. Having examined this system, we observed differences in the nickel cluster stability, depending on the solvent polarity and the stabiliser concentration.
To study the influence of solvent polarity, we compared acetonitrile (MeCN), tetrahydrofuran (THF) and dimethylformamide (DMF). In each run, all the conditions were the same as in the standard procedure. The cluster stability increased in the order MeCN < THF < DMF. When MeCN was used, the clusters aggregated already after a few hours. Conversely, in THF, we were able to keep the dispersion stable for several days, and in DMF the clusters remained stable and did not aggregate even after four weeks.
MeCN is a very polar solvent (polarity parameter ET = 45.6), whereas THF and DMF are considerably less polar with (ET = 37.4 and ET = 30, respectively).28,29 We saw that aggregation was faster in more polar solvents, in good agreement with the results of Helbig.30 This may be due to a reduced electrostatic interaction between the metal nanoparticles and the TOAB in the more polar surrounding. In a less polar solvent, the stabilizing shell would shield the nanoparticles more effectively, preventing further growth.
To examine the effects of the stabilizer concentration, we repeated the process using 0.01 M, 0.1 M, and 1.0 M TOAB under otherwise identical conditions. Significantly, we observed that with increasing TOAB concentrations the solution was stable for a longer period. In the case of 0.01 M TOAB, the nickel particles precipitated immediately after the electrolysis was stopped. We also observed a green layer on the platinum cathode, assumed to be a nickel(II) species (possibly nickel oxide particles formed by reaction with traces of water). This indicates that the concentration of ammonium ions near the cathode was not enough to stabilize all the adatoms. Accordingly, this green layer was not observed when 0.1 M or 1.0 M TOAB was used. With 1.0 M stabilizer, the Ni cluster suspension did not aggregate even after four weeks! However, this suspension was less active in catalysis, perhaps because the cluster surface was covered by ammonium ions that impeded the access of the reactants. Both the solvent polarity and concentration of stabiliser are therefore important factors in optimising the cluster stability and activity.
The deposition of the Pd shells on the Ni cores is driven by the different standard reduction potentials (eqns (1) and (2)) allowing the reduction of Pd(II) by Ni(0). As shown by Miyake and co-workers,23 this process can be catalysed by the nickel nanoparticles themselves.
| Pd2+ + 2e− → Pd0E0 = +0.83 V | (1) |
| Ni2+ + 2e− → Ni0E0 = −0.23 V | (2) |
Hiyama cross-coupling catalysis
Cross-coupling is a versatile method for forming carbon–carbon bonds in a catalytic or stoichiometric manner.31 In particular, biaryl formation is sought after because biaryl-containing compounds are key intermediates for preparing biologically active molecules, organic semiconductors and liquid crystals.32 The Stille reaction (coupling of aryl halides with organostannanes)33 and the Suzuki–Miyaura reaction (coupling of aryl halides with organoboranes)34 are two practical routes to biaryls, though the former suffers from the toxicity of the tin by-product.35 An alternative protocol is the Hiyama cross-coupling of aryl halides and organosiliconates.36–39 The usual catalysts for Hiyama cross-coupling are {ligand-Pd(II)} complexes. Typically, 2.5–4 mol% catalyst is used, with a large excess (3–4 equivalents) of base.31Recently, we showed that transition metal clusters can catalyse ligand-free Huisgen-type cycloadditions17 as well as Suzuki11,12 and Sonogashira15 reactions. Here, we applied core–shell Ni–Pd clusters as alternative, ligand-free catalysts for the Hiyama reaction, using iodotoluene 1b and trimethoxyphenylsilane 2 (eqn (3)) as model substrates. The rationale behind using core–shell cluster catalysts is that in cluster catalysis one expects the reaction to occur at the cluster surface. The metal in the core is therefore not used. We envisaged that core–shell Ni–Pd clusters would maximise the efficiency of the Pd species (one needs a high Pd surface area, but there is no need to ‘waste Pd’ in the cluster core).
 | (3) |
 | ||
| Fig. 4 Comparison of catalytic activity for six different systems in the Hiyama cross-coupling of iodotoluene 1b and trimethoxyphenylsilane 2: Ni(OAc)2, Ni(0) clusters, Pd(OAc)2, Pd(0) clusters, Ni(0)/Pd(0) alloy clusters and Ni(0)/Pd(0) core–shell clusters. Reactions were carried out for 24 h in THF at 65 °C. | ||
We then compared the catalytic activity of Ni–Pd alloy clusters, core–shell Ni–Pd clusters, and segregated Pd and Ni clusters, as shown in Fig. 5. Control experiments confirmed that no conversion occurred without catalyst added. To further rule out the possibility that the non-reduced Pd(II) may be the active species, we also tested Pd(OAc)2 and Ni(OAc)2 separately under the same conditions.40,41 Note also that the amount of the metal catalyst was the same in each case (1 mol% Pd or Ni, depending on the experiment, relative to aryl halide).
 | ||
| Fig. 5 Photograph and schematic drawing of the home-made electrochemical cell used for the nanocluster synthesis. In the schematic, the two wire anodes are shown as solid bars and the supporting glass rods are omitted for clarity. | ||
Pure Pd clusters gave a lower yield (44%) compared to the alloy Ni–Pd ones (63%), that in turn were less active than the Ni–Pd core–shell clusters. As all these catalysts contain the same amount of palladium, this indicates that the core–shell structure results in more Pd atoms on the surface. This means more accessible catalytic sites per mole of palladium, and is reflected by the higher catalytic activity. EXAFS studies by Toshima and co-workers have shown that the total co-ordination number around Ni atoms in bimetallic clusters is usually higher than that around Pd.21 This also suggests that the Pd atoms are located preferentially on the surface. The tendency of Pd to go to the surface may explain the difference in activity between the Pd clusters and the alloy Ni–Pd clusters. No reaction took place when the Ni clusters or Ni(II) alone were used. Therefore, it is likely that only Pd is responsible for the catalysis in the case of the alloy and the core–shell clusters. The most important finding is that by combining palladium with another, non-reactive metal (in this case Ni), we can increase the activity per Pd atom (segregated Pd clusters < alloy Ni–Pd clusters < core–shell Ni–Pd clusters).
To examine the scope of this catalytic system, we tested the core–shell Ni–Pd clusters using a variety of substrates (eqn (4)). Aryl iodides with either electron-donating or electron-withdrawing substituents gave practically quantitative yields (Table 1, entries 1–6). For aryl bromides we observed good yields for electron-neutral and electron-withdrawing substituents, but much less conversion for the electron-donating bromoanisole. Further studies implementing these new catalysts in other reactions, as well as supporting the clusters on solid supports for easier separation will be the subject of future research in our laboratory.
 | (4) |
| Entry | Alkyne | Product | Yield(%)b |
|---|---|---|---|
| a Reaction conditions: 1.0 mmol substrate, 1.5 mmol phenyltrimethoxysilane, 1.5 mmol TBAF, 1.0 mol% catalyst (core–shell Ni–Pd clusters containing 0.1 mmol Ni and 0.01 mmol Pd), 5.0 mL THF, N2 atmosphere, 65 °C, 24 h. Reaction time is not optimised.b Yields are based on GC analysis, corrected for the presence of an internal standard. Side-products account for 2% or less of the aryl halides. | |||
| 1 |  |  | >99 |
| 2 |  |  | >99 |
| 3 |  |  | >99 |
| 4 |  | 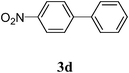 | >99 |
| 5 |  |  | >99 |
| 6 |  |  | >99 |
| 7 |  |  | 62 |
| 8 |  | 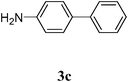 | 6 |
| 9 | 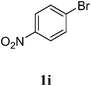 |  | 86 |
| 10 |  |  | 74 |
Conclusions
Core–shell Ni–Pd nanoparticles can be synthesised easily by combining electrochemical and ‘wet chemical’ methods, at room temperature in the presence of surfactant stabilisers. These methods yield stable and highly monodispersed particles. These bimetallic nanoparticles catalyze the Hiyama cross-coupling reaction. The catalytic activity studies show that the core–shell Ni–Pd clusters are superior to alloy bimetallic clusters and monometallic clusters, indicating an efficient use of the surface Pd atoms. We believe that this type of structuring can open new pathways in bimetallic and ligand-free catalysis.Experimental section
Materials and instrumentation
Electrochemical experiments were performed using a special home-made cell coupled to a dual current supply (bigalvanostat) with a maximum applied output of 10 V/40 mA and three terminal connections (two working electrodes and a single counter electrode). 1H NMR was measured on a Varian Mercury vx300 NMR spectrometer at 25 °C. Chemical shifts are referenced to Me4Si and internal solvent resonances. UV–visible spectra were measured using a Hewlett Packard 8453 spectrophotometer with a diode array detector. The quartz cuvette (Hellma Benelux, 3.5 mL, path length 1 cm) was mounted on a sample holder with controlled heating (Neslab constant temperature bath) and stirring (compressed air was used to turn the magnet stirrer under the cuvette). The wavelength range was 190–1100 nm, at 1 nm resolution. Transmission electron microscopy (TEM) images for nanoparticles characterization were obtained with a JEOL-TEM 1200 EXII instrument, operated at an accelerating voltage of 120 kV. Samples for TEM were prepared by placing 150 μL of cluster suspension on carbon-coated copper grids. These were then placed in vacuum oven at 50 °C at 250 mm of Hg to evaporate the solvent. GC analysis was performed using an Interscience GC-8000 gas chromatograph with a 100% dimethylpolysiloxane capillary column (DB-1, 30 m × 0.325 mm). GC conditions: isotherm at 105 °C (2 min); ramp at 30 °C min−1 to 280 °C; isotherm at 280 °C (5 min). Pentadecane was used as internal standard. Sets of 16 reactions for catalytic tests were performed using a Chemspeed Smartstart 16-parallel reactor block, modified in-house for efficient reflux and stirring. All chemicals were purchased in their dry pure form or dried using the standard drying procedure prior to use. All reactions were carried out under N2 atmosphere. All products are known compounds and were identified by comparison of their spectral properties to those of authentic samples.Electrochemical setup
All the reactions were carried out under dry N2 atmosphere and at 25 °C in a single-compartment 100 mL cell constructed in-house (see Fig. 5), a space-divided three-electrode modification of the two-electrode design published by Reetz and Helbig.42 The cell was equipped with special glass cover, bearing a tubular glass ‘finger’ supporting the coiled Pt cathode. Holes drilled in this tubular ‘finger’ ensured free movement of ions between the helical turns, enabling efficient mixing throughout the cell, while still separating the cathodic and anodic spaces. The cover also included two outlets for the anodes, a pin contact to the Pt counter electrode and inert gas inlet/outlet. Pd and/or Ni wires (3 × 0.5 cm2 surface area, 0.5 mm thickness and 99.99% purity) served as sacrificial anodes, coiled on glass rods for support. Pt wire (3 × 3 cm2 surface area, 0.5 mm thickness and 99.99% purity) was used as the inert cathode. Before use, electrodes were cleaned with scouring powder and washed with H2O, acetone and toluene. The supporting electrolyte was dry 0.1 M tetraoctylammonium bromide (TOAB) in dimethylformamide (DMF), degassed for 30 min prior to use. The yield for the cluster electrosynthesis was calculated as the current efficiency (the ratio of the theoretical charge required for the amount of product obtained to the total charge passed through the electrolysis cell during the duration of the electrolysis).43Synthesis of monometallic clusters
Pd clusters in tetrahydrofuran (THF) were similarly prepared first in DMF, followed by drying the suspension under vacuum (0.4 atm, 60 °C) for 5 h and redispersing the resulting powder in dry THF.
Synthesis of alloy Ni–Pd clusters
Ni and Pd wires as the anodes and Pt wire as the inert cathode were immersed in 60 mL of the supporting electrolyte solution (12 mmol, 3.3 g, 0.1 M (C8H17)4N+Br− in 60 mL dry DMF). A constant current density of 20 mA cm−2 was applied and the charge consumed was 2 F mol−1. The solution in the electrolysis cell was stirred for 5 h. A dark brown suspension was obtained, transferred to a Schlenk tube and kept under nitrogen. The current yield was 60–80%. The suspension was dried under vacuum (0.4 atm, 60 °C) for 5 h, and the resulting solid was redispersed in dry THF.Synthesis of core–shell Ni–Pd clusters
The Ni wire anode and the Pt wire (the inert cathode) were immersed in 60 mL of the supporting electrolyte solution (12 mmol, 3.3 g, 0.1 M (C8H17)4N+Br− in 60 mL of dry DMF). A constant current density of 20 mA cm−2 was applied. The solution in the electrolysis vessel was stirred for 2 h. A light brown suspension (the Ni cores) was obtained. Then, Pd(OAc)2 (0.16 g, 0.7 mmol, 10 mol% relative to Ni) was added to the suspension. The mixture was stirred for 2 h. A dark brown suspension was obtained, transferred to a Schlenk tube, dried under vacuum (0.4 atm, 60 °C) for 5 h, and redispersed in dry THF. The resulting suspension was kept under an inert atmosphere for characterization and catalytic studies.Procedure for cluster-catalysed Hiyama cross-coupling
Example: 4-trifluoromethyl-biphenyl (3f). A solution of phenyltrimethoxysilane 2 (1.5 mmol, 0.29 g), 4-bromobenzotrifluoride 1f (0.5 M, 2.0 mL, 1.0 mmol) and tetra-n-butylammonium fluoride (TBAF, 1.5 mmol, 0.47 g) in 5 mL THF was stirred in the reactor (magnetic stirring, 5 min). A suspension of core–shell Ni–Pd clusters with a 10 ∶ 1 Ni–Pd ratio (10.0 mM, 5.0 mL, 1.0 mol% total metal, equivalent to 0.1 mol% of Pd relative to 1) was then added in one portion, and the reaction mixture was stirred at 65 °C for 24 h. Reaction progress was monitored by GC. The product was extracted by washing the reaction mixture three times with diethyl ether/water (1 ∶ 1, 20 mL). The phases were separated and the ether phase was evaporated under vacuum to obtain 0.82 g (82 mol% based on 1) as a yellowish crystalline solid. 1H NMR (ppm, Me4Si): δ 7.72 (s, 4 H), 7.58 (d, 2 H), 7.46 (t, 2 H), 7.38 (t, 1 H). A good agreement was found with the literature values.44Acknowledgements
We thank Bert van Groen and Paul F. Collignon (both University of Amsterdam) for excellent technical assistance in building the electrochemical cell and the bi-galvanostat, and Anil V. Gaikwad for measuring the UV–visible spectra.References
- N. A. Peppas, Adv. Drug Delivery Rev., 2004, 56, 1529 CrossRef CAS.
- N. Ledbetter and T. Goswami, J. Mech. Behavior Mater., 2002, 13, 353 Search PubMed.
- H. Bönnemann and R. Richards, in Nanomaterials as Precursors for Electrocatalysts, ed. A. Wieckowski, E. R. Savinova and C. G. Vayenas, Marcel Dekker, New York, 2003 Search PubMed.
- E. Eiser, F. Bouchama, M. B. Thathagar and G. Rothenberg, ChemPhysChem, 2003, 4, 526 CrossRef CAS.
- F. Bouchama, M. B. Thathagar, G. Rothenberg, D. H. Turkenburg and E. Eiser, Langmuir, 2004, 20, 477 CrossRef CAS.
- D. H. Turkenburg, A. A. Antipov, M. B. Thathagar, G. Rothenberg, G. B. Sukhorukov and E. Eiser, Phys. Chem. Chem. Phys., 2005, 7, 2237 RSC.
- R. Richards, G. Geibel, W. Hofstadt and H. Bönnemann, Appl. Organomet. Chem., 2002, 16, 377 CrossRef CAS.
- C.-J. Zhong and M. M. Maye, Adv. Mater., 2001, 13, 1507 CrossRef CAS.
- G. Schmid, Adv. Eng. Mater., 2001, 3, 737 CrossRef CAS.
- H. Bönnemann and R. Richards, Eur. J. Inorg. Chem., 2001, 2455 CrossRef CAS.
- M. B. Thathagar, J. Beckers and G. Rothenberg, J. Am. Chem. Soc., 2002, 124, 11858 CrossRef CAS.
- M. B. Thathagar, J. Beckers and G. Rothenberg, Adv. Synth. Catal., 2003, 345, 979 CrossRef CAS.
- M. T. Reetz, R. Breinbauer and K. Wanninger, Tetrahedron Lett., 1996, 37, 4499 CrossRef.
- E. G. Ijpeij, F. H. Beijer, H. J. Arts, C. Newton, J. G. de Vries and G. J. M. Gruter, J. Org. Chem., 2002, 67, 169 CrossRef CAS.
- M. B. Thathagar, J. Beckers and G. Rothenberg, Green Chem., 2004, 6, 215 RSC.
- M. Moreno-Mañas and R. Pleixats, Acc. Chem. Res., 2003, 36, 638 CrossRef CAS.
- L. Durán-Pachón, J. H. van Maarseveen and G. Rothenberg, Adv. Synth. Catal., 2005, 347, 811 CrossRef.
- L. Guczi, Catal. Today, 2005, 101, 53 CrossRef CAS.
- M. Takanori and T. Asakawa, Appl. Catal., A, 2005, 280, 47 CrossRef.
- M. T. Reetz, M. Winter, R. Breinbauer, T. Thurn-Albrecht and W. Vogel, Chem. Eur. J., 2001, 7, 1084 CrossRef CAS.
- P. Lu, T. Teranishi, K. Asakura, M. Miyake and N. Toshima, J. Phys. Chem. B, 1999, 103, 9673 CrossRef CAS.
- T. Miyake and T. Asakawa, Appl. Catal., A, 2005, 280, 47 CrossRef.
- T. Teranishi and M. Miyake, Chem. Mater., 1999, 11, 3414 CrossRef CAS.
- R. W. J. Scott, H. C. Ye, R. R. Henriquez and R. M. Crooks, Chem. Mater., 2003, 15, 3873 CrossRef CAS.
- S. U. Son, Y. Jang, J. Park, H. B. Na, H. M. Park, H. J. Yun, J. Lee and T. Hyeon, J. Am. Chem. Soc., 2004, 126, 5026 CrossRef CAS.
- M. T. Reetz, W. Helbig and S. A. Quaiser, Chem. Mater., 1995, 7, 2227 CrossRef CAS.
- J. Wang, H. F. M. Boelens, M. B. Thathagar and G. Rothenberg, ChemPhysChem, 2004, 5, 93 CrossRef CAS.
- V. Gold, K. L. Loening, A. D. McNaught and P. Sehmi, in IUPAC Compendium of Chemical Terminology, Blackwell Scientific, Oxford, 1997 Search PubMed.
- C. Reichardt, in ‘Solvents and Solvent effects in Organic Chemistry’Weinheim, 1998 Search PubMed.
- W. Helbig, PhD Thesis, Ruhr University, Bochum, 1994.
- A. F. Littke and G. C. Fu, Angew. Chem., Int. Ed., 2002, 41, 4176 CrossRef CAS.
- A. N. Cammidge and K. V. L. Crepy, J. Org. Chem., 2003, 68, 6832 CrossRef CAS.
- J. K. Stille, Angew. Chem., Int. Ed. Engl., 1986, 25, 508 CrossRef.
- N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457 CrossRef CAS.
- Y. Arakawa, in Chemistry of Tin, ed. P. J. Smith, Blackie, London, 1998 Search PubMed.
- Y. Hatanaka and T. Hiyama, J. Org. Chem., 1988, 53, 918 CrossRef CAS.
- T. Hiyama and Y. Hatanaka, Pure Appl. Chem., 1994, 66, 1471 CrossRef CAS.
- M. E. Mowery and P. DeShong, J. Org. Chem., 1999, 64, 1684 CrossRef CAS.
- S. K. Kang and W. Y. Kim, Synth. Commun., 1998, 28, 3743 CrossRef CAS.
- M. L. Clarke, Adv. Synth. Catal., 2005, 347, 303 CrossRef CAS.
- D. Domin, D. Benito-Garagorri, K. Mereiter, J. Fröhlich and K. Kirchner, Organometallics, 2005, 24, 3957 CrossRef CAS.
- M. T. Reetz and W. Helbig, J. Am. Chem. Soc., 1994, 116, 7401 CrossRef CAS.
- G. Gritzner, G. Kreysa, G. S. Wilson, D. Landolt, V. M. M. Lobo, W. Plieth, M. Sluytersrehbach, K. Tokuda, C. P. Andrieux, Y. A. Chizmadzhev, B. E. Conway, J. Koryta, G. Kreysa, O. A. Petrii, D. Pletcher, M. J. Weaver, A. J. Arvia, T. Biegler, H. D. Hurwitz, D. Pavlov, G. Horanyi, S. K. Rangarajan, E. Gileadi, S. Trasatti, T. Watanabe, G. A. Wright, W. Paik, C. Gutierrez, D. Simonsson, M. L. Berkem, A. K. Covington and D. Drazic, Pure Appl. Chem., 1993, 65, 1009 CrossRef CAS.
- L. Liu, Y. Zhang and Y. Wang, J. Org. Chem., 2005, 70, 6122 CrossRef CAS.
| This journal is © the Owner Societies 2006 |