Excited state hydrogen transfer in fluorophenol·ammonia clusters studied by two-color REMPI spectroscopy
Norihiro
Tsuji
a,
Shun-ichi
Ishiuchi
a,
Makoto
Sakai
a,
Masaaki
Fujii
*a,
Takayuki
Ebata
b,
Christophe
Jouvet
*c and
Claude
Dedonder-Lardeux
*c
aChemical Resources Laboratory, Tokyo Institute of Technology, 4259 Nagatsuta, Midori-ku, Yokohama, 226-8503, Japan
bDepartment of Chemistry, Graduate School of Science, Hiroshima University, Higashi-hiroshima, 739-8526, Japan
cLaboratoire de Photophysique Moléculaire Bat. 213, Universitde Paris-sud, 91405, Orsay Cedex, France
First published on 21st November 2005
Abstract
Two-color (1 + 1′) REMPI mass spectra of o-, m- and p-fluorophenol·ammonia (1 ∶ n) clusters were measured with a long delay time between excitation and ionization lasers. The appearance of NH4(NH3)n−1+ with 100 ns delay after exciting the S1 state is a strong indication of generation of long-lived species via S1. In analogy with the phenol·ammonia clusters, we conclude that an excited state hydrogen transfer reaction occurs in o-, m- and p-fluorophenol·ammonia clusters. The S1-S0 transition of o-, m- and p-fluorophenol·ammonia (1 : 1) clusters were measured by the (1 + 1′) REMPI spectra, while larger (1 ∶ n) cluster (n = 2–4) were observed by monitoring the long-lived NH4(NH3)n−1 clusters action spectra. The vibronic structures of m- and p-fluorophenol·ammonia clusters are assigned based on vibrational calculations in S0. The o-fluorophenol·ammonia (1 : 1) cluster shows an anharmonic progression that is analyzed by a one-dimensional internal rotational motion of the ammonia molecule. The interaction between the ammonia molecule and the fluorine atom, and its change upon electronic excitation are suggested. The broad action spectra observed for the o-fluorophenol·ammonia (1 : n) cluster (n ≧ 2) suggest the excited state hydrogen transfer is faster than in m- and p-fluorophenol·ammonia clusters. The different reaction rates between o-, m- and p-fluorophenol·ammonia clusters are found from comparison between the REMPI and action spectra.
1. Introduction
Excited state hydrogen transfer (ESHT) has been attracting many researchers because it provides new possibilities for the interpretation of well known photochemical reactions. This reaction channel was discovered through the generation of long-lived species from the photo-excited phenol·(NH3)n cluster.1–6 When the two-color (1 + 1′) resonant ionization via S1 state is applied to the phenol·ammonia cluster, NH4(NH3)n−1+ ions are generated in addition to the parent phenol·ammonia cluster cations. The parent cluster ions cannot be observed when a long delay (200 ns) is introduced between the excitation and ionization lasers, because of the short lifetime in S1 (a few ns to 50 ps). Yet, the NH4(NH3)n−1+ ions are still observed at such long delays. Besides, these NH4(NH3)n−1+ ions are not detected when the clusters are directly ionized by vacuum UV light from the ground state, which indicates that they are due to a reaction occurring in the excited state. NH4(NH3)n−1+ ions must then be produced by direct ionization of long-lived species generated in the S1 state of the phenol·(NH3)n clusters. The electronic and vibrational transitions of the long-lived species have been measured by UV-near IR-UV and UV-IR-UV ion dip spectroscopy that allowed to identify the long-lived species to neutral NH4(NH3)n−1 radicals.3–6 The generation mechanism of neutral radicals was completely different from the excited state proton transfer that was expected for the photo-excited phenol and corresponds to a hydrogen atom transfer from the excited phenol to the ammonia cluster (ESHT). Several other examples of ESHT or excited state hydrogen loss reactions have been now reported in indole·ammonia clusters,7 pyrrole·ammonia clusters,8 and amino-acid cations.9–11 In aromatic molecules containing OH or NH groups, ESHT seems to be a general reaction that needs to be further investigated and understood.Theoretically, the mechanism of the ESHT reaction points to the importance of a πσ* state localized on the OH group and dissociative along this coordinate (SDDJ model).12 In the free molecule the πσ* state lies higher in energy than the S1 ππ* state and crosses both the excited and ground states, which results in a competition between H loss and fast internal conversion. In clusters, the πσ* state is stabilized by the hydrogen-bond formation and still crosses the S1 ππ* state, inducing the transfer of the hydrogen atom to the solvent cluster but the crossing with the ground state is removed. This πσ* model that explains qualitatively many different photochemical processes in aromatic acids should be tested in a more systematic way. In that view, we have studied the ESHT reaction in fluorophenol·ammonia clusters. Fluorine substitution is one of the most benign perturbations for phenol because the number of vibrational modes remains the same after substitution and the electronic state is weakly perturbed. The fluorine substituent has an induction effect that attracts the electron in the σ orbital. Furthermore, the strength of the perturbation is expected to vary with the substituent position (ortho-, meta- and para-) according to the OH group. Therefore the o-, m- and p-fluorophenol·(NH3)n clusters provide important data to validate the πσ* model. In the present work, as a first step of a systematic study, we have measured the two-color resonant enhanced two-photon ionization (1 + 1′ REMPI) spectra and the action spectra obtained by monitoring the reaction products NH4(NH3)n−1 for o-, m- and p-fluorophenol·(NH3)n clusters. The vibronic structures are discussed based on the quantum chemical calculation in the neutral ground state. The difference of reaction efficiency among the three fluorophenol isomers will be discussed.
2. Experimental
The molecular beam chamber combined with the linear time of flight (TOF) mass spectrometer used in this work has been previously reported.3–6 Fluorophenol·(NH3)n (1 : n) clusters were generated in a supersonic-jet expansion of a gas mixture of fluorophenol vapor (at room temperature) and NH3/Ne (0.5%) premixed gas through a pulsed valve (General Valve). The supersonic jet was skimmed (Beam Dynamics skimmer, 2-mm-diameter) before entering the detection chamber. The clusters were excited to the S1 state by the first laser ν1. After an appropriate delay (typically 100 ns), the remaining clusters and their reaction products were ionized by the ionization laser ν2. The generated ions were accelerated and extracted into in the TOF mass spectrometer and detected by an Even-cup detector.13 The signal was integrated by a digital boxcar integrator after amplification by a preamplifier (NF BX-31A), and was recorded by a personal computer. The output of a dye laser (Lumonics: HD-500) pumped by the 355 nm radiation of a Nd3+:YAG laser (Spectra Physics: GCR-170) was frequency-doubled in a KDP crystal (Inrad: AUTO TRACKER III) and used as the first laser light ν1 to excite the clusters to S1. The ionization laser ν2 was the third harmonic (355 nm) of a Nd3+:YAG laser (Spectra Physics: INDI-40) or the frequency-doubled output (310 nm) of a dye laser pumped by the second harmonics of the Nd3+:YAG laser (HOYA Continuum, PowerLite 8010). The samples were purchased from Tokyo Kasei and used after purification by vacuum sublimation (p-fluorophenol) or vacuum distillation (o- and m-fluorophenol).3. Theoretical calculation
All calculations have been performed with the Turbomole program package.14 The starting geometries for the cluster isomers have been selected in analogy with previous calculations performed for phenol·(NH3)n clusters.6 For fluorophenol clusters with one and two ammonia molecules, ground state geometry optimization and frequency calculations were performed with the RIDFT method implemented in Turbomole (resolution-of-the-identity approximation with the BP86 functional) with the triple-zeta valence plus polarization (TZVP) basis sets, in order to estimate the low frequency intermolecular vibrations. For the o-fluorophenol·ammonia complex, the ground state frequencies have also been calculated with the B3LYP functional and at the SCF/MP2 level, still with the TZVP basis set. The frequencies calculated with the three different methods are compared in Table 1. Since the vibrational frequencies obtained with the different methods are in reasonable agreement, the frequency calculations for other 1 ∶ 1 and 1 ∶ 2 clusters have been performed with the fastest RIDFT/TZVP method.| RIDFT (B-P86)/TZVP | DFT (B3-LYP)/TZVP | SCF/RI-MP2/TZVP | |
|---|---|---|---|
| 23 | 26 | 29 | Out of plane bending |
| 55 | 46 | 78 | NH3 torsion around H⋯N axis |
| 110 | 119 | 101 | In plane bending |
| 197 | 187 | 188 | Stretching OH⋯N |
| 289 | 274 | 274 | Wagging |
| 298 | 308 | 300 | Rocking (in plane) |
4. Results and discussion
4.1 Mass-spectrum
Fig. 1 shows the mass spectrum of p-fluorophenol·(NH3)n clusters obtained by delaying the ionization laser by 5 ns (Fig. 1a) and 100 ns (Fig. 1b) after the excitation to S1, the exciting laser ν1 and the ionization laser ν2 are fixed to 285 nm and 310 nm, respectively. The mass spectrum at 5 ns delay shows the peaks of the p-fluorophenol·(NH3)n cations (1 : n) (n = 0–4) and NH4(NH3)n−1+ (n = 2–5) clusters. At long delays (Fig. 1b), the p-fluorophenol·(NH3)n mass peaks become very weak while the NH4(NH3)n−1 peaks remain as intense as for short delays. The decrease of the p-fluorophenol·(NH3)n clusters ion signal is expected since the typical lifetime of excited phenol and derivatives is no more than a few nanoseconds. Thus the decay of the signal can be understood as the decay of S1. In contrast, the stable signal of NH4(NH3)n−1+ at long delay suggests that these ions are not generated from the p-fluorophenol·(NH3)n cations, but from long-lived species originating from S1. The same phenomenon, NH4(NH3)n−1+ surviving for long delays between excitation and ionization, has been found in phenol·(NH3)n clusters where the long-lived species have been shown to be neutral NH4(NH3)n−1 radicals generated by an excited state hydrogen transfer (ESHT) reaction in S1. Likewise, we conclude that an ESHT reaction also occurs in p-fluorophenol·(NH3)n clusters, leading to long-lived NH4(NH3)n−1 radicals ionized with laser ν2 at 310 nm. The same NH4(NH3)n−1+ are also observed when o- and m-fluorophenol·(NH3)n clusters are excited to S1. Thus ESHT reactions occur in the excited state of o-, m- and p-fluorophenol clustered with ammonia. | ||
| Fig. 1 Mass-spectrum of p-fluorophenol·(NH3)n clusters obtained by introducing the ionization laser ν2 with (a) 5 ns and (b) 100 ns delay after the excitation to S1. The exciting laser ν1 and the ionization laser ν2 are fixed to 285 nm and 310 nm, respectively. NH4(NH3)n−1 radicals are known to be much more stable than free NH426 and their lifetimes have been measured by Fuke et al. to be 3 μs and 7 μs for n = 2 and 3, respectively whereas NH4 is a very short-lived species (15 ps).26–28 The ionization potentials for NH4(NH3)n−1 clusters decrease from 3.88 eV for n = 2, 3.31 eV for n = 3, to 2.97 eV for n = 4,27,28 so they can be ionized with 310 nm photons (4 eV). | ||
4.2 Calculated geometry and vibrations
The fluorophenol·NH3 (1 : 1) complexes all adopt a near planar geometry in the ground state, with the ammonia hydrogen bonded to the hydrogen of the hydroxyl group, like in the phenol-ammonia complex. The lowest ground state intermolecular vibrations (RIDFT/TZVP) in the (1 : 1) clusters are all very similar as can be seen in Table 2. The calculation gives the frequency of out-of-plane bending mode around 20–30 cm−1. The torsional motion of the ammonia molecule is the second lowest vibration with a frequency around 50–60 cm−1.| Species | Out of plane bending | Torsion of NH3 about the H⋯N axis | In plane bending | OH⋯N stretching |
|---|---|---|---|---|
| a cis-conformation. | ||||
| o-fluorophenol/NH3a | 22 | 55 | 110 | 197 |
| p-fluorophenol/NH3 | 26 | 58 | 64 | 191 |
| m-cis-fluorophenol/NH3 | 36 | 59 | 66 | 195 |
| m-trans-fluorophenol/NH3 | 28 | 49 | 67 | 186 |
For (1 : 2) clusters, as in phenol, two close geometries are found, a cyclic H⋯NH3⋯NH3⋯O structure and an open structure where the ammonia dimer is out of plane.6,15 For p-fluorophenol·(NH3)2 clusters, the geometries are very similar to those found for phenol·(NH3)2, and the cyclic isomer is slightly lower in energy (170 cm−1). The ground state vibrational frequencies are not very different for the two isomers and will not enable their discrimination.
The cyclic and open structures are shown in Fig. 2a in the case of the m-fluorophenol·(NH3)2 (1 : 2) clusters. Since for the bare m-fluorophenol molecule, the trans- and cis-isomers are present in the jet, four structures have been calculated for the 1 : 2 clusters and their energies are all within 150 cm−1. The cyclic structures are very similar for the cis- and trans-isomers, but in the open structure the second ammonia molecule gets much closer to the fluorine atom in the cis-isomer. The calculated low frequencies vibrational modes are listed in Table 3. In any conformation, the butterfly motion is the lowest frequency, followed by the waging and in-plane bending. Stretching vibrations are found around 190 cm−1 (between the two ammonia molecules) and 220 cm−1 (between the hydroxyl and the closest NH3 molecule).
 | ||
| Fig. 2 Calculated geometries for the different isomers of the (a) m-, (b) o- and p-fluorophenol·(NH3)2 (1 : 2) cluster. | ||
| m-Fluorophenol cis isomer | m-Fluorophenol trans isomer | p-Fluorophenol | ||||
|---|---|---|---|---|---|---|
| Vibrational mode | Cyclic | Open | Cyclic | Open | Cyclic | Open |
| a Ammonia 1 is the ammonia molecule linked to the hydroxyl group, ammonia 2 is linked to ammonia 1. | ||||||
| Butterfly motion | 9 | 17 | 8 | 24 | 6 | 22 |
| Wagging | 44 | 27 | 33 | 34 | 37 | 29 |
| In plane bend for cyclic isomer/ | 46 | 47 | 46 | |||
| bending for the open isomer | 55 | 62 | 77 | |||
| Stretching of the ammonia dimer versus ring/ | 83 | 89 | 89 | |||
| wagging (NH3)2 and fluorophenol | 82 | 83 | 60 | |||
| Torsion of ammonia 2a | 132 | 70 | 131 | 100 | 124 | 89 |
| Out of plane fluorophenol deformation | 197 | 199 | 200 | 187 | 147 | 147 |
| Stretching ammonia 1..and ammonia 2a | 196 | 192 | 195 | 195 | 193 | 194 |
| Coupled torsion of the 2 ammonia molecules | 206 | 212 | 202 | 216 | 215 | 212 |
| Stretching of OH…ammonia 1 | 223 | 223 | 213 | 219 | 218 | 221 |
The geometries found for o-fluorophenol·(NH3)2 (1 : 2) clusters are slightly different: in the cyclic geometry, the H⋯NH3⋯NH3⋯O moiety is in a plane that makes a 50° angle with the fluorophenol plane, whereas the m and p-fluorophenol·(NH3)2 (1 : 2) clusters are quasiplanar; in the open geometry, the second ammonia molecule interacts with the fluorine atom and is thus farther from the aromatic ring (Fig. 2b).
4.3 REMPI and action spectra of p-fluorophenol·(NH3)n clusters
The two-color action spectra of o-, m- and p-fluorophenol·(NH3)n cluster have been measured by monitoring the NH4(NH3)n−1+ ion signal which is the photoionization signature of the ESHT reaction product NH4(NH3)n−1. The (1 + 1′) resonant enhanced multiphoton ionization (REMPI) spectra were also recorded by monitoring the p-fluorophenol·(NH3)n parent ion signal. Fig. 3a and b show mass-selected (1 + 1′) REMPI spectra of p-fluorophenol·ammonia (1 : 1) and (1 : 2) clusters. For convenience, the REMPI spectrum obtained by monitoring the p-fluorophenol·(NH3)n ion is indicated in the figure as para (1 : n), while the action spectrum obtained by monitoring NH4(NH3)n−1 are indicated as para (0∶n). The (1 : 1) cluster shows well-resolved vibronic structures, while no signal was found for the (1 : 2) cluster. On the other hand, the action spectra of p-fluorophenol·(NH3)n obtained by monitoring NH4(NH3)n−1+ (n = 2–4) exhibit clear and well-resolved spectra (Fig. 3c–e). Each action spectrum obtained by monitoring NH4(NH3)n−1+ shows different spectral features and reflects the vibronic structures of S1 for the corresponding p-fluorophenol·(NH3)n (1 : n) cluster. | ||
| Fig. 3 Mass-selected (1 + 1′) REMPI spectra of p-fluorophenol·(NH3)n for (a) the (1 : 1) and (b) the (1 : 2) clusters. The ionization laser ν2 was fixed to 310 nm for both measurements. The action spectra of p-fluorophenol·(NH3)n obtained by monitoring NH4(NH3)n−1+ (n = 2–4) are shown in c–e, respectively. For n = 2, ν2 was fixed to 310 nm (4 eV), while for n = 3,4, it was 355 nm (3.5 eV) because of the NH4(NH3)n−1 ionization potential.27,28 The delay between the excitation laser ν1 and the ionization laser ν2 was 5 ns for the REMPI measurements and 100 ns for the action spectra. | ||
For the p-fluorophenol·ammonia (1 : 1) cluster (Fig. 3a), the lowest-frequency peak at 34 382 cm−1 is assigned to the S1 origin. The band at 430 cm−1 from the origin is assigned to the first quantum of the intramolecular vibrational mode 6a (6a1) in S1 from comparison with the free p-fluorophenol.16 These two bands are associated with a low frequency band of 186 cm−1, assigned to the intermolecular stretching vibration σ between p-fluorophenol and ammonia, calculated at 191 cm−1 in the ground state (Table 2). In the (0 : 2) action spectrum obtained by monitoring NH4(NH3)+ (Fig. 3c), low-frequency progressions with spacing of 18 ∼ 20 cm−1 are observed all over the investigated spectral range. The calculations suggest that the low-frequency vibration is an out of plane motion of the ammonia molecules with respect to the fluorophenol ring (butterfly motion, Table 3). The lowest member of the most red progression at 34 189 cm−1 is assigned to the S1 origin. Similarly, the bands at 34 604 and 34 996 cm−1, located 415 and 807 cm−1 above the origin are assigned to 6a1 and 11 intramolecular vibrations. The correspondence to the vibronic structure of the (1 ∶ 1) cluster are indicated by broken lines. The bands at 34 400 and 34 817 cm−1, i.e. 210 cm−1 above the origin and 6a1 bands can be assigned to the intermolecular stretching σ1 and its combination band with 6a1 (6a1σ1), in analogy with the ground state stretching frequency between the OH group and the first ammonia calculated at 219 and 221 cm−1 in the cyclic and open isomers, respectively.
The vibronic structure of the (0 ∶ 3) action spectrum obtained by monitoring the NH4(NH3)2+ product ion also shows low-frequency progressions in the higher frequency side, though the structure is not clear in the lower region (see Fig. 3d). The progression with 15 cm−1 spacing starting around 34 939 cm−1 is assigned to the S1(11) transition of the (1 : 3) cluster and the weak bands having 15 cm−1 interval around 34 610 cm−1 are assigned to a progression built on the 6a1 in analogy to the (0 : 2) action spectrum. The (0 ∶ 4) action spectrum obtained by monitoring NH4(NH3)3+ presents long harmonic progressions of 14 cm−1 spacing at 34 160, 34 580 and 34 985 cm−1 as shown in Fig. 3e. Those bands are assigned to the S1 origin, 6a1 and 11 transitions of the corresponding (1 ∶ 4) cluster. We tentatively assigned the low frequency vibrations in (0 ∶ 3) and (0 ∶ 4) action spectra to the butterfly motions (out of plane bending motions).
4.4 REMPI and action spectra of m-fluorophenol·(NH3)n clusters
It has been known that m-fluorophenol and its hydrogen-bonded clusters have cis- and trans-isomers according to the orientation of OH group.17–19 The hydrogen-bonded clusters with ammonia can also be expected to have cis- and trans-isomers. Fig. 4a shows the mass-selected REMPI spectrum of m-fluorophenol·ammonia (1 : 1) cluster where the two sharp peaks at 36 045 and 36 277 cm−1 are assigned to the origins of cis- and trans- isomers, respectively. The red-shifts from the origin of free cis- and trans-m-fluorophenol are 578 and 553 cm−1, respectively. The larger red-shift of the cis-(1 : 1) cluster suggests an effect of the position of the fluorine substituent on the intermolecular interaction. The ground state calculations indicate that the complex with the cis-isomer has a slightly lower energy than the complex with the trans-isomer (50 cm−1). This is an indication of a different stabilization interaction in the cis (1 : 1) and trans (1 : 1) isomers. | ||
| Fig. 4 Mass-selected (1 + 1′) REMPI spectra of m-fluorophenol·(NH3)n for (a) the (1 : 1) and (b) the (1 : 2) clusters. The ionization laser ν2 was fixed to 310 nm for both measurements. The action spectra of m-fluorophenol·(NH3)n obtained by monitoring NH4(NH3)n−1+ (n = 2–4) are shown in c–e, respectively. ν2 was fixed at 310 nm for n = 2, and at 355 nm for n = 3 and 4. The delay between the excitation laser ν1 and the ionization laser ν2 was 5 ns for the REMPI measurements and 100 ns for the action spectra. | ||
The S1–S0 transition of the (1 : 2) cluster has been measured by both mass-selected REMPI spectrum (Fig. 4b, indicated as meta (1 : 2)) and action spectrum obtained by monitoring NH4NH3+ (Fig. 4c, meta (0 : 2)). The REMPI spectrum shows sharp peaks on a broad background signal in the low frequency region from 35 800 to 36 200 cm−1, but no clear peak is found in the region above 36 400 cm−1. In contrast, the action spectrum shows clear and sharp peaks up to 36 700 cm−1. The peak positions in the low frequency region coincide in both spectra, thus this difference suggests that the ESHT reaction is accelerated when the excess energy in S1 increases.
Two low-frequency progressions of 12 and 13 cm−1 spacing start at 35 819 cm−1. The band at 35 819 cm−1 is assigned to the origin of the cis-(1 : 2) cluster. The calculated vibrations for the cis-isomer in the ground state (cyclic or open) indicate that the low frequencies correspond to the bending motions of the ammonia molecules out of the plane defined by the aromatic ring (9 and 44 cm−1 for the cyclic isomer, 17 and 27 cm−1 for the open isomer, respectively).
A progression of 17 cm−1 interval is found at around 36 100 cm−1, and the lowest member at 36 108 cm−1 is assigned to the trans-(1 : 2) cluster. This low frequency vibration is assigned to out of plane bending of the ammonia molecules (24 cm−1 calculated for the open isomer, 8 cm−1 for the cyclic one in S0).
The progressions around 36 600 cm−1 have intervals similar to those of the cis-isomer and are assigned to the 11 vibronic band of the cis-isomer. The red-shifts of the cis- and trans-(1 : 2) clusters are 226 and 169 cm−1 from the cis- and trans-(1 : 1) clusters. As in the (1 : 1) clusters, the red-shift is larger for the cis-(1 : 2) isomer than for the trans-isomer. It may be due to an ammonia-fluorine interaction in the excited state of the cis-isomer since in the case of an open structure, the second ammonia molecule is closer to the fluorine atom.
Fig. 4d and 4e shows the action spectra obtained by monitoring NH4(NH3)n−1+ (n = 3 and 4), which are denoted as meta (0 : 3) and meta (0 : 4). Sharp structures are found in the action spectrum of meta (0 : 3), and the lowest band at 35 892 cm−1 is assigned to the S1 origin of the cis-(1 : 3) cluster. As for the cis-(1 : 2) cluster, two low-frequency progressions are found around the origin, and will be assigned to similar vibrational modes, out of plane bendings of the ammonia molecules. A short progression with 10 cm−1 interval is found at around 36 100 cm−1, and the lowest member at 36 118 cm−1 is assigned to the origin of the trans-(1 : 3) cluster. The progression at ∼36 600 cm−1 is considered to be built on the 11 vibronic band of the cis-(1 : 3) cluster. The action spectrum of meta (0 : 4) is broad with a maximum at 36 060 cm−1. No evidence is found for the co-existence of isomers. The large ammonia cluster of four molecules may cause a stronger steric hindrance, and one of the isomer may not be stable enough to exist under this experimental condition.
4.5 REMPI and action spectra of o-fluorophenol·(NH3)n clusters
Fig. 5a shows the mass-selected REMPI spectrum of the o-fluorophenol·NH3 (1 : 1) cluster that presents well-resolved vibronic structure, and the lowest band at 36 192 cm−1 is assigned to the S1 origin. Two progressions arise from the origin. One is a short progression with 40 cm−1 interval. The second is a long anharmonic progression with an interval increasing in higher vibrational state from 30 to 42 cm−1. This increasing spacing suggests that this progression originates from an internal rotational motion, rather than simple low frequency vibration. The quantum chemical calculation for the o-fluorophenol·NH3 cluster in S0 gives two vibrational modes lower than 60 cm−1 in the ground state. One is the out-of-plane bending vibration between ammonia and o-fluorophenol molecules (22 cm−1, see Table 2) and the other is the torsion (or pseudo-rotation) of the ammonia molecule around the hydrogen-bonding, which is calculated as 55 cm−1. We have assigned the long anharmonic progression to the pseudo-rotation and the short progression of 40 cm−1 to the out-of-plane bending.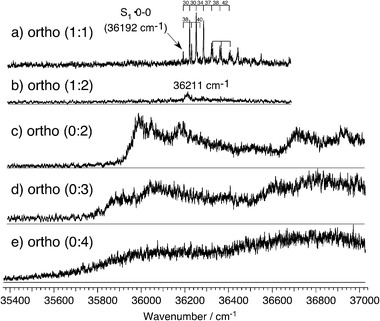 | ||
| Fig. 5 Mass-selected (1 + 1′) REMPI spectra of o-fluorophenol·(NH3)n for (a) the (1 : 1) and (b) the (1 : 2) clusters. The ionization laser ν2 was fixed to 310 nm for both measurements. The action spectra of o-fluorophenol·(NH3)n obtained by monitoring NH4(NH3)n−1+ (n = 2–4) are shown in c–e, respectively. ν2 was fixed at 310 nm for n = 2, and at 355 nm for n = 3 and 4. The delay between the excitation laser ν1 and the ionization laser ν2 was 5 ns for the REMPI measurements and 100 ns for the action spectra. | ||
To confirm the assignment of the anharmonic progression, the observed vibrational levels can be analyzed by a one-dimensional internal rotational motion. This model has been successfully applied to various internal rotational motions.20–23 The potential curve for internal rotation is described by the following function,
 | (1) |
 | (2) |
The internal rotational motion has not been observed in m- and p-fluorophenol·ammonia (1 : 1) clusters, though the same internal rotation of ammonia is possible in these clusters. It suggests that the internal rotational potential curve is not changed much by the electronic excitation in m- and p-fluorophenol·ammonia (1 : 1) clusters. In both clusters, the ammonia molecule is located far from the fluorine atom, no strong interaction is expected, and the ammonia molecule should be a nearly free rotor, regardless of the electronic excitation. In contrast, in the o-fluorophenol·ammonia (1 : 1) cluster, the ammonia molecule is just beside the fluorine substituent and a cyclic hydrogen-bond among –OH⋯N–H⋯F– is expected in the quantum chemical calculation. Such interaction will prevent the ammonia rotation and this validates the one-dimensional rotor analysis with three- and six-fold barriers. This hindrance will be changed drastically by the electronic excitation thus the potential curve for the ammonia rotation can also be changed by the electronic excitation. Therefore the appearance of the ammonia rotational levels suggests the existence of a ammonia-fluorine interaction and its change upon electronic excitation.
The REMPI spectrum obtained by monitoring the ion signal of the o-fluorophenol·(NH3)2 (1 : 2) cluster shows no signal except for a small broad band at 36 211 cm−1 (see Fig. 5b), lying slightly higher in energy than the origin of the (1 : 1) cluster. It is difficult to believe that the larger hydrogen-bonded cluster gives a blue-shift of the origin. At the present time, we have no good explanation for this peak but it should not be assigned to the o-fluorophenol·(NH3)2 cluster. On the other hand, the action spectrum obtained by monitoring NH4(NH3)+ (ortho (0 : 2)) presents broad features in the region lower than the origin of the (1 : 1) cluster (Fig. 5c). We assigned this spectrum to the o-fluorophenol·(NH3)2 cluster and the broad peak at 35 990 cm−1 is assigned to the S1 origin. Broadened peaks are observed at 55, 190, 730 and 920 cm−1 above the origin, and are tentatively assigned to intermolecular bending, intermolecular stretching σ1, intramolecular 11 and 11σ1 combination band, respectively.
The action spectra obtained by monitoring NH4(NH3)n−1+ (n = 3 and 4), denoted ortho (0 : 3) and ortho (0 : 4), are also broad, as shown in Fig. 5d and 5e. The onset of the broad signal is red-shifted in both spectra from the spectrum of the (1 : 2) cluster. In the p-fluorophenol·(NH3)n clusters, sharp vibronic structures are observed not only in the small clusters but also in the larger (1 : 3) and (1 : 4) clusters. For the m-fluorophenol·(NH3)n clusters, the (1 : 4) cluster presents a broadened spectrum but smaller clusters exhibit sharp vibronic structures. The spectral broadening may be related with the excited state hydrogen transfer reaction rate, suggesting that it is faster in o-fluorophenol·(NH3)n clusters.
4.6 Fluorination effect on ESHT and its dependence on the substituent position
We have shown that excited o-, m- and p-fluorophenol clustered with ammonia undergo an hydrogen transfer (ESHT) reaction from the fluorophenol to the ammonia cluster. The fluorine substituent has an induction effect on the σ orbital, that may perturb the ESHT reaction rate by changing the energy of the πσ* state, which promotes the ESHT through a conical intersection with the ππ* state. Here, we will discuss qualitative changes of the ESHT reaction by comparing the REMPI and action spectra in the three fluorophenol·(NH3)2 (1 : 2) clusters. The REMPI spectrum of m-fluorophenol·(NH3)2 shows the same peaks as those in the (0 : 2) action spectrum obtained by monitoring NH4NH3+. It means that the ESHT reaction rate is comparable to the ionization rate, which is determined by the ionization cross-section from S1 and the photon density of the ionization laser. In contrast, the REMPI spectra monitoring the o- and p-fluorophenol·(NH3)2 cluster ions did not show the vibronic structures of the cluster, which can be interpreted as a faster rate for ESHT than for ionization. We used the same intensity for the ionization laser, so the photon density is roughly the same in all the measurements. Thus, if we assume that the ionization cross sections are the same for the three fluorophenol·(NH3)n clusters, ESHT in m-fluorophenol·(NH3)n is thought to be the slowest. The action spectra of m- and p-fluorophenol·(NH3)n show sharp vibronic structures, while that of o-fluorophenol·(NH3)n is broad. If the broadening is due to ESHT, the reaction rate in o-fluorophenol·(NH3)n is faster than that in p-fluorophenol·(NH3)n clusters. Therefore, we roughly estimate that the ESHT reaction between excited fluorophenol and ammonia clusters becomes faster in the order meta-, para- and ortho-.5. Conclusion
Two-color (1 + 1′) REMPI mass spectra of o-, m- and p-fluorophenol·ammonia (1 : n) clusters were measured with a long delay time between the two lasers. The generation of a long-lived species via S1 was assessed by the observation of NH4(NH3)n−1+ with a 100 ns delay after exciting the S1 state. As in the case of phenol·ammonia clusters, excitation to S1 induces an excited state hydrogen transfer reaction from the o-, m- and p-fluorophenol molecule to the ammonia clusters. The S1–S0 transition of o-, m- and p-fluorophenol·ammonia (1 : 1) clusters were measured by the (1 + 1′) REMPI spectra, while larger (1 : n) cluster (n = 2–4) were observed by action spectra that monitor the ionized reaction products NH4(NH3)n−1+. The vibronic spectra observed in m- and p-fluorophenol·ammonia (1 : 1) clusters are tentatively assigned on the basis of vibrational calculations in the ground state. The o-fluorophenol·NH3 (1 : 1) cluster shows an anharmonic progression that is analyzed in terms of one-dimensional rotational motion of the ammonia molecule. The interaction between the ammonia and the fluorine atom, and its change upon electronic excitation are suggested. The broad action spectra of the o-fluorophenol·ammonia (1 ∶ n) clusters (n ≧ 2) suggest the excited state hydrogen transfer is faster than in m- and p-fluorophenol·ammonia clusters. To obtain more details on the changes in ESHT reaction rates, experiments using time-resolved spectroscopy are now in progress. The different reaction rates between o-, m-and p-fluorophenol·ammonia clusters could be due to an effect of the geometry on the electronic states, especially on the πσ* state. Preliminary calculations of the excited state geometries suggest a significant out of plane distortion of the o-fluorophenol·ammonia cluster, which can promote an efficient state mixing between ππ* and πσ* state. Higher level calculations are necessary to understand the different reaction rate of fluorophenols and are now in progress.Acknowledgements
The authors thank Professor Kenro Hashimoto in Tokyo Metropolitan University, Dr Kota Daigoku in Aoyama University and Dr Pierre Carçabal for valuable discussions and helpful comments. This work has been supported in part by a joint Centre National de la Recherche Scientifique/Japan Society for Promotion of Science bilateral program.References
- G. A. Pino, C. Dedonder-Lardeux, G. Grégoire, C. Jouvet, S. Martrenchard and D. Solgadi, J. Chem. Phys., 1999, 111, 10747 CrossRef CAS.
- G. A. Pino, G. Grégoire, C. Dedonder-Lardeux, C. Jouvet, S. Martrenchard and D. Solgadi, Phys. Chem. Chem. Phys., 2000, 2, 893 RSC.
- S. Ishiuchi, M. Saeki, M. Sakai and M. Fujii, Chem. Phys. Lett., 2000, 322, 27 CrossRef CAS.
- S. Ishiuchi, M. Sakai, K. Daigoku, T. Ueda, T. Yamanaka, K. Hashimoto and M. Fujii, Chem. Phys. Lett., 2001, 347, 87 CrossRef CAS.
- S. Ishiuchi, K. Daigoku, M. Saeki, M. Sakai, K. Hashimoto and M. Fujii, J. Chem. Phys., 2002, 117, 7077 CrossRef CAS.
- S. Ishiuchi, K. Daigoku, M. Saeki, M. Sakai, K. Hashimoto and M. Fujii, J. Chem. Phys., 2002, 117, 7083 CrossRef CAS.
- C. Dedonder-Lardeux, D. Grosswasser, C. Jouvet and S. Martrenchard, PhysChemComm, 2001, 1 RSC.
- O. David, C. Dedonder-Lardeux, C. Jouvet, H. Kang, S. Martrenchard, T. Ebata and A. L. Sobolewski, J. Chem. Phys., 2004, 120, 10101 CrossRef CAS.
- H. Kang, C. Dedonder-Lardeux, C. Jouvet, S. Martrenchard, G. Gregoire, C. Desfrancois, J. P. Schermann, M. Barat and J. A. Fayeton, Phys. Chem. Chem. Phys., 2004, 6, 2628 RSC.
- H. Kang, C. Jouvet, C. Dedonder-Lardeux, S. Martrenchard, G. Gregoire, C. Desfrancois, J. P. Schermann, M. Barat and J. A. Fayeton, Phys. Chem. Chem. Phys., 2005, 7, 394 RSC.
- H. Kang, C. Dedonder-Lardeux, C. Jouvet, G. Gregoire, C. Desfrancois, J. P. Schermann, M. Barat and J. A. Fayeton, J. Phys. Chem. A, 2005, 109, 2417 CrossRef CAS.
- A. L. Sobolewski, W. Domcke, C. Dedonder-Lardeux and C. Jouvet, Phys. Chem. Chem. Phys., 2002, 4, 1093 RSC.
- D. Bahat, O. Cheshnovsky, U. Even, N. Lavie and Y. Magen, J. Phys. Chem., 1987, 91, 2460 CrossRef CAS.
- R. Ahlrichs, M. Bär, M. Häser, H. Horn and C. Kölmel, Chem. Phys. Lett., 1989, 162, 165 CrossRef CAS.
- M. Schmitt, C. Jacoby, M. Gerhards, C. Unterberg, W. Roth and K. Kleinermanns, J. Chem. Phys., 2000, 113, 2995 CrossRef CAS.
- C. Ratzer, M. Nispel and M. Schmitt, Phys. Chem. Chem. Phys., 2003, 5, 812 RSC.
- A. Oikawa, H. Abe, N. Mikami and M. Ito, J. Phys. Chem., 1984, 88, 5180 CrossRef CAS.
- M. Ito and A. Oikawa, J. Mol. Struct., 1985, 126, 133 CrossRef CAS.
- A. Oikawa, H. Abe, N. Mikami and M. Ito, Chem. Phys. Lett., 1985, 116, 50 CrossRef CAS.
- H. Ikoma, K. Takazawa, Y. Emura, S. Ikeda, H. Abe, H. Hayashi and M. Fujii, J. Chem. Phys., 1996, 105, 10201 CrossRef CAS.
- K. Suzuki, Y. Emura, S. Ishiuchi and M. Fujii, J. Electron Spectrosc. Relat. Phenom., 2000, 108, 13 CrossRef CAS.
- K. Suzuki, S. Ishiuchi and M. Fujii, Faraday Discuss., 2000, 115, 229 RSC.
- K. Suzuki, S. Ishiuchi, M. Sakai and M. Fujii, J. Electron Spectrosc. Relat. Phenom., 2005, 142, 215 CrossRef CAS.
- J. P. Lewis and J. Laane, J. Mol. Struct., 1972, 12, 427 CrossRef CAS.
- S. J. Humphrey and D. W. Pratt, J. Chem. Phys., 1996, 104, 8332 CrossRef CAS.
- G. I. Gellene and R. F. Porter, J. Phys. Chem., 1984, 88, 6680 CrossRef CAS.
- K. Fuke, R. Takasu and F. Misaizu, Chem. Phys. Lett., 1994, 229, 597 CrossRef CAS.
- K. Fuke and R. Takasu, Bull. Chem. Soc. Jpn., 1995, 68, 3309 CAS.
| This journal is © the Owner Societies 2006 |