A combined Raman, DFT and MD study of the solvation dynamics and the adsorption process of pyridine in silver hydrosols†
Marco
Pagliai
a,
Luca
Bellucci
a,
Maurizio
Muniz-Miranda
a,
Gianni
Cardini
*ab and
Vincenzo
Schettino
ab
aLaboratorio di Spettroscopia Molecolare, Dipartimento di Chimica, Università di Firenze, Via della Lastruccia 3, 50019, Sesto Fiorentino, Florence, Italy
bEuropean Laboratory for Nonlinear Spectroscopy (LENS), Via Nello Carrara 1, 50019, Sesto Fiorentino, Florence, Italy. E-mail: gianni.cardini@unifi.it
First published on 17th October 2005
Abstract
The adsorption of pyridine onto silver colloids has been investigated by Raman spectroscopy experiments and by ab initio DFT and MP2 calculations. The solvation dynamics of the pyridine in water has been studied by a molecular dynamics simulation. The results are compared with the latest available experimental and theoretical data. It is found that the pyridine is essentially hydrogen bonded to one solvent molecule. Calculations based on pyridine–water and pyridine–Ag+ complexes allow the reproduction of the experimentally observed Raman features and explain the adsorption process of the ligand in silver hydrosols.
1. Introduction
Metal nanoparticles dispersed in aqueous media are able to adsorb several different species from the solution, in particular organic molecules. The adsorption is favored by the presence of heteroatoms such as N or O in the molecular structure. The adsorption process in a Ag hydrosol can be monitored by various spectroscopic techniques. In the UV-Vis spectrum an intense band usually occurs around 400 nm, due to the plasmon resonance of the electrons localized at the surface of Ag colloidal particles with 10–20 nm average diameters, as shown in Fig. 1A. The presence of a ligand adsorbed onto the Ag nanoparticles is able to induce a colloidal aggregation, which is detected by the occurrence of a secondary plasmon band at longer wavelengths (Fig. 1B). This spectral feature is more evident for molecules that are chemically adsorbed on the Ag surface.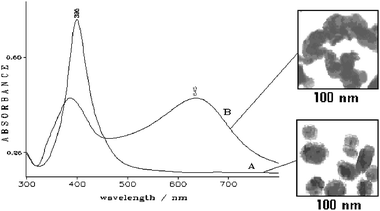 | ||
| Fig. 1 UV-visible absorption spectra of Ag colloids in the absence of ligand (A) and with 10−3 M pyridine (B). The corresponding transmission electron microscopy (TEM) images are reported as insets. | ||
The adsorption of organic ligands can be studied by means of the surface-enhanced Raman scattering (SERS) effect. The molecules adsorbed on nanostructured surfaces of Ag, Au or Cu undergo a huge intensification of their Raman signals. This effect is generally explained on the basis of two different mechanisms:1 the electromagnetic enhancement, and the charge-transfer effect between the molecule and metal. In the first case a giant enhancement of the electric field occurs near the surface of the metal nanoparticles, in the second a resonance effect involves the energy levels of both the metal and molecule. The electromagnetic mechanism is estimated to provide enhancement factors with a magnitude of 106 for the Raman signals of molecules approaching the metal surface, even without a chemical interaction with the latter. The charge-transfer effect provides a further Raman enhancement that can reach a factor of 102 for chemisorbed molecules. Consequently, these exhibit more intense SERS spectra in comparison with physisorbed molecules. In addition, chemisorbed molecules show significant frequency shifts for some bands compared with the corresponding normal Raman bands of the free molecule. This effect is due to the chemisorption that modifies some geometrical parameters and the electron density of the molecule with a consequent perturbation of the force constants. On the other hand, frequency shifts of vibrational bands are also induced by interactions with molecules in the surrounding environment, mainly with those of the solvent. When a molecule is dissolved in aqueous solution, it is solvated by water molecules, but in the presence of suspended metal particles it can adsorb onto the metal surface through a chemical interaction that successfully competes with the aqueous medium. The aim of this work is to evaluate the possible chemisorption of organic ligands onto a Ag hydrosol by a computational approach based on the density functional theory (DFT).2,3 Here, DFT calculations are employed to reproduce the frequency shifts observed in the Raman spectra of aqueous solutions and in the SERS spectra in Ag hydrosols with respect to the normal Raman spectra of the free molecules, i.e. without interactions with the solvent molecules as well as with the Ag nanoparticles. On the basis of a satisfactory evaluation of these frequency shifts, it is possible to find a suitable explanation of the different capabilities of organic ligands to adsorb onto a Ag hydrosol, instead of remaining bound to the water molecules. Actually, DFT calculations allow the evaluation of the structural changes of the molecule upon interaction with water or Ag, as well as the charge-transfer effects. In both solvation and chemisorption processes the formation of charge-transfer complexes has to be taken into account. If the charge-transfer is larger in the molecule–metal interaction than in the molecule–water interaction, a more stable complex is formed on the metal surface. The analysis of the SERS spectra of some organic molecules has been previously performed with ab initio calculations without taking solvent effects into account, and only comparing the frequency shifts on moving from the free organic molecule to the complex with the metal or metallic clusters, both in their ionic or neutral form.4–7 Although this procedure usually allows a correct reproduction of the Raman spectra, the information on the solvent effects is completely neglected. In this work the solvation dynamics of pyridine (Py), which was the first molecule to exhibit SERS enhancement in both an electrochemical cell8 and a Ag hydrosol,9 have been studied by a molecular dynamics (MD) simulation. Although the structural reorganization of the water close to the pyridine molecule has been studied through Monte Carlo simulations,10,11 a dynamical view is still lacking. A detailed description of the H-bond dynamics is essential for characterizing the stability and strength of the H-bond with the solvent and to set up a simple model to explain the Raman spectra of pyridine in water. This model represents a starting point for understanding the SERS spectra of pyridine by DFT calculations. The SERS spectra of pyridine in Ag colloids activated by coadsorbed chloride anions are quite similar to the normal Raman spectrum of the Ag(I) coordination compound,9 suggesting that the interaction with the metal surface closely resembles that of pyridine interacting with a Ag ion. In activated Ag colloids, the presence of active sites constituted by positively-charged Ag atoms has been previously ascertained.12,13
2. Experimental
Stable Ag hydrosols were prepared by the reduction of AgNO3 (99.9999% purity, Aldrich) with an excess of NaBH4 (99.9% purity, Aldrich) in extra-pure distilled water according to Creighton’s procedure,14 and taking precautions to avoid reduction products.15 The addition of 10−3 M NaCl (99.999% purity, Aldrich) ensured a stronger Raman enhancement of pyridine (99.9+% purity, Aldrich) at a concentration of 10−3 M in the Ag hydrosols. Pyridine (1M) was also dissolved in an aqueous and a CCl4 (99.9+% purity, Aldrich) solution. Raman measurements were performed using a Jobin-Yvon HG-2S monochromator, a cooled RCA-C31034A photomultiplier and the 514.5 nm exciting line was supplied by an Ar+ laser with a power of 50 mW. Power density measurements were performed with a power meter (model 362, Scientech, Boulder, CO), giving an ∼5% accuracy in the 300–1000 nm spectral range.Absorption spectra in the 200–800 nm region were measured with a Cary5 Spectrophotometer in order to detect the surface plasmon bands of the Ag colloidal particles. TEM measurements of Ag colloids in the absence and presence of pyridine were performed using a Philips EM 201 instrument with an electron beam emitted at 80 kV, after placing a drop of the colloidal sample onto a carbon–Cu grid.
3. Computational details
The DFT calculations2,3 have been performed with the Gaussian 98 rev. A.7 suite of programs,16 using a combination of the BLYP,17,18 B3LYP19–21 or B3PW9119,22–24 exchange and correlation functionals along with the LANL2DZ25–27 or CEP-31G28–30 basis sets. The LANL2DZ basis set consists of the Dunning–Huzinaga31 full double-ζ contraction on first row elements and Los Alamos pseudopotentials for core electrons in conjunction with a double-ζ contraction for the other elements, whereas the CEP-31G basis set consists of effective core pseudopotentials in conjunction with a double-ζ contraction for the valence electrons. MP232 calculations were also performed with both the CEP-31G and LANL2DZ basis sets, using the Gaussian 98 rev. A.7 suite of programs.16 Structure optimizations, with a very tight criterion, and normal frequencies calculations have been performed using an improved grid in the numerical evaluation of the integrals, INTEGRAL(GRID = 199974). The calculated frequencies have been uniformly scaled using a 0.983 factor for the CEP-31G basis set and a 0.972 factor for the LANL2DZ basis set for both the B3LYP and B3PW91 calculations. For the BLYP calculations, 1.022 and 1.018 scaling factors have been adopted for the CEP-31G and LANL2DZ basis sets, respectively. For all of the MP2 calculations, a 1.01 scaling factor was used. The Raman intensities of the vibrational modes, computed on the basis of the double harmonic approximation, i.e. without taking into account the electric and mechanical anharmonicity, correspond to spatially averaged values according to the usual formulae reported in standard textbooks.33In order to estimate the number of water molecules H-bonded to the N atom of pyridine, a classical molecular dynamics (MD) simulation has been performed, adopting the standard OPLS 11 site potential10 for the solute and modeling the water molecules with the TIP4P potential.34,35 The model system, consisting of one pyridine and 345 solvent molecules in a periodic cubic box with sides of 21.8828 Å (at the experimental density of water, 0.9966 g cm−3), has been thermalized at ∼298 K for ∼50 ps by velocity scaling. The trajectory has been collected for 200 ps, saving the atomic coordinates every 2 fs, with a time step of 0.2 fs in the NVE ensemble. The MD simulations, when keeping all of the molecules rigid, have been performed with the Moldy program.36
4. Raman spectra of pyridine
Fig. 2 shows the Raman spectra of pyridine as a pure liquid, in aqueous and in CCl4 solutions, in comparison with the SERS spectrum of the molecule adsorbed on Ag colloids activated by the addition of chloride anions. All of the Raman spectra are dominated by two very intense bands around 1000 and 1030 cm−1, attributed to the ring breathing and ring trigonal mode, respectively.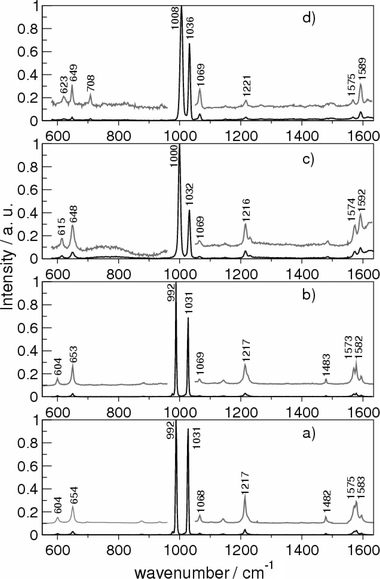 | ||
| Fig. 2 Raman spectra of pyridine in: (a) CCl4 solution, (b) pure liquid; (c) aqueous solution; (d) Ag hydrosol. Excitation line: 514.5 nm. The Raman intensity has been enhanced and reported (grey line) for a better visualization of the spectral features. | ||
Significant frequency shifts of some bands are detected upon going from the liquid to water solutions or to a Ag hydrosol, whereas no appreciable frequency shift occurs in the Raman spectrum in CCl4 solution. This indicates that the interactions with the molecules of an apolar solvent like CCl4 or with other pyridine molecules in the pure liquid can be considered similar when compared with those occurring when pyridine interacts with water molecules or Ag particles. Hence, it is reasonable to compare the DFT calculated frequencies of pyridine in aqueous solutions or Ag hydrosols with the vibrational data in the liquid. Moreover, as shown in Fig. 2, the frequency shifts observed in the SERS spectrum are larger than those observed in the Raman spectrum of the aqueous solution, indicating a stronger interaction with the Ag surface. Finally, the intensity ratio (1.49) between the two strongest bands observed in the Raman spectrum of the liquid at 992 and 1031 cm−1 markedly changes in the Raman spectrum of the aqueous solution (2.06) and is only slightly affected in the SERS (1.45). The DFT calculations are employed here to reproduce the frequency shifts, as well as the relative intensities of the observed bands.
5. Results and discussion
The adsorption of pyridine molecules onto Ag colloids occurs in an aqueous environment and the process is in competition with the pyridine–water solvation. The analysis of the SERS spectra of pyridine (frequency shifts, relative intensities) should be made in comparison with the spectra in water. Therefore, a classical molecular dynamics simulation has initially been performed in order to evaluate the structure and dynamics of pyridine solvation as a basis for the construction of a reliable model for the subsequent DFT calculations. The pair radial distribution functions and the running integration number obtained from 200 ps simulations are reported in Fig. 3.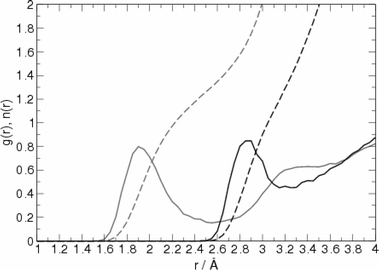 | ||
| Fig. 3 Radial distribution functions (unbroken line) and running integration number (dashed line) for the H⋯N (grey) and O⋯N (black) intermolecular contacts. | ||
The first peak position and coordination number obtained from the H⋯N and O⋯N pair radial distribution functions are in perfect agreement with the latest experimental measurements of Bakó et al.37 In fact, the first H⋯N peak position is in the 1.8–1.9 Å range, in agreement with experiment, and the coordination numbers are 1.3 and 1.4 for the H⋯N and O⋯N contacts, respectively. For comparison, the measured value is 1.2 ± 0.1. This result is a consequence of the presence of only one water molecule directly bound to the N atom of pyridine, with at least another water molecule close to this, as shown in Fig. 4 where a snapshot of the MD simulation is reported.
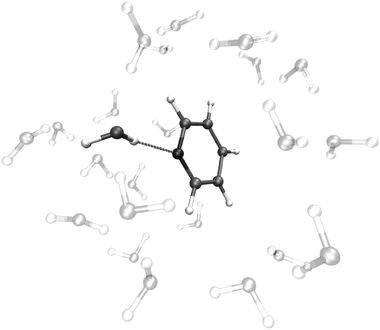 | ||
| Fig. 4 Snapshot of the pyridine molecule H-bonded to a water molecule extracted from the MD simulation. | ||
A specific analysis has been performed to characterize the pyridine–water H-bond in depth, starting from the pair angular distribution function. In fact, the H-bond is a directional interaction38 and its strength depends not only on the interatomic distances but also on the distribution of the HO⋯N angle.
The angular distribution g(θ) related to all of the contacts (top panel of Fig. 5) and the H-bonded molecule (bottom panel of Fig. 5), selected on the basis of H⋯N and O⋯N distances lower than the first peak minimum in their g(r), does not show a minimum at 30° as hypothesized by Fileti et al.,11 but only a flex. The choice of 30° as a limiting value of HO⋯N angle appears to be restrictive. In fact, as can be seen in Fig. 5 (lower panel), the HO⋯N angle distribution extends up to 50°, a quite large value which is, however, very close to that found in pure water.39 Starting from these results it is possible to describe the configurational H-bond space and to obtain the average lifetime related to the formation and breaking of the H-bond. To this end the H-bond can be described by the traditional classical40 approach based on the following geometrical criteria:
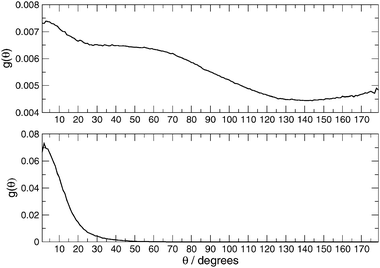 | ||
| Fig. 5 Angular distribution function of the HO⋯N angle. Top panel, all of the contacts. Bottom panel, only H⋯N and O⋯N distances lower than the position of the first minimum in the respective g(r). | ||
1. r(H⋯N) < 2.6 Å
2. r(O⋯N) < 3.3 Å
3. HO⋯N < 50°
Alternatively, the H-bond could be characterized by the function introduced recently to describe the H-bond network in liquid methanol41 and the Cl− + CH3Br reaction in water42
| FjHB = A(r(t)) × B(θ(t)) | (1) |
The values of the parameters re, θe, σr and σθ are directly extracted from the non-normalized pair radial, h(r), and angular distribution function, h(θ).
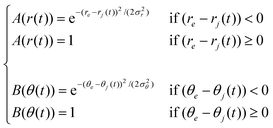
In the above definitions, re is the position of the first peak in the h(r)H⋯N function and θe is the position of the first peak in the h(θ) function, whereas σr and σθ are the half width at half maximum in the h(r) and h(θ) functions, respectively.
Fig. 6 shows the configurational H-bond space described by the two approaches. It is clear from this result that the HO⋯N angle extends to at least 30°. However, larger values of the angle should also be considered according to the classical approach. These arise from the continuous exchange of water molecules that enter or leave the H-bond region.
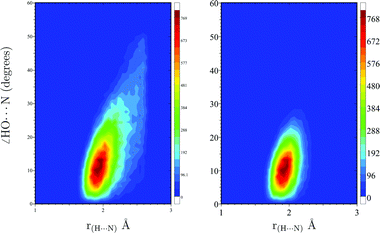 | ||
| Fig. 6 H-bond configurational space obtained with the geometrical criteria (left surface) and with the new weighted function (right surface). A color image of this figure is available in the html. | ||
The continuous exchange of the water molecules bound to pyridine is depicted in Fig. 7 where the H-bonded water molecules, out of 345 molecules of the simulated sample, are identified at each time of the simulation. The lifetime of the H-bond to the pyridine has been calculated to be 359 and 306 fs according to the classical and alternative approach, respectively. These values are the averages of a number of processes with different lifetimes.
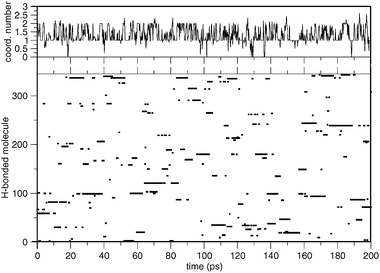 | ||
| Fig. 7 Coordination number averaged every 0.2 ps (upper panel) and water molecules H-bonded to the solute during the simulation (lower panel). All of the solvent molecules in the sample are labeled from 1 to 345. The figure identifies the molecules directly H-bonded to the pyridine during the simulation. | ||
The solvent reorganization in the neighborhood of the N atom during the simulation can be easily observed from Fig. 8, showing that the surface spanned by the O atom of the water molecules closest to the N atom is approximately a spherical cap with a large extension.
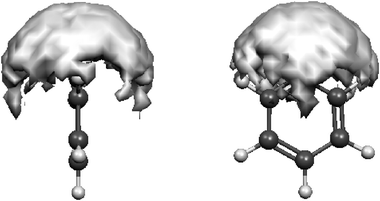 | ||
| Fig. 8 Distribution of the water molecules around the N atom for values of the H⋯N and O⋯N distances and HO⋯N angle that satisfy the geometrical criteria for the presence of the H-bond. | ||
The molecular dynamics simulation described above clearly shows that the pyridine molecule is directly bound to a single water molecule by a strong H-bond. This suggests the adoption of a bimolecular pyridine–water complex as a model to mimic the solvent effect on the structural and vibrational properties of the system. The starting point to validate this model is represented by the recent results of Dkhissi et al.43 and Schlücker et al.44 showing that the B3LYP exchange and correlation functionals in conjunction with the 6-31++G(d,p) basis set satisfactorily reproduce both the structure and the energy of the pyridine–water H-bonded complexes. The accuracy of the B3LYP functional in describing the structural and electronic properties of H-bonded systems has been recently assessed and described by Rabuck and Scuseria.45 In order to compare the frequency shifts moving from the pyridine to the water system and to the pyridine–Ag+ complex, it is not possible to adopt the same computational approach, and a more appropriate basis set is required. To further validate the model and assess the charge-transfer influence on both the vibrational frequencies and Raman intensities, the calculations have also been performed on the pyridine–Ag(0) complex, although the surface active sites of the nanoparticles are considered to be positively charged by the adsorption of Ag+ ions counterbalanced by coadsorbed anions.12,13 Therefore, DFT calculations have been carried out for the above systems using different functionals and the CEP-31G and LANL2DZ basis sets and it turns out that these reproduce the frequency shifts accurately. This further justifies the importance of the direct bonding of a water molecule to the N atom of pyridine.
The optimized structures show the N atom of pyridine involved in the interaction with the water molecules as well as with the Ag ion, as expected when considering the fact that the conjugation between the lone-pair and the π electrons of the aromatic ring rather weakens the basicity of the N atom. The DFT calculation of pyridine bound to one water molecule, even given the simplicity of the model, leads to a H⋯N H-bonding distance (∼1.80 Å) comparable with the first peak position (1.85 Å) obtained from the MD calculations shown in Fig. 3. The calculated Ag–N distance in the pyridine–Ag+ complex (2.17 Å) is similar to that in the Ag(I) coordination compound (2.16 Å),46 whereas the calculated Ag–N distance in the pyridine–Ag(0) complex is rather longer, as reported in Table 1.
| MP2 | BLYP | B3LYP | B3PW91 | |||||
|---|---|---|---|---|---|---|---|---|
| CEP | LANL | CEP | LANL | CEP | LANL | CEP | LANL | |
| H⋯N/Å | 1.877 | 1.871 | 1.811 | 1.805 | 1.803 | 1.800 | 1.765 | 1.770 |
| Ag+⋯N/Å | 2.200 | 2.240 | 2.168 | 2.174 | 2.170 | 2.176 | 2.158 | 2.164 |
| Ag(0)⋯N/Å | 2.378 | 2.447 | 2.428 | 2.483 | 2.434 | 2.484 | 2.400 | 2.454 |
The calculated Ag+⋯N distances are very close to those reported by Wu et al.47 (2.198 Å) and Yang et al.48 (2.196 Å), from DFT and MP2 calculations, respectively.
The vibrational frequencies of pyridine, pyridine–water and pyridine–Ag+ have been calculated using the CEP-31G and LANL2DZ basis set and different functionals. The results are reported in Tables S1 and S2 of the ESI.† The vibrational frequencies calculated at the MP2 level are reported in Table S3 of the ESI.† The average discrepancy between calculated and observed frequencies of pyridine νobs − νcalc is summarized in Table 2, showing that the DFT approach is more efficient, and that the LANL2DZ basis set seems to work better than the CEP-31G basis set.
| CEP-31G/cm−1 | LANL2DZ/cm−1 | |
|---|---|---|
| BLYP | 12.3 | 10.2 |
| B3LYP | 12.8 | 9.3 |
| B3PW91 | 13.3 | 7.4 |
| MP2 | 13.3 | 27.5 |
In Tables 3 and 4 the frequency shifts, observed in the Raman spectrum of the aqueous solutions and in the SERS spectrum with respect to the Raman spectrum of the liquid, are compared with the results of DFT calculations. It can be seen that the general trend of the frequency shift is pretty well reproduced by the approach of this paper. The frequencies of the pyridine–Ag(0) complex are not reported in Tables 3 and 4, but are instead reported in Table S4 of the ESI†, since they do not reproduce the observed frequency shifts and relative Raman intensities.
| Δν liq. Py → water | Δν liq. Py → colloid | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
|
Raman
cm −1 |
Obs.
cm −1 |
Calc. A
cm −1 |
Calc. B
cm −1 |
Calc. C
cm −1 |
Calc. D
cm −1 |
Obs.
cm −1 |
Calc. A
cm −1 |
Calc. B
cm −1 |
Calc. C
cm −1 |
Calc. D
cm −1 |
|
| A1ν(10) | 604 w/m | +11 | +14 | +13 | +15 | +13 | +19 | +38 | +37 | +39 | +39 |
| B2ν(22) | 653 m | −4 | −2 | −2 | −3 | −3 | −4 | −7 | −6 | −6 | −4 |
| A1ν(9) | 992 vvs | +8 | +19 | +17 | +21 | +19 | +16 | +33 | +30 | +34 | +34 |
| A1ν(8) | 1031vs | +1 | +2 | +1 | +1 | +6 | +5 | +13 | +14 | +11 | +31 |
| A1ν(7) | 1069 w | 0 | 0 | 0 | 0 | −1 | 0 | −2 | 0 | +3 | −3 |
| A1ν(6) | 1217 m | −1 | +3 | +3 | +1 | +3 | +4 | +13 | +12 | +12 | +18 |
| A1ν(5) | 1483 w | −3 | +7 | +6 | +6 | +8 | +4 | +15 | +15 | +15 | +21 |
| B2ν(16) | 1573 w/m | +1 | +1 | +2 | +2 | +3 | +2 | −3 | -4 | −3 | +6 |
| A1ν(4) | 1582 m | +10 | +12 | +12 | +9 | +12 | +17 | +26 | +26 | +27 | +36 |
| Δν liq. PY → water | Δν liq. PY → colloid | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
|
Raman
cm −1 |
Obs.
cm −1 |
Calc. A
cm −1 |
Calc. B
cm −1 |
Calc. C
cm −1 |
Calc. D
cm −1 |
Obs.
cm −1 |
Calc. A
cm −1 |
Calc. B
cm −1 |
Calc. C
cm −1 |
Calc. D
cm −1 |
|
| A1ν(10) | 604 w/m | +11 | +6 | +15 | +17 | +16 | +19 | +30 | +40 | +42 | +40 |
| B2ν(22) | 653 m | −4 | −14 | −2 | −2 | −1 | −4 | −19 | −6 | −6 | −2 |
| A1ν(9) | 992 vvs | +8 | +12 | +17 | +20 | +20 | +16 | +26 | +30 | +34 | +35 |
| A1ν(8) | 1031vs | +1 | −4 | +2 | +3 | +6 | +5 | +7 | +17 | +15 | +30 |
| A1ν(7) | 1069 w | 0 | −4 | 0 | 0 | +10 | 0 | −6 | −2 | +1 | +22 |
| A1ν(6) | 1217 m | −1 | −4 | +1 | 0 | +2 | +4 | +6 | +8 | +8 | +14 |
| A1ν(5) | 1483 w | −3 | 0 | +5 | +6 | +8 | +4 | +8 | +11 | +12 | +17 |
| B2ν(16) | 1573 w/m | +1 | +1 | +2 | +3 | +3 | +2 | −3 | −3 | −2 | +4 |
| A1ν(4) | 1582 m | +10 | +12 | +15 | +9 | +11 | +17 | +26 | +22 | +23 | +34 |
The calculated Raman spectra of pyridine, pyridine–water and pyridine–Ag+ using B3LYP with the LANL2DZ basis set are shown in Fig. 9. These should be compared with the experimental spectra of Fig. 2. It can be seen that the agreement is quite satisfactory for not only the peak positions but also their relative intensities. In particular the intensity ratio of the 992 and 1031 cm−1 peaks changes in the B3LYP/LANL2DZ calculation in pretty close agreement with experiment. The calculated Raman spectrum of pyridine–Ag(0) has been shown in the upper panel of the Fig. 9 for comparison. Although the main features of the Raman spectra are accurately reproduced, it is not possible to reproduce the absolute Raman intensities, since the effects due to the atoms that make up the Ag nanoparticles are missing in the present approach.
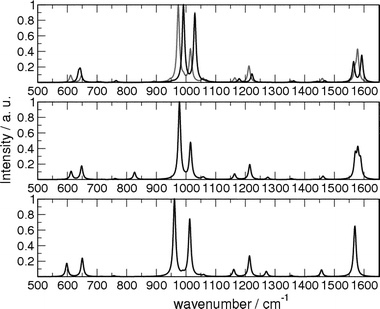 | ||
| Fig. 9 Calculated Raman spectra of (from top to bottom) pyridine–Ag+, pyridine–water and pyridine. The DFT calculations have been performed using the LANL2DZ basis set with the B3LYP functional. The Raman spectrum of pyridine–Ag(0) is also shown (grey line). | ||
The overall results on the frequencies, frequency shifts and relative intensities show that the simple models of the pyridine–water and pyridine–metal interactions are rather appropriately used in a DFT approach to describe the processes of pyridine solvation and chemisorption on metal particles.
In both cases, charge-transfer processes are rather important; in the case of pyridine the charge-transfer effect to the metal particles is much larger than that to the water molecules. The charge-transfer obtained by the natural bond orbital (NBO)49 analysis is summarized in Table 5. It is seen that with the B3LYP functional there is a predicted charge-transfer of 0.130 e− and 0.047 e− to Ag and water, respectively. On the contrary, for the complex with the metal atom, the charge-transfer is slightly lower (0.036 e−) than that in water.
| MP2 | BLYP | B3LYP | B3PW91 | |||||
|---|---|---|---|---|---|---|---|---|
| CEP | LANL | CEP | LANL | CEP | LANL | CEP | LANL | |
| Δq(H2O) /e− | 0.037 | 0.038 | 0.051 | 0.051 | 0.047 | 0.047 | 0.054 | 0.052 |
| Δq(Ag+) /e− | 0.078 | 0.075 | 0.178 | 0.175 | 0.130 | 0.131 | 0.122 | 0.122 |
| Δq(Ag(0)) /e− | −0.004 | 0.001 | 0.048 | 0.052 | 0.034 | 0.036 | 0.029 | 0.031 |
These results give a picture of the chemisorption process of an organic ligand such as pyridine onto colloidal Ag. The tendency to adsorb on Ag, removing the coordinated water molecule, is linked to the formation of a more stable complex with the metal surface. In this respect the stabilization energy related to the formation of the pyridine–water and pyridine–Ag+ complexes are −32.8 and −229.8 kJ mol−1, respectively. Since the binding energy for the pyridine–Ag(0) complex is only −19.71 kJ mol−1, the chemisorption process will occur on positively charged sites of the colloid, as experimentally hypothesized.12,13 As a consequence, the variation of the electronic structure is much more pronounced in pyridine–Ag+ than in pyridine–water. This behavior is illustrated in Fig. 10, where the electron density differences for the two complexes show that the electronic structure of the ring is strongly perturbed by presence of the Ag+ ion.
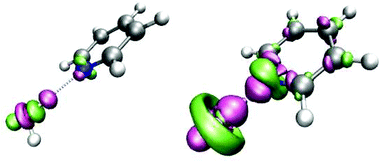 | ||
| Fig. 10 Electron density differences for the pyridine–water (left) and pyridine–Ag+ complexes (right) at the B3LYP/LANL2DZ level of theory with an isosurface value of 0.005 a.u. A color image of this figure is available in the html. | ||
If charge-transfer and chemisorption are closely related, the in-plane ring deformation modes should undergo the largest frequency shifts, since the atoms of the ring are those most involved in the electron rearrangement. As shown in Tables 3 and 4, the largest frequency shifts really occur for these vibrational modes, i.e. ring bending, ν(10); ring breathing, ν(9); trigonal ring deformation, ν(8); and quadrant ring deformation, ν(4).
6. Conclusions
In the past, the interpretation of the SERS effect has been mainly based on the modeling of the metal surface, where the ligand molecules were considered to be physisorbed.50,51 On the other hand, DFT calculations have been successfully employed in the study of the Raman enhancement related to the chemisorption process, which results in the formation of complexes between the ligand and active sites on the metal surface.6,43,47,48,52–56 In the present work we have shown that the DFT method is not only a convenient approach for correctly interpreting the SERS data, but it also allows the description of the possible chemisorption of a ligand in a Ag hydrosol, thus exhibiting Raman enhancements on the basis of the molecule–metal ion charge-transfer effect.It has been observed that water binding and the model adopted for the description of the solvation play a central role in the understanding of both frequency shifts and chemisorpton processes. Molecular dynamics simulations in conjunction with ab initio calculations have allowed the development and justification of a simple model that reproduces the experimental findings.
References
- A. Otto, Light Scattering in Solids, Springer-Verlag, Berlin, 1984, vol. IV Search PubMed.
- W. Kohn and L. J. Sham, Phys. Rev., 1965, 140, A1133 CrossRef.
- P. Hohenberg and W. Kohn, Phys. Rev., 1964, 136, B864 CrossRef.
- R. F. Aroca, R. E. Clavijo, M. D. Halls and H. B. Schlegel, J. Phys. Chem. A, 2000, 104, 9500–9505 CrossRef CAS.
- G. Cardini and M. Muniz-Miranda, J. Phys. Chem. B, 2002, 106, 6875–6880 CrossRef CAS.
- G. Cardini, M. Muniz-Miranda and V. Schettino, J. Phys. Chem. B, 2004, 108, 17007–17011 CrossRef CAS.
- M. Baia, L. Baia, W. Kiefer and J. Popp, J. Phys. Chem. B, 2004, 108, 17491–17496 CrossRef CAS.
- M. Fleischmann, P. J. Hendra and A. J. McQuillan, Chem. Phys. Lett., 1974, 26, 163–166 CrossRef CAS.
- M. Muniz-Miranda, Chem. Phys. Lett., 2001, 340, 437–443 CrossRef CAS.
- W. L. Jorgensen and N. A. McDonald, J. Mol. Struct. (THEOCHEM), 1998, 424, 145–155 CrossRef CAS.
- E. E. Fileti, K. Coutinho, T. Malaspina and S. Canuto, Phys. Rev. E, 2003, 67, 061504 CrossRef.
- H. Wetzel and H. Gerisher, Chem. Phys. Lett., 1980, 76, 460–464 CrossRef CAS.
- M. Muniz-Miranda and G. Sbrana, J. Raman Spectrosc., 1996, 27, 105–110 CrossRef CAS.
- J. A. Creighton, C. G. Blatchford and M. G. Albrecht, J. Chem. Soc., Faraday Trans. 2, 1979, 75, 790–798 RSC.
- M. Muniz-Miranda, N. Neto and G. Sbrana, J. Mol. Struct., 1986, 143, 275–278 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, V. G. Zakrzewski, J. A. Montgomery, Jr., R. E. Stratmann, J. C. Burant, S. Dapprich, J. M. Millam, A. D. Daniels, K. N. Kudin, M. C. Strain, O. Farkas, J. Tomasi, V. Barone, M. Cossi, R. Cammi, B. Mennucci, C. Pomelli, C. Adamo, S. Clifford, J. Ochterski, G. A. Petersson, P. Y. Ayala, Q. Cui, K. Morokuma, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. Cioslowski, J. V. Ortiz, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. Gomperts, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, C. Gonzalez, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, J. L. Andres, C. Gonzalez, M. Head-Gordon, E. S. Replogle and J. A. Pople, GAUSSIAN 98 (Revision A.7), Gaussian Inc., Pittsburg, PA, 1998 Search PubMed.
- A. D. Becke, Phys. Rev. A, 1988, 38(6), 3098–3100 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B, 1988, 37(2), 785–789 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, J. Phys. Chem., 1994, 98, 11623–11627 CrossRef CAS.
- R. H. Hertwig and W. Koch, Chem. Phys. Lett., 1997, 268, 345–351 CrossRef CAS.
- J. P. Perdew and Y. Wang, Phys. Rev. B, 1992, 45, 13244–13249 CrossRef.
- J. P. Perdew, J. A. Chevary, S. H. Vosko, K. A. Jackson, M. R. Pederson, D. J. Singh and C. Fiolhais, Phys. Rev. B, 1993, 48, 4978 CrossRef CAS.
- J. P. Perdew, J. A. Chevary, S. H. Vosko, K. A. Jackson, M. R. Pederson, D. J. Singh and C. Fiolhais, Phys. Rev. B, 1992, 46, 6671–6687 CrossRef CAS.
- P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 270–283 CrossRef CAS.
- W. R. Wadt and P. J. Hay, J. Chem. Phys., 1985, 82, 284–298 CrossRef CAS.
- P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 299–310 CrossRef CAS.
- W. J. Stevens, H. Basch and M. Krauss, J. Chem. Phys., 1984, 81, 6026–6033 CrossRef.
- W. J. Stevens, M. Krauss, H. Basch and P. G. Jasien, Can. J. Chem., 1992, 70, 612–630 CAS.
- T. R. Cundari and W. J. Stevens, J. Chem. Phys., 1993, 98, 5555–5565 CrossRef CAS.
- T. H. Dunning, Jr and P. J. Hay, Modern Theoretical Chemistry, Plenum, New York, 1976, vol. 3 Search PubMed.
- C. Møller and M. S. Plesset, Phys. Rev., 1934, 46, 618–622 CrossRef CAS.
- E. B. Wilson, Jr, J. C. Decius and P. Cross, Molecular Vibrations, Dover Publications Inc., New York, NY, 1980, p. 10014 Search PubMed.
- W. L. Jorgensen, J. Chandrasekhar, J. D. Madura, R. W. Impey and M. L. Klein, J. Chem. Phys., 1983, 79, 926–935 CrossRef CAS.
- W. L. Jorgensen and J. D. Madura, Mol. Phys., 1985, 56, 1381–1392 CAS.
- K. Refson, Comput. Phys. Commun., 2000, 126, 309–328.
- I. Bakó, G. Pálinkás, J. C. Dore, H. Fischer and P. Jóvári, Chem. Phys. Lett., 2004, 388, 468–472 CrossRef CAS.
- G. R. Desiraju and T. Steiner, The Weak Hydrogen Bond, Oxford University Press, Oxford, New York, Tokyo, 1999 Search PubMed.
- P. Raiteri, A. Laio and M. Parrinello, Phys. Rev. Lett., 2004, 93, 087801 CrossRef.
- D. C. Rapaport, Mol. Phys., 1983, 50, 1151–1162 CAS.
- M. Pagliai, G. Cardini, R. Righini and V. Schettino, J. Chem. Phys., 2003, 119, 6655–6662 CrossRef CAS.
- M. Pagliai, S. Raugei, G. Cardini and V. Schettino, J. Mol. Struct. (THEOCHEM), 2003, 630, 141–149 CrossRef CAS.
- A. Dkhissi, L. Adamowicz and G. Maes, J. Phys. Chem. A, 2000, 104, 2112–2119 CrossRef CAS.
- S. Schlücker, R. K. Singh, B. P. Asthana, J. Popp and W. Kiefer, J. Phys. Chem. A, 2001, 105, 9983–9989 CrossRef.
- A. D. Rabuck and G. E. Scuseria, Theor. Chem. Acc., 2000, 104, 439–444 CrossRef CAS.
- S. Menchetti, G. Rossi and V. Tazzoli, Rend. Ist. Lomb. Accad. Sci. Lett., A: Sci. Mat. Fis. Chim. Geol., 1970, 104, 309–316 Search PubMed.
- D.-Y. Wu, B. Ren, Y.-X. Jiang, X. Xu and Z.-Q. Tian, J. Phys. Chem. A, 2002, 106, 9042–9052 CrossRef CAS.
- Y.-S. Yang, W.-Y. Hsu, H.-F. Lee, Y.-C. Huang, C.-S. Yeh and C.-H. Hu, J. Phys. Chem. A, 1999, 103, 11287–11292 CrossRef CAS.
- A. E. Reed, L. A. Curtiss and F. Weinhold, Chem. Rev., 1988, 88, 899–926 CrossRef CAS.
- S. Corni and J. Tomasi, Chem. Phys. Lett., 2001, 342, 135–140 CrossRef CAS.
- S. Corni and J. Tomasi, J. Chem. Phys., 2002, 116, 1156–1164 CrossRef CAS.
- T. Tanaka, A. Nakajima, A. Watanabe, T. Ohno and Y. Ozaki, Vib. Spectrosc., 2004, 34, 157–167 CrossRef CAS.
- A. Vivoni, R. L. Birke, R. Foucault and J. R. Lombardi, J. Phys. Chem. B, 2003, 107, 5547–5557 CrossRef CAS.
- D. Y. Wu, M. Hayashi, Y. J. Shiu, K. K. Liang, C. H. Chang, Y. L. Yeh and S. H. Lin, J. Phys. Chem. A, 2003, 107, 9658–9667 CrossRef CAS.
- D. Y. Wu, M. Hayashi, S. H. Lin and Z. Q. Tian, Spectrochim. Acta, Part A, 2004, 60, 137–146 CrossRef CAS.
- T. Osaki, T. Yoshikawa, Y. Satoh and R. Shimada, J. Raman Spectrosc., 2005, 36, 199–207 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Calculated and measured vibrational frequencies of pyridine, pyridine–water, pyridine–Ag+ and pyridine–Ag(0) (Tables S1–S4). See DOI: 10.1039/b509976e |
| This journal is © the Owner Societies 2006 |